Perfused multiwell plate for 3D liver tissue engineering†
Karel
Domansky
a,
Walker
Inman
ab,
James
Serdy
b,
Ajit
Dash
a,
Matthew H. M.
Lim
c and
Linda G.
Griffith
*ab
aDepartment of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA. E-mail: griff@mit.edu
bDepartment of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
cDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Cambridge, UK
First published on 22nd October 2009
Abstract
In vitro models that capture the complexity of in vivo tissue and organ behaviors in a scalable and easy-to-use format are desirable for drug discovery. To address this, we have developed a bioreactor that fosters maintenance of 3D tissue cultures under constant perfusion and we have integrated multiple bioreactors into an array in a multiwell plate format. All bioreactors are fluidically isolated from each other. Each bioreactor in the array contains a scaffold that supports formation of hundreds of 3D microscale tissue units. The tissue units are perfused with cell culture medium circulated within the bioreactor by integrated pneumatic diaphragm micropumps. Electronic controls for the pumps are kept outside the incubator and connected to the perfused multiwell by pneumatic lines. The docking design and open-well bioreactor layout make handling perfused multiwell plates similar to using standard multiwell tissue culture plates. A model of oxygen consumption and transport in the circulating culture medium was used to predict appropriate operating parameters for primary liver cultures. Oxygen concentrations at key locations in the system were then measured as a function of flow rate and time after initiation of culture to determine oxygen consumption rates. After seven days of culture, tissue formed from cells seeded in the perfused multiwell reactor remained functionally viable as assessed by immunostaining for hepatocyte and liver sinusoidal endothelial cell (LSEC) phenotypic markers.
Introduction
In vitro models of liver are an important tool in pharmaceutical drug development and in understanding liver pathophysiology.1 Many biological responses in liver arise from complex interactions between different cell types and vary with position along the capillary bed.2,3 Microfluidic platforms that recapitulate the physiological cellular microenvironment of the hepatic capillary bed, with perfusion flow that allows development of physiological oxygen gradients, may be useful in understanding liver toxicity, disease, inflammation, and drug metabolism.4–14 Although many types of perfusion bioreactors for 3D culture have been developed, they are generally limited in throughput and often complicated to use. The approach reported in this paper adapts 3D models to an easy-to-use, multiwell plate format suitable for higher throughput applications. Ease of use is achieved by designing the bioreactors as an array of open wells covered by a conventional tissue culture lid and by employing docking stations that facilitate handling the perfused multiwell plates.We describe the design and function of a perfused multiwell plate containing 12 fluidically isolated bioreactors that each accommodate 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–600000 cells. Perfusion of the tissue is achieved by pneumatically actuated micropumps embedded in the plate. This type of actuation was selected because it offers significant advantages for operation inside humidified CO2 incubators maintained at 37 °C, a harsh environment for electrically driven micropumps. Pneumatic actuation permits the necessary electronic timing components to be kept outside the incubator. This arrangement can make perfused multiwell plates inexpensive or even disposable if mass-produced. Additionally, life science research laboratories in academia and industry typically have distributed outlets for vacuum and compressed air which make sources for pneumatic actuation readily available.
000–600000 cells. Perfusion of the tissue is achieved by pneumatically actuated micropumps embedded in the plate. This type of actuation was selected because it offers significant advantages for operation inside humidified CO2 incubators maintained at 37 °C, a harsh environment for electrically driven micropumps. Pneumatic actuation permits the necessary electronic timing components to be kept outside the incubator. This arrangement can make perfused multiwell plates inexpensive or even disposable if mass-produced. Additionally, life science research laboratories in academia and industry typically have distributed outlets for vacuum and compressed air which make sources for pneumatic actuation readily available.
Three dimensional tissue cultures in the perfused multiwell system are initiated by pipetting a cell suspension directly onto extracellular matrix (ECM)-coated scaffolds. Microscale perfusion is started immediately after seeding to control the local oxygen concentration and biomechanical stimuli (i.e., shear stress) during the critical time period of tissue formation. This arrangement addresses some of the issues that have been reported in seeding microscale perfusion systems such as seeding efficiencies and local control of oxygen during the initial static incubation required for cell attachment.15–18
Oxygen is a key regulator of cell survival and function and thus its level in cell culture medium must be adequately controlled. We developed a model of oxygen transport and reaction in our recirculating system and used it to predict oxygen concentration gradients across the tissue and profiles in the bioreactor as a function of system parameters (e.g., flow rate, flow direction, and medium volume). Finally, we demonstrate that this perfused multiwell plate system is amenable to long-term maintenance of differentiated hepatocytes and LSECs.
