Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody†
Jason P.
Gleghorn
a,
Erica D.
Pratt
b,
Denise
Denning
c,
He
Liu
d,
Neil H.
Bander
d,
Scott T.
Tagawa
e,
David M.
Nanus
e,
Paraskevi A.
Giannakakou
e and
Brian J.
Kirby
*a
aSibley School of Mechanical & Aerospace Engineering, College of Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: bk88@cornell.edu; Tel: +6072554379
bDepartment of Biomedical Engineering, College of Engineering, Cornell University, Ithaca, NY 14853, USA
cDepartment of Physical Sciences, Dublin City University, Dublin, Ireland
dDepartment of Urology, Weill Cornell Medical College, Cornell University, New York, NY 10065, USA
eDivision of Hematology & Oncology, Weill Cornell Medical College, Cornell University, New York, NY 10065, USA
First published on 16th November 2009
Abstract
Geometrically enhanced differential immunocapture (GEDI) and an antibody for prostate-specific membrane antigen (PSMA) are used for high-efficiency and high-purity capture of prostate circulating tumor cells from peripheral whole blood samples of castrate-resistant prostate cancer patients.
Prostate cancer circulating tumor cells (PCTCs) are often found in the blood of patients suffering from metastatic prostate cancer.1,2 While these PCTCs are rare, as few as one cell per 109 hematologic cells in blood,3,4 they are theorized to contribute to metastatic progression.3,5 Currently, the enumeration of PCTCs is used clinically as a prognostic indicator of patient survival.2,6,7 Capture of peripheral blood PCTCs may enable early clinical assessment of metastatic process and chemotherapeutic response, as well as genetic and pharmacological evaluation of cancer cells.
Current approaches to isolate circulating tumor cells are complex and produce low yields and purity.5 Existing commercial and research devices for the immunocapture of rare cancer cells use EpCAM antibodies,2,8,9 which capture many circulating endothelial cells and large numbers of leukocytes. As a result, purity of captured cells is widely variable and often below 50%. In addition, while previous devices use 3D antibody-coated surfaces for immunocapture,8,9 these devices are not designed to induce a size-dependent collision frequency. Devices focused on size-dependent particle transport are typically focused on sorting,10 separation,11,12 or filtration.13
In this communication, we demonstrate high-efficiency and high-purity capture of PCTCs from peripheral blood samples of castrate-resistant prostate cancer patients using an antibody for prostate-specific membrane antigen (PSMA), a highly prostate-specific cell-surface antigen.14 In addition, we describe a theoretical framework for the use of staggered obstacle arrays to create size-dependent particle trajectories that maximize PCTC–wall interactions while minimizing the interactions of other blood cells. We term this technique geometrically enhanced differential immunocapture (GEDI). Glass and silicon devices were fabricated and chemically functionalized to localize a monoclonal antibody (J591) that has high binding avidity to and specificity for epitopes on the extracellular PSMA domain15 and minimal nonspecific binding with PSMA negative cells. Cell capture efficiency and purity were determined via capture of cultured prostate cancer cell populations spiked in PBS and in whole blood and capture of PCTCs from patient blood samples.
Prior to cell capture, 2D experiments were conducted to determine capture specificity of the surface immobilized immunochemistry to PSMA expressing cells. Glass coverslips were functionalized with an amine-terminated surface via a two-step process using 4% (v/v) MPTMS (3-mercaptopropyl trimethoxysilane) [Sigma-Aldrich, St. Louis, MO] in ethanol solution for 45 min followed by incubation (20 min) with a 1 mM GMBS (N-γ-maleimidobutyryloxy succinimide ester) [Pierce Biotechnology, Rockford, IL] in ethanol solution. Next, a layer of NeutrAvidin [Pierce Biotechnology] was covalently attached to the surface by incubating (45 min) with 25 ng ml−1 in phosphate-buffered saline (PBS). Lastly, we immobilized (10 μg mL−1 for 30 min) a biotinylated monoclonal antibody, J591, for prostate circulating tumor cell capture. The resulting J591 mAb functionalized coverslips were incubated with one of three different cell suspensions in PBS: a prostate cancer cell line expressing PSMA [LNCaP], a prostate cancer cell line that does not express PSMA [PC3], or peripheral blood mononuclear cells isolated from healthy control patients [PBMC]. Following the 15 min incubation, the coverslips were gently rinsed and imaged using a microscope. Capture specificity for PSMA+ cells was verified by counting 20 distinct observation areas with 15 ± 3 LNCaPs captured per field compared with 1 ± 0.5 PC3 and 2 ± 1 PBMCs (Fig. S1, ESI†).
