Precompetitive preclinical ADME/Tox data: set it free on the web to facilitate computational model building and assist drug development
Sean
Ekins
*abc and
Antony J.
Williams
*d
aCollaborations in Chemistry, 601 Runnymede Ave, Jenkintown, PA 19046, USA. E-mail: ekinssean@yahoo.com; Fax: +1 215 481 0159; Tel: +1 269 930 0974
bDepartment of Pharmaceutical Sciences, University of Maryland, Baltimore, MD 21202, USA
cDepartment of Pharmacology, University of Medicine and Dentistry of New Jersey, Robert Wood Johnson Medical School, Piscataway, NJ 08854, USA
dRoyal Society of Chemistry, 904 Tamaras Circle, Wake Forest, NC 27587, USA. E-mail: antony.williams@chemspider.com; Fax: +1 919 300 5321; Tel: +1 919 201 1516
First published on 10th November 2009
Abstract
Web-based technologies coupled with a drive for improved communication between scientists have resulted in the proliferation of scientific opinion, data and knowledge at an ever-increasing rate. The increasing array of chemistry-related computer-based resources now available provides chemists with a direct path to the discovery of information, once previously accessed via library services and limited to commercial and costly resources. We propose that preclinical absorption, distribution, metabolism, excretion and toxicity data as well as pharmacokinetic properties from studies published in the literature (which use animal or human tissues in vitro or from in vivo studies) are precompetitive in nature and should be freely available on the web. This could be made possible by curating the literature and patents, data donations from pharmaceutical companies and by expanding the currently freely available ChemSpider database of over 21 million molecules with physicochemical properties. This will require linkage to PubMed, PubChem and Wikipedia as well as other frequently used public databases that are currently used, mining the full text publications to extract the pertinent experimental data. These data will need to be extracted using automated and manual methods, cleaned and then published to the ChemSpider or other database such that it will be freely available to the biomedical research and clinical communities. The value of the data being accessible will improve development of drug molecules with good ADME/Tox properties, facilitate computational model building for these properties and enable researchers to not repeat the failures of past drug discovery studies.
Introduction
Biomedical research is fast moving towards a collaborative network of chemists and biologists and making knowledge available to the masses, enabling rapid sharing of information.1–4 Yet, pharmaceutical scientists (biologists and chemists in particular) commonly find themselves overwhelmed by the availability of information on the web, in primary commercial databases such as CAS Scifinder (http://www.cas.org/), journals and, commonly, a plethora of internally developed systems inside their companies. From another perspective, that of the academic or those in the financially constrained developing countries, biology and chemistry information has long been limited by the tolls associated with accessing commercial databases. Even the calculation of relatively simple molecular properties (such as lipophilicity) has, up until very recently, required knowledge and ownership of informatics software. Structure searching of such chemically aware databases used to be restricted to specialists but now with user friendly web-based tools, even the biologist or biomedical researcher (who is likely to be chemistry naïve) can find such tools of value for searching for interesting molecules from any of the commercial vendors. Today we find a major limitation in the availability of biological information related to chemical structures. For example the understanding of absorption, distribution, metabolism, excretion and toxicity (ADME/Tox) data5–7 for drugs and molecules evaluated as drug candidates is provided in individual commercial databases such as Symyx' Metabolite and Toxicity (http://www.symyx.com/products/databases/bioactivity/index.jsp), Prous Ensemble (http://www.prous.com/products/) and Aureus Auroscope databases (http://www.aureus-pharma.com/Pages/Products/Aurscope.php) as representative examples.8As an example, in the world of chemistry there are tens if not hundreds of chemical structure databases, many containing molecules of biological interest, yet until recently there was no single way to search across them. There are databases of curated literature data, chemical vendor catalogs, molecular properties, environmental data, toxicity data, analytical data, etc. The only way to know whether a specific piece of information is available for a chemical structure is to have simultaneous access to all of these databases as well as journals and other commercial resources for mining and integrating them. Since many of these databases are commercial there is no way to easily determine the availability of information either within these or in the open access databases. The availability of molecule databases such as PubChem (http://pubchem.ncbi.nlm.nih.gov/) has changed scientists’ expectations of web-based databases in many ways but only goes part way to inform us about our chemical universe, and in particular those molecules we might be interested in for their pharmaceutical properties. For example, while the web has provided improved access to chemistry-related information there has not been an online central resource allowing integrated chemical structure combined with biology data-searching of chemistry or biology databases, chemistry articles, patents and web pages such as blogs and wikis. For example the commercial company Collaborative Drug Discovery, Inc. (CDD, http://www.collaborativedrug.com)3 has integrated data from the NIMH Psychoactive Drug Screening Program (PDSP, http://pdsp.med.unc.edu/indexR.html).9–11 This dataset includes >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compound structures associated with the biology data in one place. Previously there had been links from the PDSP database to PubChem only. This suggests to us an enormous opportunity to link the diverse biological data residing in other databases, patents and publications, with molecular structures.
000 compound structures associated with the biology data in one place. Previously there had been links from the PDSP database to PubChem only. This suggests to us an enormous opportunity to link the diverse biological data residing in other databases, patents and publications, with molecular structures.