Experimental
Concept of perfused multiwell plates
The perfused multiwell is an array of open-well bioreactors (Fig. 1a), each consisting of a reactor well, a reservoir well, and an integrated micropump. The reactor well contains an ECM-coated scaffold where cells self-assemble into an array of 3D microtissue units. The reservoir well holds cell culture medium that is continually circulated between the two wells and through the scaffold by a diaphragm micropump (Fig. 2). Similar to static multiwell plates, all bioreactors are fluidically isolated and covered with a single lid. Ridges around the outside of the plate and around each bioreactor reduce the risk of airborne contamination. The open-well design is conducive to manual or automated cell seeding and fluid handling, and the center-to-center spacing between bioreactors (18 mm) is set to allow use of multichannel pipettors with alternating tips omitted.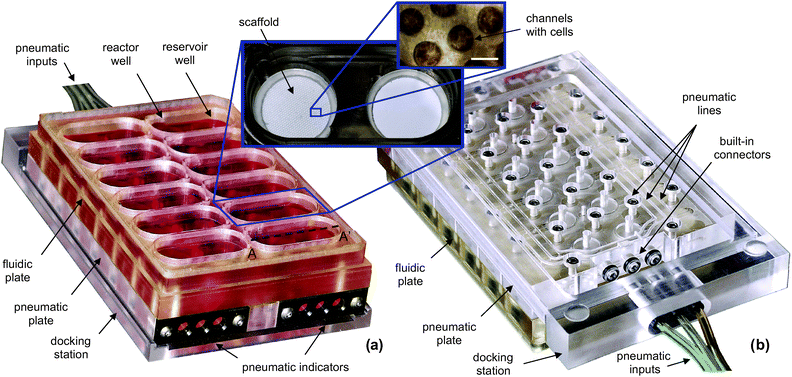 | ||
| Fig. 1 Photographs of a perfused multiwell with an array of 12 bioreactors. The size of the assembled multiwell plate is ∼127.8 × 85.5 × 34 mm. The top view (a) includes inserted photographs of a bioreactor and a scaffold. The size of the white bar in the scaffold photograph is 300 µm. The bottom view (b) of a partially docked perfused multiwell shows the built-in connectors and pneumatic lines distributing positive and negative air pressure to individual valves and pump chambers. | ||
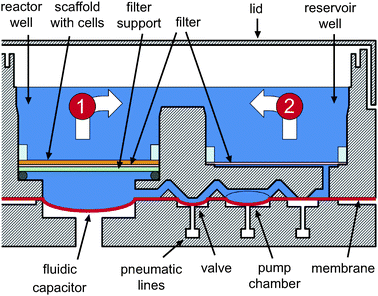 | ||
| Fig. 2 A schematic cross-section of a bioreactor (section A–A′ from Fig. 1a). Maintenance and post-seeding flow directions are indicated by arrows (1) and (2), respectively. Diameter of the reactor and reservoir well is 15 mm. Centers of the wells are 20 mm apart. Diameter of the scaffold is 14.9 mm. When there are 3 mL of culture medium in a bioreactor (a typical total volume), top of the scaffold is under ∼7 mm of fluid. The valves, the pump chamber, and other features are not drawn to scale. | ||
Micropumps are actuated in parallel from pneumatic inputs connected to the perfused multiwell through a dock (Fig. 1b). Docks are kept in standard CO2 incubators for cell culture and in sterile hoods for seeding. Pneumatic lines run from the docks to small electronic controllers outside the sterile environment. The docking design makes handling a perfused multiwell similar to handling a standard tissue culture plate. Additionally, no electronics are contained in the perfused multiwell.
The scaffold provides the 3D physical support for cell attachment and tissue formation. Its design principles are constrained by several requirements that have been discussed previously.8 In the perfused multiwell described herein, the scaffold is a circular ECM-coated polymer wafer with an array of 769 microchannels, each 0.24 mm deep and 0.34 mm in diameter. It is backed by a filter with 5 µm pores and a filter support. The filter captures cells in the scaffold immediately after seeding. There is also a filter in the reservoir well. The presence of filters in both wells prevents the cells from entering the valves and pumps.
Tissue perfusion system
Each bioreactor has an integrated pneumatic micropump comprising a pump chamber, inlet and outlet valves, and a fluidic capacitor.19,20 Culture medium is pumped by opening the inlet valve, filling the pump chamber, reversing the state of the valves, and draining the pump chamber. Approximately 1 µL is pumped in each cycle. Capacitors convert pulsatile flow generated by the pump to constant flow through the tissue, thereby eliminating disruptive oscillatory movement of cells in channels during cell attachment and contributing to an appropriate physiological flow pattern during long-term operation. (Blood flow pulses due to the heartbeat are damped in the microcirculation in vivo.) The pumping sequence can be run bidirectionally in order to flow up or down through the scaffold. Flow rate varies linearly with the pumping frequency of up to 1.2 mL min−1 (∼25 Hz). The effectiveness of the capacitor, which increases with increasing flow rate, has been demonstrated for flows as low as 0.05 mL min−1.20 The operating range of the pump was targeted to span the range of physiological and pathological flows observed in vivo. Shear stress in the liver sinusoid in vivo has been reported between 10 and 50 mPa.21 Based on shear stress calculations previously reported8 and a flow rate of 0.25 mL min−1 (0.3 µL min−1 channel−1), we estimate that shear stress in the scaffold should not exceed 30 mPa.The pumps, valves, and capacitors are made by sandwiching a thin (∼25 µm), highly flexible polyurethane membrane between the top (fluidic) and the bottom (pneumatic) plates. Air pressure drives the pumps by actuating the membrane. A valve is opened when negative air pressure pulls the membrane towards the pneumatic plate. Positive air pressure closes the valve by pressing the membrane against the valve seat in the fluidic plate. The pump chamber fills when negative pressure is applied to the membrane and drains in response to positive air pressure. The fluidic capacitor is created by allowing a section of the membrane (between the pumps and tissue) to flex. The membrane also provides a partition between the sterile upper half of the plate (containing the cell cultures) and the non-sterile lower half (containing the pneumatics).