The GEDI μdevice geometry was designed to maximize streamline distortion and thus bring desired cells in contact with the immunocoated obstacle walls for capture. Blood is a dense heterogeneous cell suspension consisting of cells of various sizes ranging from approximately 4 to 18 μm in size.16 PCTCs, in contrast, are larger and range from 15 to 25 μm in diameter.16 Relative obstacle alignment was chosen so that the displacement caused by cell impact with obstacles (which ranges from zero to one cell radius) increases the likelihood of future cell impacts for large cells more than for small cells. Thus when cell-obstacle impact does not lead to capture, larger cells are displaced onto streamlines that impinge onto the next obstacle, while smaller cells are displaced onto streamlines that do not impinge (Fig. 1A). Cell advection was modeled in silico (computational details in the ESI†) to determine obstacle array geometries that optimize PCTC–wall interactions and minimize wall shear forces to maximize PCTC capture. For a given obstacle geometry, the frequency of cell–wall collisions is a function of cell size (Fig. 1B). Obstacle shape and/or array geometries determine a tunable cell diameter threshold whereby larger cells have significantly more cell–wall collisions compared with smaller cells. This feature of the GEDI μdevice may increase cell capture population purity by decreasing opportunities for non-target blood cells to interact with immunocoated surfaces. The GEDI μdevice designs used in these studies consist of approximately 5000 circular or octagonal posts (80 μm diameter) in a 100 μm deep by 8 mm wide by 25 mm long channel. The posts form a regular array with 100 μm gaps and each subsequent row is shifted by 7 μm.
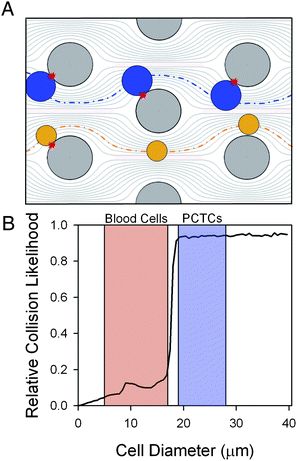 | ||
| Fig. 1 In geometrically-enhanced differential immunocapture (GEDI), obstacles distort streamlines, leading to cell–wall impact. Positioning of obstacles (large grey circles) is chosen to differentially cause cell–wall impacts for target cells (A). Larger cells (PCTCs and LNCaPs, black circles) are displaced onto streamlines that impinge on the next obstacle, while smaller cells (small grey circles) convect through the device with fewer interactions. In silico simulation of cell advection through the flow field predicts the frequency of cell–wall collisions over a range of cell sizes (B). Shaded regions indicate the general size range for hematologic cells and PCTCs respectively. | ||
In order to characterize the GEDI μdevice performance, target cell capture efficiency and purity was determined for experiments using fluorescently labeled model prostate tumor cell populations (detailed cell handling methods in the ESI†). LNCaPs were fluorescently labeled with a standard cell labeling kit [Invitrogen, Eugene, OR], counted on a hemacytometer, and resuspended in either PBS supplemented with 1% bovine serum albumin (BSA) or whole blood from healthy donors at concentrations of 150–220 cells mL−1. The labeled cell suspensions were processed through the GEDI μdevice at 1 mL h−1 followed by a PBS rinse at the same flow rate for 10 min. Captured cells were then enumerated by scanning the entire device area using fluorescence microscopy. The GEDI μdevices captured 97 ± 3% of the spiked LNCaP cells from PBS and 85 ± 5% from blood (Fig. 2). In addition to performing with capture efficiencies from unprocessed whole blood that are approximately 40% higher than other one-step microfluidic immunoreactive devices,8 the μdevice also captured a cell population from whole blood that was 68 ± 6% pure. The 37% increase in capture purity with the GEDI μdevice compared to similar capture methods may be attributed to the combined effects of using a high specificity mAb, J591, and the geometry of the GEDI μdevice.