There are many freely available chemical compound databases on the web and they assume different forms.2,12 These files generally contain the chemical identifiers in the form of chemical names (systematic and trade) and registry numbers. Since the files are assembled in a heterogeneous manner the resulting data are plagued with inconsistencies and data quality issues. Such an approach to gathering and merging data is a far cry from that taken by commercial database vendors who manually gather and curate data. While the commercial databases offer curated data there is certainly a price barrier to accessing the information. A number of the free online resources are also manually curated and, as will be discussed later, can offer as high a quality as the commercial offerings. These resources are, however, constructed with a specific focus in mind and therefore commonly number in the low thousands of structures rather than the millions available in the larger online databases. Meanwhile, there are several large online database resources offering access to valuable data and knowledge.
The quality of chemical information in the public domain is generally quite low. This does not mean that the data are not of value but that care needs to be taken in the nature of the provider as an authority. There is, of course, no central body responsible for the quality of data in the public domain. Databases of chemical structure information besides PubChem include ChemIDPLus (http://chem.sis.nlm.nih.gov/chemidplus/) and ChembioFinder (http://chembiofinder.cambridgesoft.com/chembiofinder/SimpleSearch.aspx) which are commonly looked upon as authorities in terms of reliable information. However, these sources are also aggregators of information and are at risk of perpetuating errors from the original public data and depositions. Errors in structure–identifier pairs are common and inaccurate structure representations, specifically with regard to stereochemistry, proliferate across many databases. A definitive description of the challenges regarding quality in public domain databases and the rigorous processes required to aggregate quality data were provided by Richard et al.13 During their assembly of the EPA DSSTox databases (http://www.epa.gov/NCCT/dsstox/) they assembled the chemical structures, chemical names and CAS registry numbers for over 8000 chemicals from numerous toxicity databases. The data they extracted were carefully curated and validated using multiple public information sources.
The creation, hosting and support of a curated compound database containing structures of chemical and biological interest with integrated content is an expensive enterprise. Historically these databases have been built as a result of hundreds if not thousands of man years of rigorous and exacting human effort and then, for some of the original founders in this domain, migrated onto computer systems. In the development of these systems some host organizations have created sizeable revenues. The hosting of large databases, the text-based searching of immense amounts of data and the ability to disseminate complex forms of graphical information via standard protocols provide an opportunity for future disruptive offerings in this domain whereby online offerings can also become authorities and, with the support and input of the community, can offer the benefits of crowdsourcing for enhancing the data.
Certain areas of the scientific literature, while still of high value, can become antiquated fairly quickly. With the capabilities of Internet-based searching and direct access to abstracts for the majority of publishers, even a rudimentary text search can expose articles previously unavailable except through an abstracting service. Search engines will increasingly be utilized for first level searches specifically because they are simple to use, they are fast and are free. With chemically searchable patents also available online, at no charge, the landscape for scientists searching for information is more open than ever. We believe if there are data of interest to be located then Internet search engines will enable it.
The premier curated database offerings of today have an interesting if not challenging future ahead of them. Their value-added enhancements of the distributed data must be significant enough to warrant an investment in their services. As expressed earlier, the quality of the data resulting from curation is significant but the longevity of that distinguishing factor moving forward is questionable. Roboticized recognition and conversion of chemical names to chemical structures can dramatically shift this domain and efforts have already been demonstrated in applications with patents and publications. Should the quality of these efforts reach a sufficient standard then today's publishers’ business models will definitely be at risk, as free content will be greatly expanded compared with today.
Examples of free key databases with molecule information
The following represent examples of some free databases containing molecule information of interest to chemists and biologists in drug discovery:PubChem
The highest profile online database is certainly PubChem which was launched by the NIH in 2004 to support the ‘New Pathways to Discovery’ component of their roadmap initiative.14 PubChem archives and organizes information about the biological activities of chemical compounds into a comprehensive biomedical database and is the informatics backbone for the initiative, intended to empower the scientific community to use small molecule chemical compounds in their research. PubChem Compound contains over 25 million unique structures and provides biological property information for each compound. The majority of databases discussed in this article now use two primary identifiers in their systems—the CAS registry number (commercial) and a PubChem ID number (non-commercial). This alone indicates a shift in equality of commercial versus public compound repositories. For now, PubChem remains focused on its initial intent to support the National Molecular Libraries Initiative.DSSTox
The EPA distributed structure-searchable toxicity (DSSTox) database project15,16 provides a series of documented, standardized and fully structure-annotated files of toxicity information. The initial intention for the project was to deliver a public central repository of toxicity information to allow for flexible analogue searching, SAR model development and the building of chemical relational databases. In order to ensure maximum uptake by the public and allow users to integrate the data into their own systems the DSSTox project adopted the use of the common standard file format (SDF) to include chemical structure, text and property information. The DSSTox datasets are among the most highly curated public datasets available and likely the reference standard in publicly available structure-based toxicity data.eMolecules (http://www.emolecules.com/)
This website offers a free online database of almost 8 million unique chemical structures. The database is assembled from data supplied by over 150 suppliers and provides a path to identifying a vendor for a particular chemical compound. Their database was recently enhanced by providing access to NMR, MS and IR spectra from Wiley-VCH for over 500000 compounds via ChemGate, a fee-based service. eMolecules also provides links to many sources of data for spectra, physical properties and biological data.