Pneumatic signals are distributed in parallel to all 12 pumps by three pneumatic lines (Fig. 1b). The lines interface with the controller through a docking station. Connectors in the docking station have spring-loaded shutoff valves (Colder Products, USA) that automatically close when the perfused multiwell is unplugged. This prevents discharge of non-sterile air into the incubator or tissue culture hood and introduction of the humidified air from the incubator into the pneumatic system. Flexing membranes at the ends of the pneumatic lines indicate proper connections and functioning pneumatics (Fig. 1a). Compressed air and vacuum are sourced from laboratory distribution outlets. Air is filtered, dried with desiccant, and regulated to 40 kPa.
The pumps were designed to be self-priming, bubble-tolerant, and insensitive to changes in head pressure and pneumatic pressure within their operating range. Reliability of the pumps was achieved by implementing a number of key design features. For example, sealing pressure between the plates was maximized by using narrow bosses in the pneumatic plate around the pump features. Also, a very thin membrane was used to minimize membrane penetration into the pump features. Dowell pins ensure proper alignment of the pumps. A channel was cut across the top of the pump chamber to prevent the membrane from throttling the outlet before all of the fluid is ejected. Similarly, a channel was milled into the bottom of the pump chamber to ensure that the membrane does not prematurely seal off the pneumatic input when vacuum is applied. Additionally, fluidic connections between the pump features were created by drilling angled channels rather than milling troughs (see Fig. 2). This prevents pneumatic leakage between valves and pumps. Finally, at least one valve is closed at all times to prevent backflow.
We tested durability of the polyurethane pump membrane by continuously operating 12 bioreactors (eight filled with fluid but without the cells, four dry) at the flow rate of 0.2 mL min−1 for six months (several hundred thousand cycles). None of the valves or pumps leaked or failed during this stress test. Thus, these pumps appear to be well suited for long-term perfusion of cell cultures.
Fabrication, sterilization and assembly
The majority of parts were fabricated by CNC machining. Polystyrene and polycarbonate scaffolds were fabricated by microdrilling. The polyether aromatic polyurethane membrane (ST-625FS, Stevens Urethane, USA, and MT 2001, American Polyfilm, USA) was cut by laser machining. The 127.8 × 85.5 mm footprint of an assembled perfused multiwell complies with standard dimensions of the 96-well microplates established by the Society for Biomolecular Screening.22Following sterilization, scaffolds were coated with collagen and air-dried. PVDF filters for the reactor wells were coated with 1% bovine serum albumin. The pneumatic plate and fluidic plate were assembled with screws fitted with Belleville disc springs to provide constant sealing pressure on the thin membrane for the duration of an experiment. The bioreactors were primed with culture medium. After priming, the reactor wells were fitted with O-rings, filter supports, filters, and scaffolds. Filters were added to the reservoir wells. These components were secured by tightly fitting retaining rings. Additional details about fabrication and sterilization are provided in ESI†.
Liver cell isolation, culture initiation and maintenance
Hepatocyte-enriched populations (hepatocytes) were isolated from Fischer rats using a modification of Seglen's two-step collagenase perfusion procedure.23 LSEC-enriched non-parenchymal cell populations were isolated from enhanced green fluorescent protein (EGFP)-positive Sprague-Dawley rats. This LSEC-enriched fraction comprises ∼80% LSEC and includes stellate and Kupffer cells.23Prior to seeding, priming medium in the reservoir and reactor wells was replaced with a fresh medium. Cells were seeded by pipetting the cell suspension above the scaffold in the reactor well. Monocultures of hepatocytes were seeded with ∼800000 cells, and since non-parenchymal cells are smaller than hepatocytes, cocultures were seeded with ∼500000 hepatocytes and ∼500000 LSEC-enriched population. Immediately after seeding, flow was initiated downward through the scaffold at 0.25 mL min−1 (∼0.3 µL min−1 channel−1) and culture medium was added to obtain a total volume of ∼3 mL. The perfused multiwell was then moved inside a humidified 5% CO2 (air balance) incubator maintained at 37 °C.
Cells attach to the scaffold within the first few hours, and, for hepatocyte cultures, result in a stable population of ∼400000 to 600000 cells (as measured by total protein bicinchoninic acid (BCA) assay). This is consistent with typical plating efficiencies in 2D cultures. After 8 h the direction of flow was reversed to dislodge dead cells and debris. Flow up through the scaffold at 0.25 mL min−1 was maintained throughout the remaining culture period.
Hepatocyte cultures were initiated and maintained in serum-free hepatocyte growth medium (HGM).23 Cocultures of hepatocytes combined with the LSEC-enriched population were initiated with a mixture containing HGM and endothelial growth medium in a 1 : 1 volume ratio for the first 24 h, and then maintained in HGM. Culture medium was changed every 48 h during long-term culture. During each exchange, flow was paused and approximately 80% of the medium was replaced. The remaining medium kept the cells submerged and the pumps primed.
Cell viability was assessed with the live/dead viability kit. Synthesis of albumin was demonstrated by staining with an antibody for rat albumin (AbCam, USA), and DRAQ-5 was used to stain cell nuclei (Biostatus Limited, USA). Antibodies for SE-1 were obtained from IBL America, USA. More details about the cells and culture medium are provided in ESI†.