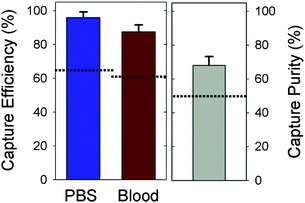 | ||
| Fig. 2 Fluorescently labelled PSMA positive LNCaP cells were spiked into either phosphate-buffered saline (PBS) or whole blood. Cell suspensions were processed by GEDI μdevices and capture efficiency (# captured cells/spiked cells × 100) and capture purity (# target cells/captured cells × 100) was calculated. Data displayed as mean +/− SD, n = 7. Dotted reference lines represent the maximum values from one-step microfluidic immunocapture processes for prostate cancer CTCs found in the literature.8 | ||
Following performance validation, human PCTCs from whole peripheral blood samples of patients diagnosed with castrate-resistant prostate cancer were captured and enumerated. GEDI devices processed 1 mL samples of whole blood obtained under university IRB-approved protocols. The devices were rinsed with PBS for 10 min to remove unbound cells, and FITC-conjugated J591 was used to fluorescently label PSMA+ captured cells in situ. Cells were enumerated with a microscope as described above. While direct comparison of PCTC density and purity between individual patients is impossible due to the variability in individual patient cancer progression, PCTCs were found in 90% of samples tested (n = 20) (Fig. 3B). To assess the repeatability of the μdevice to capture native PCTCs from patients, one blood sample was well mixed and split into 4 aliquots of one millilitre each. Each aliquot was processed by different devices, with the number of captured PCTCs averaging 27 ± 4 cells and a capture purity of 62 ± 2% (Fig. 3A). This constitutes a PCTC enrichment of approximately 109 as compared to the other cells in the system and demonstrates high reproducibility of PCTC capture from whole blood using J591 mAb and the current geometry.
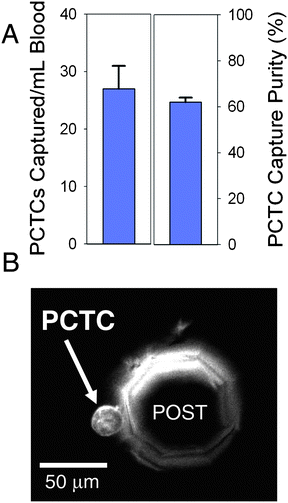 | ||
| Fig. 3 Peripheral whole blood samples taken from a patient with castrate-resistant prostate cancer were processed with GEDI μdevices. PCTCs were enumerated and the capture purity was calculated (A). Data is displayed as mean +/− SD, n = 4 aliquots of an individual patient sample. PCTCs were captured for 90% of patient samples analyzed (n = 20). A representative image is shown of a GEDI μdevice with a single PCTC captured on the wall of an octagonal post (B). | ||
In summary, this work describes a GEDI array functionalized with prostate-specific J591 mAb and demonstrates its use for the high-efficiency capture of PCTCs from peripheral blood samples of castrate-resistant prostate cancer patients. Leveraging the fluid mechanical design to control cell transport combined with high antibody specificity to target cells has demonstrated both exceptionally high cell capture efficiencies and highly pure captured cell populations. Compared to reported work,2,8 this approach yields a 40% improvement in both capture efficiency and capture purity producing significant enrichment of rare cell populations from unprocessed clinical whole blood samples. While the isolation of PCTCs has been demonstrated in these studies, the design of GEDI arrays can be applied to any transport problem that requires differential particle–surface interaction frequencies within heterogeneous-sized colloidal suspensions. Potential applications for the μdevices described herein include capture of PCTCs to study the molecular mechanisms of clinical drug resistance, profiling of individual patients' PCTCs in order to generate personalized chemotherapeutics, as well as real-time monitoring of chemotherapeutic efficacy using a non-invasive blood-based assay.