DrugBank (http://www.drugbank.ca/)
This is a manually curated resource17 assembled from the collection information of a series of other public domain databases and enhanced with additional data generated within the laboratories of the hosts. The database aggregates both bioinformatics and cheminformatics data and combines detailed drug data with comprehensive drug target (i.e. protein) information. The database contains FDA approved small molecule and biotech drugs as well as experimental drugs. Each record in the database, known as a DrugCard, has >80 data fields. The information is split into drug/chemical data and drug target or protein data and many data fields are linked to other databases. The database supports extensive text, sequence, chemical structure and relational query searches.PharmGKB (http://www.pharmgkb.org/)
This database brings together human genetic variation data that impact drug response, including curated primary genotype and phenotype data, variants and gene–drug–disease relationships from the literature, along with key genes and drug pathways.18 The database also contains drugs with annotations related to some pharmacokinetic (PK) properties but it does not appear that you can query by these properties, molecular structure, or output the data in a format needed for computational modeling.Wikipedia (http://en.wikipedia.org/wiki/Main_Page)
This certainly represents an important shift in the future access of information associated with small molecules. At present there are approximately 6000 articles with a chembox or drugbox. The detailed information offered regarding a particular chemical or drug can be excellent. The advantage of a wiki is that changes can be made within a few keystrokes and the quality is immediately enhanced. This community curation process makes Wikipedia a very important online chemistry resource whose impact will only expand with time.ZINC (http://zinc.docking.org/index.shtml)
This is a free database of commercially available compounds for virtual screening.19,20 The library contains over 10 million molecules, each with a 3D structure and gathered from the catalogs of compounds from vendors. All molecules in the databases are assigned biologically relevant protonation states and annotated with molecular properties. The database is available for free download in several common file formats and a web-based search page, including a molecular drawing interface which allows the database to be searched.SureChem (http://www.surechem.org/)
This site provides chemically intelligent searching of a patent database containing over 8 million US, European and World patents. Using extraction heuristics to identify chemical and trade names and conversion of the extracted entities to chemical structures using a series of name to structure conversion tools, SureChem has delivered a database of over 10 million individual chemical structures. The free access online portal allows scientists to search the system based on structure, substructure or similarity of structure, as well as the text-based searching expected for patent inquiries.ChemSpider (http://www.chemspider.com/)
ChemSpider2,12 was initially developed as a hobby project by a small group of dedicated cheminformatics specialists. The intention was to aggregate and index available sources of chemical structures and their associated information into a single searchable repository and make it available to everybody, at no charge. ChemSpider was unveiled to the public in March 2007 with the intention of “building a structure centric community for chemists”. ChemSpider has grown into a resource containing over 21 million unique chemical structures. The data sources have been gathered from chemical vendors as well as commercial database vendors and publishers and members of the Open Notebook Science community. ChemSpider has also integrated the SureChem patent database collection of structures to facilitate links between the systems. The database can be queried using structure/substructure searching and alphanumeric text searching of both intrinsic and predicted molecular properties. The ChemSpider developers also added virtual screening results using the LASSO similarity search tool to screen the ChemSpider database against all 40 target families from the Database of Useful Decoys (DUD) dataset.ChemSpider has enabled unique capabilities relative to the primary public chemistry databases. These include real-time curation of the data, association of analytical data with chemical structures, real-time deposition of single or batch chemical structures (including with activity data) and transaction-based predictions of physicochemical data. The system developers have also made available a series of web services to allow integration to the system for the purpose of searching the system as well as generation of InChI identifiers and conversion routines.
The system also integrates text-based searching of open access (OA) articles. The index is expected to increase dramatically as they extract chemical names from OA articles and convert the names to chemical structures using name to structure conversion algorithms. These chemical structures will be deposited back to the ChemSpider database thereby facilitating structure and substructure searching in concert with text-based searching.
ChemSpider has a focus on, and commitment to, community curation and ease of use (Fig. 1). The social community aspects of the system demonstrate the potential of this approach. The team have committed to the release of a Wiki-like environment for further annotation of the chemical structures in the database, a project they term WiChempedia. They will utilize both available Wikipedia content and deposited content from users to enable the ongoing development of community curated chemistry.
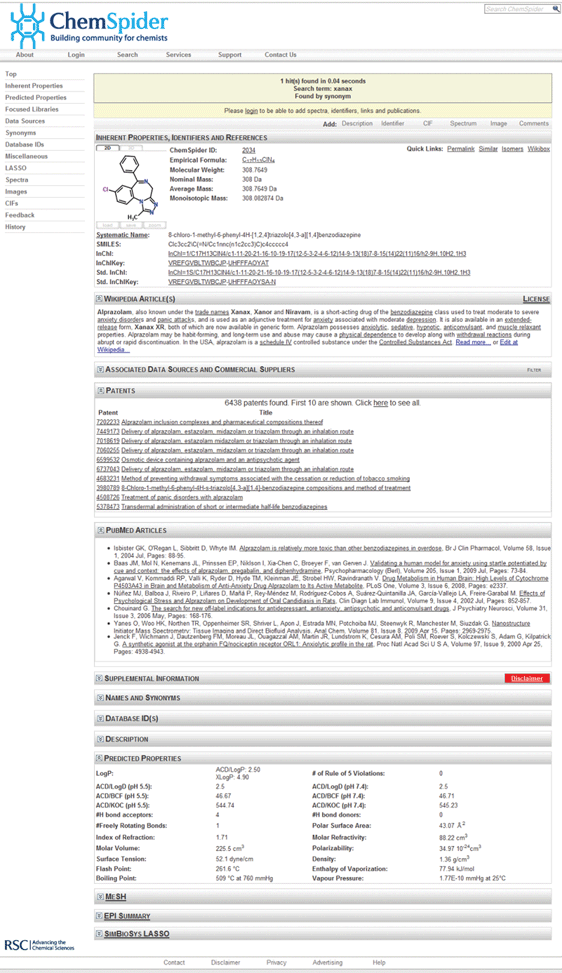 | ||
| Fig. 1 Screenshot of a molecule record in ChemSpider. The record for Xanax shows the header of the related Wikipedia article, links to 10 patents (with over 6400 available), a series of PubMed articles (the long list is truncated for this figure) and a series of predicted properties. The full record is available here: http://www.chemspider.com/2034. | ||
ChemSpider was acquired in May 2009 by the Royal Society of Chemistry and will continue to grow in its reach into the chemistry, biology and biomedical research communities with a number of specific missions:
(1) Improving the quality of available information. With millions of indexed compounds ChemSpider has enabled a community-based curating process to help in improving the association between a chemical compound and a set of identifiers (systematic names, trade names, synonyms and registry numbers).