Oxygen sensors
Oxygen measurements were performed using custom fiber optic probes mounted on a custom lid. Oxygen is detected using a 2 mm diameter ruthenium-based sensing layer (PreSens, Germany), where luminescence of ruthenium molecules in an excited state is quenched by collisions with molecular oxygen. The probes are connected to a 4-channel fiber optic meter (OXY-4 by PreSens, Germany) and luminescence decay time is measured by the phase modulation technique.The custom lid contains 24 ports centered on each well in the plate. Each port contains a connector and an O-ring used to vertically position the oxygen probe. All probes are submerged to a depth of ∼2 mm above the scaffold. Optical fibers coming vertically out from the lid are supported by a second platform ∼10 cm above the lid. The second platform holds unions that connect the probes with the optical fibers coming from the OXY-4 meter. This arrangement facilitates adjustment of the probe depth and allows quick connections between the probes and the optical fibers. Prior to experiments, oxygen sensors are calibrated with two calibration standards. Sterilization of the probes was verified not to interfere with the calibration.
Computational simulation of oxygen transfer
During bioreactor operation, cell culture medium circulates between the reactor and reservoir. Oxygen consumed by cells in the scaffold is replenished by diffusive and convective flux from the air–liquid interface. This generates a multidimensional pattern of oxygen concentration in the bioreactor. To simulate how operating parameters such as medium flow rate and total medium volume influence oxygen transport and ensuing concentrations of oxygen at the upstream side of the tissue, and to predict appropriate locations for measuring oxygen concentration, we developed a model of oxygen transport (with fixed inlet concentration) and applied it to predict oxygen concentration profiles in the reactor. Following the guidance of the model, we placed the oxygen probes into the bioreactor with cells and performed several measurements of oxygen concentration. Using the experimental measurements, we developed a model of oxygen consumption by liver tissue maintained in the reactor and then coupled it with the model of oxygen transport.Oxygen transport and consumption were simulated with a 3D finite differences model. To understand the model development, consider the path of fluid flowing clockwise in Fig. 2, starting at the bottom of the reservoir. Fluid velocity exiting the reservoir is constant due to a high-impedance filter; however, the oxygen concentration varies with position. As fluid passes through the pump we assume it is completely mixed and oxygen transfer into culture medium through the low-permeability polyurethane membrane can be neglected. We calculate the mixed concentration from flux across the reservoir filter. Thus, fluid enters the lower face of the scaffold (i.e., scaffold inlet) at a uniform concentration, Cin. Fluid velocity entering the scaffold is also uniform due to a filter below the scaffold. Oxygen is consumed by the cells as fluid passes upward through the scaffold. Experimentally, no significant differences are observed in the cell distributions at different regions of the scaffold, hence, it is reasonable to assume that concentration exiting the scaffold, Cout, is constant everywhere on the upper face of the scaffold. As fluid progresses upward, velocity and concentration profiles evolve in response to the boundary conditions on the walls, convection, and diffusion from the gas–liquid interface. Fluid then flows across an open channel into the reservoir generating a non-uniform oxygen concentration profile in the reservoir, where the cycle is completed.
The control volume for the model extends from the air–liquid interface down to the scaffold in the reactor well and the filter in the reservoir well. Fig. 3 shows the results of a model calculation for a flow rate of 0.25 mL min−1 upward through the scaffold and Cout of 50 µM, a concentration in the range of perivenous blood plasma oxygen concentration, hence a reasonable value to expect experimentally. The color of fluid below the scaffold represents the concentration after mixing occurs in the pump. The oxygen concentration profile indicates that probes placed in the center of the wells ∼2 mm above the scaffold measure Cin and Cout with an estimated error of less than 5% (see below in Results and discussion). When the flow is reversed, the positions of Cin and Cout are likewise reversed, and the ensuing concentration profiles mirror those in Fig. 3. The appropriate location of probe placement remains the same. Note that oxygen concentration profiles can be non-dimensionalized and thus are independent of the value of Cout specified. Therefore, the appropriate probe location is independent of outlet concentrations.
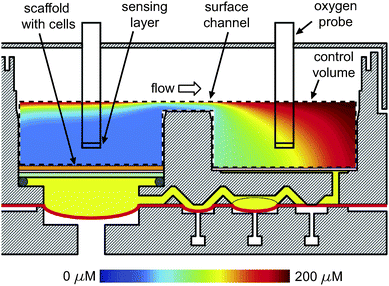 | ||
| Fig. 3 A simulation of oxygen transport. The control volume is marked by a dashed line. Oxygen probes are sketched as black rectangles in the middle of the reactor and reservoir wells. The depth of culture medium in the channel was 1 mm corresponding to a total volume of ∼3 mL. The flow rate was 0.25 mL min−1. | ||
The oxygen transport model was based upon existing MATLAB code24 with an added mass transport equation. The model first calculates a flow profile from Navier–Stokes and continuity equations and iterates solutions until the profile stabilizes. Next it solves for oxygen concentration using convection–diffusion equations, and concentration profiles are iterated until steady state. Details of the geometry, boundary conditions, and code elements are provided in ESI†.