Acknowledgements
The authors thank Sung Hoon Hong for acquisition of clinical samples. This work was performed in part at the Cornell NanoScale Facility, which is supported by the National Science Foundation (Grant ECS-0335765) and the Cornell Nanobiotechnology Center. Funding for these studies was provided by a Prostate Cancer Foundation Creativity Award, the New York State Foundation for Science, Technology, and Innovation NYSTAR grant, the NIH Clinical & Translational Science Center, the David H. Koch Foundation, and the Robert H. McCooey Cancer Research Fund.References
- W. Allard, J. Matera, M. Miller, M. Repollet, M. Connelly, C. Rao, A. Tibbe, J. Uhr and L. Terstappen, Clin. Cancer Res., 2004, 10, 6897–6904 CrossRef.
- D. Danila, G. Heller, G. Gignac, R. Gonzalez-Espinoza, A. Anand, E. Tanaka, H. Lilja, L. Schwartz, S. Larson, M. Fleisher and H. Scher, Clin. Cancer Res., 2007, 13, 7053–7058 CrossRef CAS.
- E. Racila, D. Euhus, A. Weiss, C. Rao, J. McConnell, L. Terstappen and J. Uhr, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4589–4594 CrossRef CAS.
- R. Krivacic, A. Ladanyi, D. Curry, H. Hsieh, P. Kuhn, D. Bergsrud, J. Kepros, T. Barbera, M. Ho, L. Chen, R. Lerner and R. Bruce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10501–10504 CrossRef CAS.
- V. Zieglschmid, C. Hollmann and O. Bocher, Crit. Rev. Clin. Lab. Sci., 2005, 42, 155–196 Search PubMed.
- H. Scher, X. Jia, J. de Bono, M. Fleisher, K. Pienta, D. Raghavan and G. Heller, Lancet Oncol., 2009, 10, 233–239 CrossRef CAS.
- J. de Bono, H. Scher, R. Montgomery, C. Parker, M. Miller, H. Tissing, G. Doyle, L. Terstappen, K. Pienta and D. Raghavan, Clin. Cancer Res., 2008, 14, 6302–6309 CrossRef CAS.
- S. Nagrath, L. Sequist, S. Maheswaran, D. Bell, D. Irimia, L. Ulkus, M. Smith, E. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. Balis, R. Tompkins, D. Haber and M. Toner, Nature, 2007, 450, 1235 CrossRef CAS.
- A. Adams, P. Okagbare, J. Feng, M. Hupert, D. Patterson, J. Gottert, R. McCarley, D. Nikitopoulos, M. Murphy and S. Soper, J. Am. Chem. Soc., 2008, 130, 8633–8641 CrossRef CAS.
- B. Hawkins, A. Smith, Y. Syed and B. Kirby, Anal. Chem., 2007, 79, 7291–7300 CrossRef CAS.
- L. Huang, E. Cox, R. Austin and J. Sturm, Science, 2004, 304, 987–990 CrossRef CAS.
- A. Mohan and P. Doyle, Phys. Rev. E, 2007, 76(4 Pt 1) Search PubMed.
- N. Wong, H. Kahn, L. Zhang, S. Oldfield, L. Yang, A. Marks and M. Trudeau, J. Clin. Oncol., 2005, 23, 32S–32S.
- S. Chang, Rev. Urol., 2004, 6(Suppl 10), S13–18 Search PubMed.
- S. Chang, V. Reuter, W. Heston, N. Bander, L. Grauer and P. Gaudin, Cancer Res., 1999, 59, 3192–3198 CAS.
- S. Zheng, H. Lin, J. Liu, M. Balic, R. Datar, R. Cote and Y. Tai, J. Chromatogr., A, 2007, 1162, 154–161 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental details and Fig. S1. See DOI: 10.1039/b917959c |
| This journal is © The Royal Society of Chemistry 2010 |