(2) Increased access to chemistry-related information. There are many types of data and information that can be associated with chemical compounds and made available to the benefit of the chemistry community. As an example of this the association of analytical data and the integration to patent searches have been demonstrated and the integration to QSAR-based modeling is presently in progress.
(3) Provide access to online tools and services. ChemSpider already serves up the online prediction of certain chemical properties for chemists to take advantage of, and a number of software algorithms provided by collaborators will be added into the system. Web services such as the recently exposed InChI and OpenBabel services will continue to be made available as a service to the community.
ChemSpider is acknowledged by scientists as a valuable resource for understanding chemistry.21 ChemSpider can be linked with other software and databases from other groups (academia or industry). For example Collaborative Drug Discovery, Inc. (http://www.collaborativedrug.com) recently provided links to ChemSpider for molecules in this database. This enables the users to resource more information about their molecules (Fig. 2).
 | ||
| Fig. 2 A screenshot from the Collaborative Drug Discovery database showing the ChemSpider link below a molecule in the EPA ToxCast dataset. | ||
There are a multitude of other examples of databases and Wikis linking to ChemSpider. These include Wikipedia, PubChem and many others. Other databases such as WikiProteins and GeneWiki are presently developing their integration links to ChemSpider. There have also been several applications of ChemSpider for generating structure–activity relationships. An example of this application22 used ChemSpider to provide structures and molecule properties for a human drug metabolizing enzyme. It should be noted that ChemSpider allows the user to download structures of interest and molecular properties so these could be used in other computational or analysis software (Fig. 1). A second example used ChemSpider to derive molecular properties for machine learning models to predict biopharmaceutical characteristics of drugs.23 A third application used ChemSpider to follow-up a molecule selected by pharmacophore searching of vendor databases as a potential pregnane X receptor antagonist (a potential target for modulating anticancer drug metabolism, transport, etc.). Substructure searching in ChemSpider indicated additional molecules of interest for testing which were validated in vitro and shown to have activity.24 The above examples illustrate how the content in ChemSpider is useful to the scientific community involved in drug discovery and how free connectivity between tools via the web may enable a much broader impact. It is likely that collaborative software in this space will also require links to ChemSpider as a minimum. ChemSpider has seen good growth in the number of users considering there has been no investment in publicity, now averages over 6000 unique visitors per day and continues to be described frequently in publications and presentations.1,2,12,25
One current use for ChemSpider could be to find out as much about a compound as possible as researchers can eliminate undesirable leads early in the lead generation process by quickly accessing information on the pharmacological effects, side effects and drug–drug interactions for similar compounds or compound classes of interest, as well as their corresponding metabolites. For example for the compound gefitinib (Iressa®), what preclinical information exists? A search was initiated through ChemSpider and produced one hit with the results shown in Fig. 3. The results display for gefitinib includes the chemical structure, a series of intrinsic and predicted properties, links to a number of original data sources for associated information and a number of alphanumeric identifiers, some of which are validated. A number of names, database IDs and synonyms connect to Wikipedia via the [Wiki] link, links to patents are immediately viewable and any articles containing gefitinib or other synonyms in the title or abstract are linked through to PubMed.