Results and discussion
Measured values of dissolved oxygen indicate physiologically relevant oxygen gradients across the tissue
Measured values of dissolved oxygen in the reactor and reservoir wells during the first 20 h of culture are shown in Fig. 4a. The culture medium is air-saturated at the time cells are seeded in the bioreactor. After flow is initiated, cellular oxygen consumption causes the oxygen concentration to decline for several hours until equilibrium is reached between cellular consumption and oxygen transport at the air–liquid interface. During the first 8 h, a flow rate of 0.25 mL min−1 was maintained in a downward direction through the scaffold, and Cout reached a steady state value of ∼60 µM with a corresponding Cin value of 145 µM. Because the predicted oxygen concentrations are the mirror image of those shown in Fig. 3, the oxygen concentration varies along the surface of the scaffold when the flow is in the downward direction. The model predicts that the oxygen concentration in the medium entering the scaffold ranges from ∼90 µM at the innermost location to ∼190 µM at the outer edge during the first 8 h of operation, when flow is in the downward direction.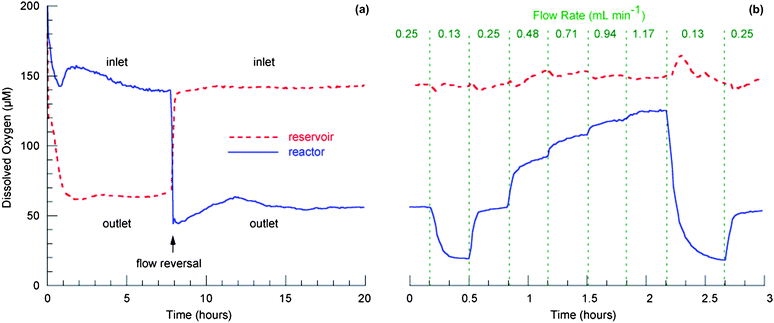 | ||
| Fig. 4 Measured concentration of dissolved oxygen in reservoir and reactor wells seeded with rat hepatocytes: (a) as a function of time post-seeding with a flow rate of 0.25 mL min−1 and a sampling rate of 0.2 measurements per minute; (b) as a function of flow rate (immediately following the time course measurement) with a sampling rate of 1 measurement per minute. | ||
After 8 h the flow direction was reversed while maintaining a constant flow rate. Immediately following the flow reversal, oxygen-depleted medium below the scaffold and in the reservoir was passed directly back through the cells. During this time (∼10 min with the flow of 0.25 mL min−1) the cells were exposed to relatively low inlet concentrations, and a concomitant drop of ∼25% in the outlet concentration was observed with a longer transient (∼2 h). When the regime of stable concentrations is reached, the concentration of oxygen entering the scaffold is constant and uniform across all channels in the scaffold. Due to oxygen consumption by the cells, there are gradients of oxygen in the tissue.
To determine a range of acceptable operating parameters, and to estimate cellular oxygen consumption rates in this perfused culture, inlet and outlet oxygen concentrations were measured as a function of flow rate. Following the initial 20 h plotted in Fig. 4a, flow rate was incrementally changed as shown in Fig. 4b. The inlet oxygen concentration in this experiment fluctuates modestly around ∼145 µM. Although this concentration is higher than values reported for the human in vivo periportal (sinusoidal entry) region (84–91 µM), it is within arterial blood oxygen concentration ranges (104–146 µM) and below hyperoxic concentration levels.25 Oxygen concentration downstream of the tissue increases with increasing flow rate, from ∼19 µM to ∼125 µM. In human, the reported in vivo perivenous (sinusoidal exit) region dissolved free oxygen concentration is 42–49 µM.25 A tissue outlet concentration of ∼50 µM is observed at a flow rate of 0.25 mL min−1, hence, this flow rate provides a reasonable approximation of physiological gradients. Gradients are steeper than those in vivo because culture medium does not contain hemoglobin, which serves as a depot of oxygen.
Oxygen consumption rates within the perfused tissue
Oxygen consumption in intact liver and whole liver cells is regulated by oxygen concentration well above the Michaelis constant Km for mitochondrial enzymes,26,27 resulting in an apparent Km of 175 µM reported for perfused rat liver.2,3 Oxygen consumption rates and concentrations of metabolic enzymes vary significantly within the sinusoid in vivo.25 Along the direction of flow, oxygen consumption decreases two-fold from periportal to pericentral regions in perfused liver. The system described here exhibits similarities including gradients of oxygen along the flow path. Additionally, oxygen gradients inherently exist between the main conduits for convective flow through the tissue and the surrounding 3D tissue structures. Thus the average oxygen concentration in the tissue at the exit is lower than the measured value of Cout, because Cout represents the oxygen concentration in the bulk fluid. This tends to elevate the apparent Km. Given these considerations, we model oxygen uptake by the cells in the scaffold as a first-order reaction and approximate the outlet oxygen concentration by the following equation: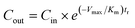 | (1) |
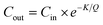 | (2) |
Total oxygen consumption throughout the tissue is found by multiplying the drop in concentration by the flow rate and dividing by the total tissue mass. Using the average tissue mass per scaffold of 0.5 mg protein, and inlet and outlet concentrations of 145 µM and 50 µM, respectively (for a flow rate of 0.25 mL min−1), we find the total oxygen consumption rate across the scaffold to be comparable to consumption rates for isolated hepatocytes and perfused rat livers (see Table 1). We can also estimate local oxygen consumption rates by multiplying the rate constant K by the local oxygen concentration and normalizing by the total cell mass in the scaffold. The values we estimate for consumption at the tissue inlet are most likely elevated because they are based on a first-order approximation, which does not inherently limit the maximum consumption rate. Our estimate for consumption at the tissue outlet is also likely higher than we could expect in the actual tissue. Since oxygen concentration deep in the tissue will be lower than the bulk concentration we measure above the scaffold, the consumption rate deep in the tissue should also be lower than those reported in Table 1.
| µmol hour−1 mg protein−1 | Periportal or tissue inlet | Pericentral or tissue outlet | Total |
|---|---|---|---|
| Perfused multiwell plate | 4.7 | 1.8 | 2.6 |
| Isolated hepatocytes29 | 2.5 | ||
| Perfused rat liver30 | 1.8 | ||
| Perfused rat liver31 | 1.2 | 0.6 | 1.0 |
Using the above rate constant K, we combined eqn (2) with the oxygen transfer model to simulate oxygen concentrations in the bioreactor. Modeled concentrations at the inlet (entering the tissue), outlet (exiting the tissue) and the location of the probe are plotted in Fig. 5 and compared to measured values. In the experiments the bioreactor contained ∼3 mL of cell culture medium, filling the wells to ∼1 mm above bottom of the surface channel. Due to a meniscus, the depth of medium in the middle of the channel is slightly less than 1 mm while the depth along the wall is higher. For this reason, simulations in Fig. 5 were performed for the channel depth of 0.5 and 1 mm. It can be seen that the depth of culture medium in the bioreactor has a significant impact on oxygen concentrations, and that the model for a depth of 0.5 mm is in better agreement with the experimental data. The model predicts that the measured value by the oxygen probe underestimates inlet oxygen concentration by less than 5% in the investigated range of flow rates.
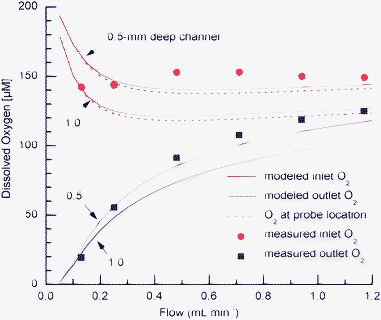 | ||
| Fig. 5 Measured and modeled inlet and outlet oxygen concentrations as a function of flow for two depths of cell culture medium in the channel. The figure also shows modeled oxygen concentration at the oxygen probe location. | ||
Since each channel in the scaffold represents an individual unit of tissue, it is desirable to define rate constant ![[K with combining macron]](https://www.rsc.org/images/entities/i_char_004b_0304.gif) on a per channel basis (K = 0.27 mL min−1 per 769 channels yields = 0.35 µL min−1 channel−1). When and flow per channel
on a per channel basis (K = 0.27 mL min−1 per 769 channels yields = 0.35 µL min−1 channel−1). When and flow per channel ![[Q with combining macron]](https://www.rsc.org/images/entities/i_char_0051_0304.gif) are used, eqn (2) scales to scaffolds with different numbers of channels.
are used, eqn (2) scales to scaffolds with different numbers of channels.
Assessment of phenotype and function of the tissue cultured in the perfused multiwell
Several assays were performed on the liver cells cultured in the perfused multiwell to assess phenotype and function. The viability of hepatocytes was determined by a fluorescent live/dead assay. Fig. 6a indicates that liver cells cultured in the perfused multiwell for seven days are predominantly viable, although there are few non-viable cells adhering to the top surface of the scaffold. To evaluate liver-specific function, we stained the tissue with anti-rat albumin antibody. Fig. 6b demonstrates retention of this hepatocyte-specific functional marker after seven days of culture.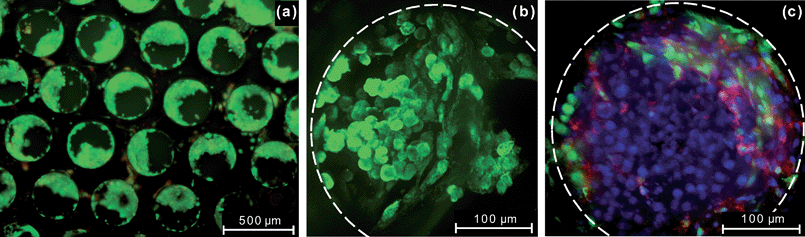 | ||
| Fig. 6 (a) Cell viability assay of rat liver cells cultured for seven days in the perfused multiwell. The image shows multiple channels in a scaffold. Live cells are stained green with calcein AM while dead cells are stained red with ethidium homodimer-1. The areas occupied by the cells are bright green and areas without cells appear dark green or black. (b) Immunostaining of rat liver cells cultured in the perfused multiwell 7 days post-seeding demonstrating retention of the hepatocyte-specific functional marker albumin (green). Staining was preformed with rat albumin antibody. This image shows a single channel with its boundary highlighted by a white dashed line. (c) Image of rat hepatocytes and sinusoidal endothelial cells cultured in the perfused multiwell plate obtained on day 13 of post-seeding. Nuclei are stained blue with DRAQ-5, non-parenchymal cells express green fluorescent protein (GFP), and the functional marker for sinusoidal endothelial cells (SE-1) is stained red. | ||
Liver tissue engineering models are increasingly moving from hepatocyte-only cultures to cocultures containing non-parenchymal cells (NPCs), such as LSECs and stellate cells in order to create more physiologically relevant systems.32,33 However, LSECs are notoriously challenging to culture under standard in vitro conditions. When cultured alone, LSECs showed signs of dedifferentiation after 1–3 days of static culture on most investigated ECMs.34 Using medium conditioned with hepatocytes, differentiated LSECs were maintained up to 6 days.35 LSECs cocultured with hepatocytes on decellularized liver-derived connective tissue matrix showed highly fenestrated phenotype (the presence of fenestrations is considered as a reliable morphological marker of LSECs) during the first 3 days of culture but showed a decline in the maintenance of fenestrated phenotype after 7 days of culture.34 An early prototype single-unit perfused bioreactor that had medium flowing through both the tissue in the scaffold and above the tissue in the direction parallel to the scaffold was shown to maintain expression of the functional marker for LSECs, SE-1, for a significantly longer time period than commonly reported in literature.23 To demonstrate suitability of the perfused multiwell (with flow through the tissue) for long-term cocultures, we seeded it with rat non-parenchymal cells and hepatocytes and followed the cultures for two weeks. At the end of these experiments, the tissue samples were fixed with 4% paraformaldehyde and immunostained with various functional markers of NPCs. Confocal microscopy of the immunostained tissue demonstrated that LSECs retain expression of the functional marker SE-1 as late as 13 days post-seeding (Fig. 6c). The preservation of the LSEC phenotype even in the absence of serum or exogenous supportive endothelial growth factors like vascular endothelial growth factor (VEGF) in the culture medium, typically needed for LSEC survival, may point to the role of the 3D cell–cell interactions and flow in the perfused multiwell. Retention of the endothelial phenotype was seen to be dependent on the flow rate and the oxygen concentration in the perfused multiwell.36 Similar staining of the functional markers with the antibodies ED-2 and GFAP for 13 days also indicated the presence of Kupffer cells and stellate cells, respectively,36 thereby establishing the system as being conducive for the culture of various non-parenchymal cells in cocultures with hepatocytes.
Conclusions
An array of bioreactors that foster maintenance of 3D liver tissue under constant microperfusion has been integrated into a multiwell plate format. The bioreactors have open wells, built-in micropumps, and the perfused multiwell plate employs a docking design that facilitates handling and integration into conventional incubation and assay tools. A model of oxygen consumption and transport in the circulating culture medium was used to predict appropriate operating parameters for liver cell cultures. Model predictions were validated by measuring oxygen concentrations in cultures of primary rat hepatocytes as a function of time and flow rate. From the measurements, we determined that consumption of oxygen by tissue can be characterized with a system-specific first-order reaction constant K = 0.27 ± 0.03 mL min−1. Considering physiological values, experimental measurements showed that tissue in the scaffold was adequately oxygenated for flow rates above 0.25 mL min−1. Consumption of oxygen by the cells in the scaffold was similar to oxygen consumption in perfused rat livers.After seven days in culture, the viability of hepatocytes was determined by the live/dead staining and liver-specific function was demonstrated by immunostaining with a rat albumin antibody. To demonstrate suitability of the perfused multiwell for long-term tissue culture, we seeded it with LSECs and rat hepatocytes and showed that LSECs, known to lose differentiated phenotype under static culture conditions, maintain expression of the functional marker SE-1 throughout 13 days of coculture with hepatocytes.
The higher throughput capability of the perfused 3D liver multiwell is beneficial for conducting assays for liver toxicology and metabolism and can be used to model hepatic disorders, cancer, and other human diseases. Although we described the design and function of an array of 24 wells, the concept is scalable to a plate with a higher number of wells, such as a 96-well plate. This format is desirable for reducing cell quantities per well (e.g., when using human cells) or increasing throughput. In the reported experiments, we chose rigid polystyrene and polycarbonate scaffolds coated with collagen, but the design provides flexibility for using scaffolds made from a wide variety of different materials such as protein matrices, hydrogels, fibers or other classes of material. Fasteners such as screws and retaining rings were used on the perfused multiwell for prototyping reasons. They facilitate testing a large spectrum of membrane and scaffold materials without the difficulties associated with joining hard-to-bond or incompatible materials. For larger scale deployment, the perfused multiwell plate can be made disposable by employing, for example, injection molding and bonding techniques. Cultures of cells from other organs and stem cell cultures also benefit from 3D environments and tissue perfusion.37 Although we designed the perfused multiwell primarily for the culture of liver cells, it can be used for perfusion culture of other high metabolically active cell types such as kidney, heart, or brain cells.
Acknowledgements
We gratefully acknowledge funding support from the NSF grant number EEC-9843342, Pfizer, the DuPont/MIT Alliance, NIH grant number 5R01ES015241, and the MIT Center for Environmental Health Sciences grant number 5P30ES002109-30. We also want to thank Stevens Urethane and American Polyfilm for generously providing us with samples of polyurethane membrane, our co-workers Bryan Owens and Romie Littrell for characterizing some functions of the perfused multiwell and isolating the rat hepatocytes, and Jean-Christophe Nave from the MIT Department of Mathematics for his help with the oxygen simulations.Notes and references
- N. J. Hewitt, M. J. Lechon, J. B. Houston, D. Hallifax, H. S. Brown, P. Maurel, J. G. Kenna, L. Gustavsson, C. Lohmann, C. Skonberg, A. Guillouzo, G. Tuschl, A. P. Li, E. LeCluyse, G. M. M. Groothuis and J. G. Hengstler, Drug Metab. Rev., 2007, 39, 159–234 CrossRef CAS.
- H. Meren, T. Matsumura, F. C. Kauffman and R. G. Thurman, Adv. Exp. Med. Biol., 1986, 200, 467–476 CAS.
- T. Matsumura, F. C. Kauffman, H. Meren and R. G. Thurman, Am. J. Physiol., 1986, 250, G800–G805 CAS.
- P. J. Lee, P. J. Hung, V. M. Rao and L. P. Lee, Biotechnol. Bioeng., 2006, 94, 5–14 CrossRef CAS.
- P. J. Lee, P. J. Hung and L. P. Lee, Biotechnol. Bioeng., 2007, 97, 1340–1346 CrossRef CAS.
- A. Sin, K. C. Chin, M. F. Jamil, Y. Kostov, G. Rao and M. L. Shuler, Biotechnol. Prog., 2004, 20, 338–345 CrossRef CAS.
- J. H. Sung and M. L. Shuler, Lab Chip, 2009, 9, 1385–1394 RSC.
- M. J. Powers, K. Domansky, M. R. Kaazempur-Mofrad, A. Kalezi, A. Capitano, A. Upadhyaya, P. Kurzawski, K. E. Wack, D. B. Stolz, R. Kamm and L. G. Griffith, Biotechnol. Bioeng., 2002, 78, 257–269 CrossRef CAS.
- K. Domansky, A. Sivaraman and L. G. Griffith, in Lab-on-Chips for Cellomics-Micro and Nanotechnologies for Life Science, ed. H. Andersson and A. Van Den Berg, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2004, pp. 319–346 Search PubMed.
- J. W. Allen and S. N. Bhatia, Biotechnol. Bioeng., 2003, 82, 253–262 CrossRef CAS.
- E. Leclerc, Y. Sakai and T. Fujii, Biochem. Eng. J., 2004, 20, 143–148 CrossRef CAS.
- S. Ostrovidov, J. Jiang, Y. Sakai and T. Fujii, Biomed. Microdevices, 2004, 6, 279–287 CrossRef CAS.
- Y.-C. Toh, C. Zhang, J. Zhang, Y. M. Khong, S. Chang, V. D. Samper, D. Noort, D. W. Hutmacher and H. Yu, Lab Chip, 2007, 7, 302–309 RSC.
- R. Chang, J. Nam and W. Sun, Tissue Eng., 2008, 14, 157–166 CAS.
- L. Kim, Y. Toh, J. Voldman and H. Yu, Lab Chip, 2007, 7, 681–694 RSC.
- A. Rotem, M. Toner, R. G. Tompkins and M. L. Yarmush, Biotechnol. Bioeng., 1992, 40, 1286–1291 CrossRef CAS.
- A. Rotem, M. Toner, S. Bhatia, B. D. Foy, R. G. Tompkins and M. L. Yarmush, Biotechnol. Bioeng., 1994, 43, 654–660 CrossRef CAS.
- C. H. Cho, J. Park, D. Nagrath, A. W. Tilles, F. Berthiaume, M. Toner and M. L. Yarmush, Biotechnol. Bioeng., 2007, 97, 188–199 CrossRef CAS.
- K. Domansky, W. Inman, J. Serdy, B. Owens, M. Whittermore, L. Vineyard and L. G. Griffith, MicroTAS, The Society for Chemistry and Micro-Nano Systems (CHEMINAS), Tokyo, Japan, 2006, pp. 951–953 Search PubMed.
- W. Inman, K. Domansky, J. Serdy, B. Owens, D. Trumper and L. G. Griffith, J. Micromech. Microeng., 2007, 17, 891–899 CrossRef.
- P. F. Lalor and D. H. Adams, J. Clin. Pathol.: Mol. Pathol., 1999, 52, 214–219 Search PubMed.
- T. W. Astle, Molecules, 1996, 1, 106–113 CAS.
- A. Hwa, R. C. Fry, A. Sivaraman, P. T. So, L. D. Samson, D. B. Stolz and L. G. Griffith, FASEB J., 2007, 21, 2564–2579 CrossRef CAS.
- G. Strang, Computational Science and Engineering, Wellesley-Cambridge Press, MA, USA, 2007 Search PubMed.
- K. Jungermann and T. Kietzmann, Hepatology, 2000, 31, 255–260 CrossRef CAS.
- N. S. Chandel, G. R. S. Budinger, S. H. Choe and P. T. Schumacker, J. Biol. Chem., 1997, 272, 18808–18816 CrossRef CAS.
- T. Matsumura, H. Yoshihara, R. Jeffs, Y. Takei, S. Nukina, T. Hijioka, R. K. Evans, F. C. Kauffman and R. G. Thurman, Am. J. Physiol. Gastrointest. Liver Physiol., 1992, 25, G645–G650.
- K. Ito and J. B. Houston, Pharm. Res., 2004, 21, 785–792 CrossRef CAS.
- P. T. Schumacker, N. Chandel and A. G. N. Agusti, Am. J. Physiol., 1993, 265, L395–L402 CAS.
- A. C. Boretto-Biazon, F. Bracht, L. Bracht, A. M. Kelmer-Bracht and A. Bracht, Acta Pharmacol. Sin., 2009, 30, 90–97 Search PubMed.
- T. Matsumura and R. G. Thurman, Am. J. Physiol. Gastrointest. Liver Physiol., 1983, 7, G656–G659.
- O. Morin and C. Normand, J. Cell. Physiol., 1986, 129, 103–110 CrossRef CAS.
- O. Loreal, F. Levavasseur, C. Fromaget, D. Gross, A. Guillouzo and B. Clement, Am. J. Pathol., 1993, 143, 538–544 Search PubMed.
- T. L. Sellaro, A. K. Ravindra, D. B. Stolz and S. F. Badylak, Tissue Eng., 2007, 13, 2301–2310 CrossRef CAS.
- P. Krause, P. M. Markus, P. Schwartz, K. Unthan-Fechner, S. Pestel, J. Fandrey and I. Probst, J. Hepatol., 2000, 32, 718–726 CrossRef CAS.
- A. Dash, PhD thesis, Massachusetts Institute of Technology, MA, USA, 2007.
- J. A. Burdick and G. Vunjak-Novakovic, Tissue Eng., 2009, 15, 205–219 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fabrication, sterilization, and MATLAB simulation. See DOI: 10.1039/b913221j |
| This journal is © The Royal Society of Chemistry 2010 |