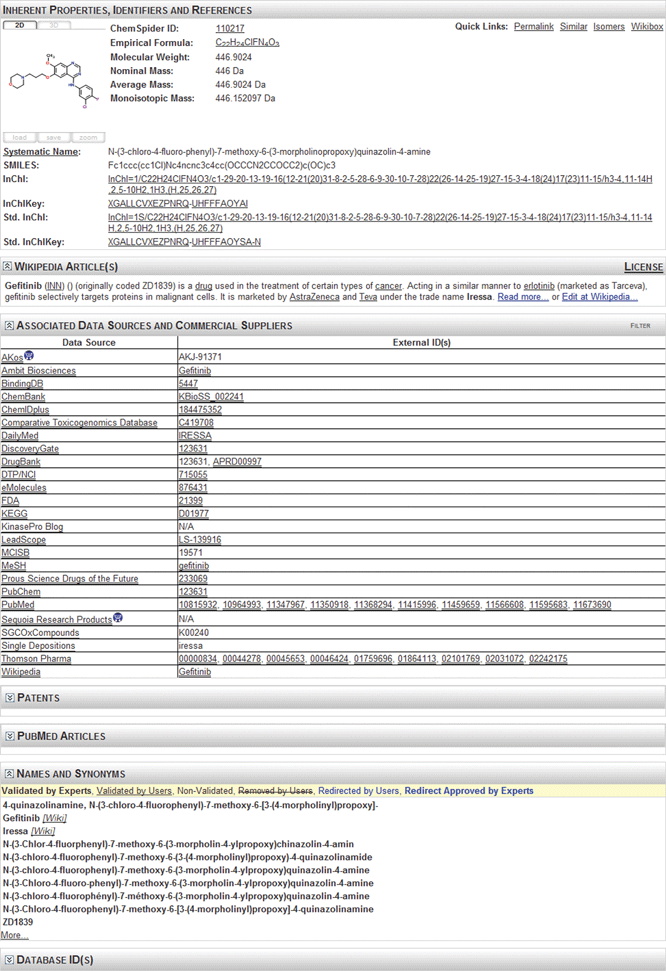 | ||
| Fig. 3 The results of a text search on “gefitinib”. The record shows the structure, intrinsic properties, systematic name and identifiers (InChI String and InChI Key) and links to multiple data sources. The header from the Wikipedia article is shown. The bolded names show manually validated identifiers. | ||
The list of data sources shown in the figure relates to various forms of information. Each is marked with the type of information associated with each data source to assist the user in deciding what data to examine. Each source listed in the data source column is hyperlinked to a description of the depositor. Where possible the entries in the external ID column have been hyperlinked to external information. By combining a search of PubChem, PubMed, DrugBank, ChemSpider and SureChem, it is possible to obtain (fairly quickly) access to a majority of the published data on this compound. This includes data such as drug safety information, toxicology, pharmacology, metabolic pathways, metabolites, synthetic routes, patents and suppliers. Interrogating the data across multiple systems is, however, challenging and time-consuming and integration would be valuable to compare/compete with commercial databases.
Commercial preclinical ADME/Tox databases and the precompetitive space
The major commercial vendors of preclinical data relating to molecules of interest include the Prous Ensemble® database which provides information on more than 127000 bioactive compounds in the drug research and development pipeline relating to over 275000 references to the biomedical and congress literature and more than 33000 patent families cited. A second product, the Aureus AurSCOPE® ADME/DDI (drug–drug interactions), is a fully annotated, structured database containing biological and chemical information on metabolic properties of drugs. The same company has a database for the potassium channel human ether-à-go-go related gene (hERG). This channel is particularly important pharmaceutically as many drugs interact and cause hERG-related cardiotoxicity. Numerous blockbuster drugs have recently been removed from the market due to QT syndrome side effects, an abnormality associated with the hERG and associated channels.26 In addition they have the AurSCOPE® Nuclear Receptor database and a pharmacological activity profiler called AurPROFILER™ which rapidly conducts thorough searches across all individual AurSCOPE Target Knowledge Databases or AurSCOPE Global Pharmacology Space to rapidly identify target, cell or drug/compound profiles. Results are displayed as interactive “heat maps” for easy visualization and navigation of the pharmacological space—the target or cell.
An additional commercial database is the PharmaPendium from Elsevier (https://www.pharmapendium.com/) which captures data from the FDA freedom of information documents and EMEA “EPAR” approval documents. This database has a large amount of preclinical and clinical data, uses the medical dictionary for regulatory activities (MedRA) standardized terminology and is structure/substructure searchable.
There is a movement towards collaborations between biomedical organizations both industrial and academic that are precompetitive in nature covering areas such as cheminformatics, toxicology, preclinical toxicology and beyond. Examples include those organized by the Health and Environmental Sciences Institute (HESI, http://www.hesiglobal.org/i4a/pages/index.cfm%3Fpageid%20%3D%203279), the Pistoia Alliance, (http://pistoiaalliance.org), the Critical Path Institute (C-Path, http://www.c-path.org/), the Drug Safety Executive Council (DSEC, http://www.drugsafetycouncil.org/pages/42_dsec_mission.cfm), Enlight Biosciences (http://www.enlightbio.com/content/about-enlight/) and Innovative Medicines Initiative (IMI, http://imi.europa.eu/index_en.html).27,28 We would argue that ADME/Tox data are also precompetitive data and should be made freely available on the web as a resource for all scientists. ADME/Tox information (we use the term broadly to include everything from in vivo and in vitro preclinical data) are data that are ultimately provided for registration with regulatory bodies and become available in package inserts for drugs or in widely distributed publications. Generating these data is costly and in many cases data are reproduced by different groups when comparing their own proprietary compounds with a competitor compound (which may not be widely known). This is entirely unnecessary. Why not share these data? It would certainly enable the industry to quickly understand ADME/Tox liabilities with different classes of compounds targeting a specific indication and enable the generation of computer models for these properties.
We propose that the scientific community should tackle the lack of public databases that contain preclinical ADME/Tox or pharmacokinetic data. This can be achieved by either creating a new database or preferably expanding the preexisting freely available ChemSpider database with all of the ADME/Tox as well as pharmacokinetic properties available from studies published in the literature (which use animal or human tissues or from in vivo studies). At the same time the number of links (and therefore connectivity) to other currently available databases available on the web should be increased.
How to create a freely available ADME/Tox database
It is one thing to propose the construction of such an ADME/Tox database and another to actually execute on this vision. For those that may take up the challenge it is perhaps worth considering at least one strategy for how the scientific community could build such a resource:(1) Identify all available publications containing ADME/Tox and PK properties data relating to molecular structures tested in animal or human tissues in vitro or in vivo. Mine the data from these publications relating to ADME/Tox and PK properties.
(2) Clean and organize data from these publications e.g. relate by species, tissue, cell types and capture experimental conditions using manual curation, create an ontology.
(3) Provide a means for other scientists to update and include new ADME/Tox and PK properties.
(4) Encourage pharmaceutical companies to publish their previously ‘unpublished preclinical data’ in exchange for access to a duplicate of the database of ADME/Tox and PK data for their own in-house efforts for internal deployment.
(5) As an example of the development and value of such a database, ADME/Tox and PK computational models could be built, validated and provided over the web for free (to the academic community).
Such data-mining could be achieved by using PubMed, Google Scholar, Highwire, SureChem and other available software to search journals, patents and the web. It will be necessary to capture the individual publications and provide links to electronic sources (whether open or commercial). We would suggest specifically annotating information like those in Table 1 for the major important enzymes like cytochrome P450s which are involved in clinically relevant drug–drug interactions, for example capturing substrate and inhibitor data.22 For nuclear receptors we could capture agonist and antagonist data which would be of most interest.24,29 A simple hierarchy could be used in data curation and also in the final data schema (Table 2). Further consultation with experts in this particular domain of schema development may be necessary for such a project. The database should be able to be updated by an array of users (the community) and also be integrated to other web-based databases such as clinical trial databases. These databases could link from a compound name to the structure in, for example, ChemSpider to show preclinical properties (enzymes inhibited, pharmacokinetic data, etc.). Providing the database behind company firewalls may be necessary for companies to use the tool for their in-house and private searches and mining rather than sending queries over the Internet.
| ADME/Tox data | Estimated amounts of data | Data types |
|---|---|---|
| Cytochrome P450s and other enzymes e.g. phase II | 1000's of data points | K m, Ki, and IC50 |
| Transporters (e.g. P-gp, BCRP) | Several hundreds to 1000 | IC50, some substrate data |
| Ion channels (hERG) | <1000 for individual channels | IC50—different cell types |
| Nuclear receptors (e.g. PXR) | <1000 for individual receptors (48 receptors in human) | EC50 and fold activation |
| Pharmacokinetics | 1000's for drugs and failed candidates in different species | AUC, Tmax, etc. |
Uses for the database
Having captured the majority of published ADME/Tox and PK data it will be possible to generate consistent datasets (e.g. for a single property like CYP3A4 inhibition) that can then be used with external algorithms to generate predictive models. These models could be automatically developed and updated30 as new data are added to the database, with tools and descriptors that can then be implemented back into web-based software as predictors. Such models could possibly be facilitated by commercial pipelining tools such as Pipeline Pilot (Accelrys, San Diego, CA) which has been widely used with Bayesian modeling methods.31,32 In addition to generating such models it may be possible to derive simple rules for some of the ADME/Tox properties. It is likely the greatest value of such a database will be as a historic reference source for scientists in drug discovery and prevent repetitive experiments on the same compound.Discussion
The technologies supporting chemistry, while immature, are fast developing to support chemical structures and reactions, analytical data support, and integration to related data sources via supporting software technologies. Communication in chemistry is already witnessing a new revolution. The diversity of information available online is expanding at a dramatic rate and a shift to publicly available resources offers significant opportunities in terms of the benefit to science and society. Biomedical researchers today have access to hundreds of thousands of chemistry, biology and clinical articles via searches on platforms including PubMed, Google Scholar and ChemSpider. While the general nature of text-based searches provides a familiar environment for chemists to search and review their results, a chemist's natural affinity for communicating via chemical structures demands the need to perform searches in their “natural language”. Ask a chemist their preferred manner for searching chemistry databases and you will generally receive a response pointing to structure-based searching. There are certainly commercial solutions to provide chemical structure-based searches of literature and patent data (CAS, Infochem and Symyx to name but a few) as well as a myriad of solutions for managing in-house organizational data collections. The challenge is finding chemistry—specifically chemical structures across the web in databases described above and in the thousands of books and journals.A number of organizations generate sizeable revenues from the creation of chemistry databases for the life sciences industry. The Chemical Abstracts Service alone generates annual revenue in excess of $250 million dollars. The total annual fees for accessing this information when other companies are included into the calculation will significantly exceed this figure. The primary advantage of commercial databases is that they have been manually examined by skilled curators, addressing the tedious task of quality data-checking. Certainly, the aggregation of data from multiple sources, both historical and modern, from multiple countries and languages and from sources not available electronically, is a significant enhancement over what is available via an Internet search alone. The question remains for how long will this remain an issue?
CAS and their CAS registry numbers (RNs) have played a dominant role in managing a curated registry of chemical entities and related chemical and biological literature. Their proprietary registration system does not link to chemical structures in the public domain and their business model is likely at risk. However, the scientific community as a whole is likely to reap increasing benefits from the growing number of free access services and content databases. While commercial vendors generally have a highly moderated release cycle of new functionality and capabilities, online services tend to move at a much faster pace adding new capabilities, resolving issues and adding fresh technologies on a rolling basis. This type of drive both excites present users and draws new users to the expanding offerings. Academics in particular are likely to have an increased focus on the use of free access databases and tools as it is demonstrative of the new found freedom of information. The benefit for them of course is reduced expenditures for the commercial offerings. This is further exaggerated in developing countries where free access systems are the primary resources for information since commercial offerings are simply out of reach due to price barriers. One could also imagine the development of such tools being funded by a micropayment system for those willing to do this.
In terms of data quality issues, the Internet generation has already demonstrated a willingness to curate, modify and enhance the quality of content as modeled by Wikipedia. With the appropriate enhancements in place, online curation and markup of the data in real-time can quickly address errors in the data as has already been demonstrated by the ChemSpider system.
Increasing access to free and open access databases of both chemistry and biological data is certainly impacting the manner by which scientists access information. These databases are additional tendrils in the web of Internet resources that continue to expand in their proliferation of freely accessible data and information, such as patents, open and free access peer-reviewed publications and software tools for the manipulation of chemistry-related data. As data-mining tools expand in their capabilities and performance, the integration of chemistry and biology databases is likely to offer even greater opportunities to benefit the process of drug discovery. As these databases grow in both their content and their quality, there may be challenging times ahead with regard to the commercial business models of publishers versus the drive towards more freely available data.
In summary, we have proposed that there should be an effort to build a structure centric community for biomedical researchers with key information relevant to drug discovery which is precompetitive. We believe a free database of preclinical properties will accelerate ADME/Tox and PK computational model building, prevent different groups from repeating the same experiments, reducing the number of animal experiments, reducing the biological and chemical reagents used and generally benefit the whole biomedical research community. While there are databases that specialize in maintaining the absolute privacy of the researchers’ data, there is a growing movement by some scientists for the open dissemination of their data (e.g. Open Notebook Science, http://usefulchem.wikispaces.com/). The provision of the ChemSpider database currently fits with this model in that data can be published to the community. It is important to consider some opportunities and limitations of such a free database. By providing actual experimental preclinical data in the database it could also be used for validation of other computational models e.g. those already integrated in ChemSpider or other databases/tools developed by third parties. These molecules could represent test sets which would also be of value to the biomedical research community in general. A major limitation is to capture the public information as most data will be in publications. Having access to many online journal subscriptions relevant to ADME/Tox and PK data, e.g. ASPET journals, Wiley, Springer, Elsevier and Nature Journals for example, will be essential. These academic and commercial publishers are likely responsible for the majority of the data that appear in this domain. It may be possible to negotiate with such publishers to get access to their historic ADME/Tox data for this database in return for links to the original data source. One could limit the data collected to the recent few decades with the assumption that more recent publications will contain more relevant data. Going forward publishers could require that authors deposit their ADME/Tox data in the database as a condition of publication. This is analogous to how protein structures are deposited in the Protein DataBank, or microarray data are deposited in various databases. Once data are uploaded and available in the database such as ChemSpider, public support, recognition and validation would be obtained via publication, web page blogs, invited oral presentations to conferences, etc. This is critically important for the information to gain maximum visibility and to be evaluated by the experts. As the target audience here is predominantly biomedical researchers it will be important to present such a database at key conferences which may raise awareness with the maximum number of researchers. This would also be a critical way to capture new and previously unpublished data. In conclusion, the ADME/Tox and PK database curation project proposed here could cost effectively and extensively leverage the existing ChemSpider database and the cheminformatics expertise built to date.
Conflicts of interest statement
SE consults for Collaborative Drug Discovery Inc. and is a member of the ChemSpider Advisory Group. AJW is employed by the Royal Society of Chemistry which owns ChemSpider and associated technologies.Acknowledgements
SE acknowledges Collaborative Drug Discovery Inc and Accelrys for providing access to their software.References
- A. J. Williams, Crowdsourcing, collaborations and text mining in a world of open chemistry, 2008, http://www.slideshare.net/AntonyWilliams/crowdsourcing-collaborations-and-text-mining-in-a-world-of-open-chemistry-presentation Search PubMed.
- A. J. Williams, Internet-based tools for communication and collaboration in chemistry, Drug Discovery Today, 2008, 13, 502–506 CrossRef CAS.
- M. Hohman, K. Gregory, K. Chibale, P. J. Smith, S. Ekins and B. Bunin, Novel web-based tools combining chemistry informatics, biology and social networks for drug discovery, Drug Discovery Today, 2009, 14, 261–270 CrossRef CAS.
- D. S. Bailey and E. D. Zanders, Drug discovery in the era of Facebook—new tools for scientific networking, Drug Discovery Today, 2008, 13, 863–868 CrossRef.
- S. Ekins, B. J. Ring, J. Grace, D. J. McRobie-Belle and S. A. Wrighton, Present and future in vitro approaches for drug metabolism, J. Pharmacol. Toxicol. Methods, 2000, 44, 313–324 CrossRef CAS.
- S. Ekins, C. L. Waller, P. W. Swaan, G. Cruciani, S. A. Wrighton and J. H. Wikel, Progress in predicting human ADME parameters in silico, J. Pharmacol. Toxicol. Methods, 2000, 44, 251–272 CrossRef CAS.
- S. Ekins and P. W. Swaan, Computational models for enzymes, transporters, channels and receptors relevant to ADME/TOX, Rev. Comput. Chem., 2004, 20, 333–415 Search PubMed.
- L. J. Jolivette and S. Ekins, Methods for predicting human drug metabolism, Adv. Clin. Chem., 2007, 43, 131–176 Search PubMed.
- R. T. Strachan, G. Ferrara and B. L. Roth, Screening the receptorome: an efficient approach for drug discovery and target validation, Drug Discovery Today, 2006, 11, 708–716 CrossRef CAS.
- K. A. O'Connor and B. L. Roth, Finding new tricks for old drugs: an efficient route for public-sector drug discovery, Nat. Rev. Drug Discovery, 2005, 4, 1005–1014 CrossRef CAS.
- B. L. Roth, E. Lopez, S. Beischel, R. B. Westkaemper and J. M. Evans, Screening the receptorome to discover the molecular targets for plant-derived psychoactive compounds: a novel approach for CNS drug discovery, Pharmacol. Ther., 2004, 102, 99–110 CrossRef CAS.
- A. J. Williams, A perspective of publicly accessible/open-access chemistry databases, Drug Discovery Today, 2008, 13, 495–501 CrossRef CAS.
- A. M. Richard, L. Swirsky Gold and M. C. Nicklaus, Chemical structure indexing of toxicity data on the Internet: Moving toward a flat world, Curr. Opin. Drug Discovery Dev., 2006, 9, 314–325 CAS.
- N.I.o.H, Office of Portfolio Analysis and Strategic Initiatives. The NIH Roadmap Initiative, 2008, http://nihroadmap.nih.gov/ Search PubMed.
- A. M. Richard and C. R. Williams, Distributed structure-searchable toxicity (DSSTox) public database network: a proposal, Mutat. Res., 2002, 499, 27–52 CrossRef CAS.
- A. M. Richard, DSSTox web site launch: Improving public access to databases for building structure-toxicity prediction models, Preclinica, 2006, 2, 103–108 Search PubMed.
- D. S. Wishart, C. Knox, A. C. Guo, S. Shrivastava, M. Hassanali, P. Stothard, Z. Chang and J. Woolsey, DrugBank: a comprehensive resource for in silico drug discovery and exploration, Nucleic Acids Res., 2006, 34, D668–672 CrossRef CAS.
- T. E. Klein, J. T. Chang, M. K. Cho, K. L. Easton, R. Fergerson, M. Hewett, Z. Lin, Y. Liu, S. Liu, D. E. Oliver, D. L. Rubin, F. Shafa, J. M. Stuart and R. B. Altman, Integrating genotype and phenotype information: an overview of the PharmGKB project. Pharmacogenetics Research Network and Knowledge Base, Pharmacogenomics J., 2001, 1, 167–170 Search PubMed.
- J. J. Irwin and B. K. Shoichet, ZINC—a free database of commercially available compounds for virtual screening, J. Chem. Inf. Model., 2005, 45, 177–182 CrossRef CAS.
- J. J. Irwin, F. M. Raushel and B. K. Shoichet, Virtual screening against metalloenzymes for inhibitors and substrates, Biochemistry, 2005, 44, 12316–12328 CrossRef CAS.
- G. Brumfiel, Chemists spin a web of data, Nature, 2008, 453, 139 CrossRef CAS.
- S. Ekins, M. Iyer, M. D. Krasowski and E. D. Kharasch, Molecular characterization of CYP2B6 substrates, Curr. Drug Metab., 2008, 9, 363–373 Search PubMed.
- A. Khandelwal, P. Bahadduri, C. Chang, J. E. Polli, P. Swaan and S. Ekins, Computational models to assign biopharmaceutics drug disposition classification from molecular structure, Pharm. Res., 2007, 24, 2249–2262 CrossRef CAS.
- S. Ekins, V. Kholodovych, N. Ai, M. Sinz, J. Gal, L. Gera, W. J. Welsh, K. Bachmann and S. Mani, Computational discovery of novel low micromolar human pregnane X receptor antagonists, Mol. Pharmacol., 2008, 74, 662–672 CrossRef CAS.
- A. J. Williams, Qualifying online information resources for chemists. Presentation at The Library of Congress, 2008, http://www.scivee.tv/node/9267 Search PubMed.
- W. J. Crumb Jr, S. Ekins, D. Sarazan, J. H. Wikel, S. A. Wrighton, C. Carlson and C. M. Beasley, Effects of antipsychotic drugs on Ito, INa, Isus, IK1, and hERG: QT prolongation, structure activity relationship, and network analysis, Pharm. Res., 2006, 23, 1133–1143 CrossRef.
- A. J. Hunter, The Innovative Medicines Initiative: a pre-competitive initiative to enhance the biomedical science base of Europe to expedite the development of new medicines for patients, Drug Discovery Today, 2008, 13, 371–373 CrossRef.
- N. Kamel, C. Compton, R. Middelveld, T. Higenbottam and S. E. Dahlen, The Innovative Medicines Initiative (IMI): a new opportunity for scientific collaboration between academia and industry at the European level, Eur. Respir. J., 2008, 31, 924–926 CrossRef CAS.
- S. Kortagere, D. Chekmarev, W. J. Welsh and S. Ekins, Hybrid scoring and classification approaches to predict human pregane X receptor activiators, Pharm. Res., 2009, 26, 1001–1011 CrossRef CAS.
- J. Cartmell, S. Enoch, D. Krstajic and D. E. Leahy, Automated QSPR through Competitive Workflow, J. Comput. Aided Mol. Des., 2005, 19, 821–833 CrossRef CAS.
- D. Rogers, R. D. Brown and M. Hahn, Using extended-connectivity fingerprints with Laplacian-modified Bayesian analysis in high-throughput screening follow-up, J. Biomol. Screen., 2005, 10, 682–686 CrossRef CAS.
- M. Hassan, R. D. Brown, S. Varma-O'Brien and D. Rogers, Cheminformatics analysis and learning in a data pipelining environment, Mol. Divers., 2006, 10, 283–299 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |