Co-culture of epithelial cells and bacteria for investigating host–pathogen interactions†
Jeongyun
Kim
a,
Manjunath
Hegde
a and
Arul
Jayaraman
*ab
aArtie McFerrin Department of Chemical Engineering, Texas A&M University, College Station, TX 77843-3122, USA. E-mail: arulj@tamu.edu; Fax: (+1) 979 845 6446; Tel: (+1) 979 845 3306
bDepartment of Biomedical Engineering, Texas A&M University, College Station, TX 77843, USA
First published on 16th October 2009
Abstract
The human gastrointestinal (GI) tract is a unique environment in which intestinal epithelial cells and non-pathogenic (commensal) bacteria co-exist. This equilibrium is perturbed by the entry of pathogens into the GI tract. A key step in the infection process is the navigation of the pathogen through the commensal bacterial layer to attach to epithelial cells. It has been proposed that the microenvironment that the pathogen encounters in the commensal layer plays a significant role in determining the extent of attachment and colonization. Current culture methods for investigating pathogen colonization are not well suited for investigating this hypothesis as they do not enable co-culture of bacteria and epithelial cells in a manner that mimics the GI tract microenvironment. Here we report the development of a microfluidic co-culture model that enables independent culture of eukaryotic cells and bacteria, and testing the effect of the commensal microenvironment on pathogen colonization. A pneumatically-actuated system was developed to form reversible islands that allow development of bacterial biofilm along with culture of an epithelial cell monolayer. The co-culture model used to develop a commensal Escherichia coli biofilm among HeLa cells, followed by introduction of enterohemorrhagic E. coli (EHEC) into the commensal island, in a sequence that mimics the sequence of events in GI tract infection. Using wild-type E. coli and a tnaA mutant (lacks the signal indole) as the commensal bacteria, we demonstrate that the commensal biofilm microenvironment is a key determinant of EHEC infectivity and virulence. Our model has the potential to be used in fundamental studies investigating the effect of GI tract signals on EHEC virulence as well as for screening of different probiotic strains for modulating pathogen infectivity in the GI tract.
1.0 Introduction
The human gastrointestinal (GI) tract lumen is colonized by hundreds of non-pathogenic (commensal) bacterial species that exist in close proximity to intestinal epithelial cells.1–3 The interactions between the host cells and the ∼1014 bacteria are critical in maintaining normal GI tract function.4,5 Introduction of pathogens such as Escherichia coli O157:H7 (EHEC) into the GI tract disturbs this balance and leads to infection. Although the numbers of pathogen are significantly smaller than the commensal bacteria (e.g., EHEC infections can occur at an infectious dose as low as 100 cfu/mL),6 pathogens often manage to out-compete commensal bacteria and attach to epithelial cells to initiate infection. While pathogen colonization typically proceeds in three distinct steps7 – (i) migration towards the intestinal epithelial cell surface, (ii) navigation through the commensal bacterial layer, and (iii) attachment and infection of epithelial cells (ESI Fig. S1†) - the ability of pathogens to navigate the commensal bacterial layer (or biofilm) and out-compete the resident bacteria is crucial for infection.It is becoming increasingly evident that pathogenic bacterial infections are strongly influenced by the GI tract microenvironment, especially the signaling molecules present in the GI tract.8–10 The commensal bacteria produce a wide range of bacterial signals such as the quorum sensing molecules autoinducer-2 and autoinducer-3, as well as other signals such as indole.11–13 The concentration of these signals in the GI tract is extremely high; for example, indole, which is a stationary-phase signal produced by commensal E. coli that constitute ∼1% of the GI tract microflora,14 has been detected at ∼1 mM in human feces.15 Similarly, the autoinducer-2 quorum sensing molecule is produced by more than 50 different GI tract commensal species,5 and is likely to be present in the commensal biofilm during pathogen colonization. The high local concentration and the close proximity of the colonizing pathogen to these signals has led to a signal-centric paradigm wherein GI tract signals are considered to be the ‘language’ through which commensals prevent pathogen colonization. Prior work from our lab has shown that commensal bacteria-produced soluble signals impact EHEC chemotaxis, biofilm formation, and attachment to epithelial cells. However, not all GI tract signals exert the same effect on pathogens; for example, we have shown that EHEC colonization is increased 2.5-fold in the presence of AI-29 but decreased 3-fold with indole.8
Microfluidic methods have enormous potential for investigating the effect of molecular signals on host–pathogen interactions as they enable localization of different cell types and investigation of signaling molecule concentration gradients on phenotypes relevant in infection.16 While several groups have pioneered the co-culture of different eukaryotic cells (e.g., hepatocytes and fibroblasts),17,18 the co-culture of eukaryotic cells and bacteria has not been described to-date. This has been attributed primarily to the difficulties involved in simultaneously maintaining both cultures because of their widely varying growth rates and the ability of bacteria to take-over eukaryotic cultures rapidly.
The goal of this work was to develop a microfluidic co-culture model of commensal bacteria and epithelial cells that can be used as a screening tool for identifying beneficial GI tract signals and screening the effectiveness of putative probiotic bacterial strains. Pneumatic trapping was used to localize bacteria and epithelial cells, and cultivate them to confluence or as a bacterial biofilm. Infection of intestinal epithelial cells with EHEC using the co-culture model, as well as the importance of EHEC interactions with commensal bacteria, was demonstrated. To our knowledge, this is the first report describing co-culture of bacteria and epithelial cells and its application to investigate pathogen colonization and infection.
2.0 Materials and methods
2.1 Bacterial strains, epithelial cells, materials and growth media
E. coli BW25113 (Yale CGSG Stock Center) was used as the prototypic commensal bacterium. The E. coli BW25113 wild-type and its isogenic mutant deficient in indole (ΔtnaA) was kindly provided by Prof. Thomas Wood. E. coli O157:H7 (CDC EDL933; referred to as EHEC) was obtained from ATCC (Manassas, VA). Plasmids pCM1819 and pDS-RedExpress (Clontech, CA) were used to constitutively express the green fluorescent protein (GFP) and red fluorescent protein (RFP), respectively. Bacteria were cultured in Luria Bertani broth (LB; 10 g L−1 tryptone, 10 g L−1 NaCl, 5 g L−1 yeast extract) for routine maintenance and in M9 minimal medium20 for seeding in the microfluidic device. Erythromycin (150 µg mL−1) and ampicillin (100 µg mL−1) were used for maintaining the GFP and RFP expression plasmids, respectively, in the bacterial strains. HeLa S3 cells (ATCC, VA) were used as the model epithelial cell line and routinely cultured and propagated in DMEM medium with 10% heat-inactivated adult bovine serum (BS) according to standard protocols (ATCC). Although not intestinal in origin, HeLa cells are an appropriate model for investigating EHEC–host cell interactions as it exhibits the morphological changes associated with attaching and effacing bacterial infections and has been extensively used for EHEC colonization studies.11,21 All other chemicals were purchased from MP Biomedicals (Irvine, CA) unless specified otherwise.2.2 Microdevice design and fabrication
Microfluidic chemotaxis devices were fabricated in the Materials Characterization Facility at Texas A&M University. Briefly, device designs were drawn in AutoCAD®2006 (Autodesk Inc.) and used to create a high-resolution (16,256 dpi) photolithography mask from Fineline-Imaging Inc (Colorado Springs, CO). Standard photolithography techniques with SU-8 2050 (Microchem Corp, MA) were used to generate imprints of the microfluidic devices on silicon wafers. The silicon-wafer templates served as negative molds to generate the co-culture device in poly(dimethyl)siloxane (PDMS, Sylgard 184, Dow Corning), using standard soft-lithography protocols.22 Channel dimensions were measured and verified using a profilometer.The PDMS device was fabricated from a molded PDMS bas-relief plate, two PDMS membranes, and a glass slide following the protocol schematically shown in ESI Fig. S2†. First, a PDMS bas-relief plate was fabricated by replica molding against the mask. Two thin PDMS membranes were fabricated by casting and curing the PDMS prepolymer between a master mold and a Teflon sheet (1 mm thick Teflon FEP, DuPont, DE).23 The PDMS membrane for the pneumatic layer was 200 µm thick (valve diaphragm thickness of 50 µm and channel height of 100 µm). The PDMS membrane for the channel layer was 150 µm thick (50 µm diaphragm and 100 µm channel height, respectively). Membranes were fabricated with PDMS posts that were removed using micro-tweezers to generate through-holes for connecting to inlet/outlet of the bacterial island. Before replicating, the mold was treated with tridecafluoro-1,2,2,2-tetrahydrooctyl-1-trichlorosilane to peel off the PDMS membrane from the SU-8 pattern without creating any defects.
The different components were assembled by sequential oxygen plasma treatment and bonding (150 mTorr, 100 W, 40 s) in a plasma etcher. The pneumatic layer membrane was first aligned and bonded to the PDMS bas-relief followed by bonding of the membrane for the channel layer. Tubing was connected to the pneumatic layer and vacuum was applied when the PDMS multilayer structure was bonded to glass to prevent binding between the PDMS island wall and the glass (which enables moving the PDMS wall to form islands). Access ports were punched into the PDMS after bonding. In order to facilitate stable operation of the valve for long time, a gas to liquid pressure converter was used in which regulated compressed air was converted to constant liquid pressure. During operation, the pneumatic channels were filled with dye solution to prevent formation of air bubbles in the channel.
2.3 Bacterial and epithelial cell culture
Commensal E. coli was grown overnight in LB medium and sub-cultured in 0.5× M9 minimal medium and grown at 37 °C with shaking. Exponential-phase (OD600 ∼1 which corresponds to ∼108 cells/mL) bacteria were introduced into the “island” region (formed by lowering a PDMS wall) through the dedicated inlet/outlet and allowed to attach for 2 h. Loosely-attached bacteria were washed out and a dilute bacterial suspension (OD600 ∼0.1) continuously perfused into the island13,24 for 48 h to promote colonization and biofilm formation in the island. The exposed region of the chamber (i.e., the glass slide except the bacterial island region) was coated with fibronectin. With the PDMS wall lowered and commensal bacteria already seeded, exponential-phase HeLa S3 cells were introduced into the microchannel and allowed to colonize without flow. The microdevice was incubated at 37 °C in a 5% CO2 incubator for 48 h to promote development of an epithelial cell monolayer. During this phase, the HeLa cell growth medium was replenished every 12 h.2.4 Commensal E. coli biofilm measurements
Biofilms were imaged using a Leica TCS SP5 Confocal Laser Scanning Microscope (Bannockburn, IL). Green and red fluorescence images, corresponding to E. coli BW25113 and EHEC, respectively, were acquired at 1 µm intervals along the biofilm depth starting from the base of the microfluidic device. Biofilm architecture was visualized using the IMARIS software as previously described.24,252.5 Pathogen colonization in the device
Pathogen colonization experiments were performed with E. coli O157:H7. Exponentially-growing EHEC was introduced into the commensal bacterial island at a cell density corresponding to a multiplicity of infection (m.o.i) of 100 : 1–200 : 1 (number of EHEC per epithelial cell). EHEC was allowed to colonize the commensal biofilm for 6 h, after which time the islands were opened by applying a negative pressure to expose HeLa cells to EHEC. EHEC attachment experiments were carried out for 6 h under no-flow conditions. After 6 h, the channel was rinsed with PBS, and stained with the Live/Dead stain (L3224, Invitrogen, CA) as per the manufacturer's protocol. Live cells (green) were imaged on a Zeiss Axiovert 200M fluorescence microscope using a FITC filter while dead cells (red) were imaged with a Texas Red filter. Fluorescence images were taken at a minimum of five locations, and the number of live and dead cells in each field of view enumerated.3.0 Results and discussion
The goal of this study was to develop a microfluidic co-culture model for investigating the effects of signaling molecules on pathogen colonization in the GI tract. The co-culture device consisted of a large chamber for culturing epithelial cells with two “islands” (1200 µm diameter) for culturing commensal bacteria and EHEC. The two islands, located 1000 µm apart, can be formed (or removed) by lowering (or raising) a 100 µm thick PDMS wall to the chamber surface using a pneumatic source. The islands also have an inlet and outlet for seeding commensal bacteria directly into the island alone and supplying appropriate growth media to the developing commensal biofilm.The overall culture scheme (Fig. 1) consisted of three steps: (i) initially introducing and culturing epithelial cells and commensal bacteria in to distinct regions of the microfluidic device without contact until the epithelial cells reach confluency and a commensal biofilm is developed, (ii) introducing a pathogen (EHEC) into the commensal region and allowing it to navigate through the commensal biofilm, thereby being exposed to signal(s) present in the commensal biofilm, and (iii) exposing epithelial cells to the pathogen for colonization. This model mimics the organization of the GI tract by ‘patterning’ islands of commensal bacteria among epithelial cells. Furthermore, by exposing pathogenic bacteria to signals present in the commensal bacterial biofilm prior to infection of epithelial cells, the developed microfluidic model also reproduced the sequence of events leading to pathogen colonization in the GI tract.
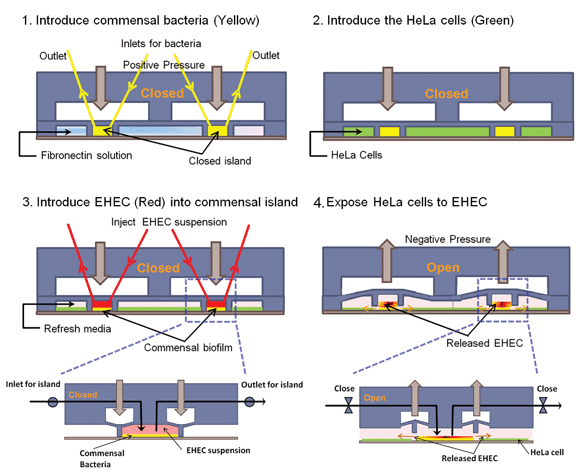 | ||
| Fig. 1 Cell seeding scheme in the co-culture model. (1) The PDMS wall is lowered to form an island, commensal bacteria are introduced into the island, and fibronectin is flowed around the island. (2) HeLa cells are seeded in the regions surrounding the island. (3) After HeLa cells reach confluence and the commensal biofilm has developed, EHEC is introduced into the island. (4) The PDMS wall is lifted up to expose HeLa cells surrounding the island to EHEC. Inset shows details of valve operation. | ||
3.1 Development of the pneumatically-controlled island trapping system
A cell-trapping scheme (Fig. 2A) conceptually similar to that described by Hsu and Folch26 was used to localize commensal bacteria in islands (formed by lowering a PDMS wall using a pneumatic control layer) among epithelial cells so that each cell type can be cultured separately on-chip prior to introduction of EHEC. This culture scheme is advantageous as it prevents the commensal bacteria from taking over the epithelial cell culture (E. coli has a doubling time of ∼30 min in rich media, whereas epithelial cells double ∼16 h). In addition, the separation of bacteria from epithelial cells also enabled use of appropriate medium conditions for bacterial seeding and commensal biofilm formation (M9 minimal medium), as well as for the growth of epithelial cells (DMEM with 10% serum). While co-culture of different eukaryotic cells (e.g., hepatocytes and fibroblasts17,27–29) has been demonstrated in microfluidic flow cells, the co-culture of bacteria and eukaryotic cells has not been previously described. Fig. 2B shows a representative microfluidic co-culture device with the different culture regions for epithelial cells and commensal bacteria highlighted by purple and green dye, respectively.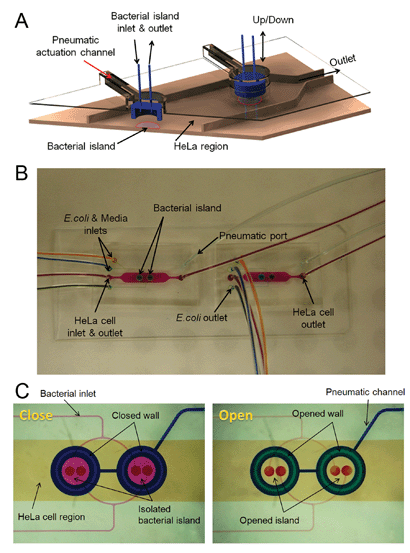 | ||
| Fig. 2 Microfluidic model for co-culture of epithelial cells and bacteria. (A) Three-dimensional rendition of the co-culture device showing pneumatically-actuated trapping regions for forming bacterial islands among epithelial cells. Each bacterial island (1200 µm diameter and 1000 µm apart) has a separate inlet and outlet for providing growth media and removing waste from the island. (B) Micrograph of the co-culture device with color dyes showing the different regions (epithelial cell zone, bacterial islands). (C) The fidelity of the pneumatic trapping system is shown by lowering the PDMS wall (left panel) using a pneumatically-activated channel (blue), introducing purple dye into the closed island islands, and flowing yellow dye around it for 48 h. When the PDMS wall is raised (right panel), the island region is exposed to the surrounding yellow dye. Scale bar represents 500 µm. | ||
Two types of pneumatically-actuated PDMS valves have been previously reported – valves comprising of only PDMS layers30,31 and hybrid valves assembled with glass and PDMS layers.32,33 While both types of valves have been extensively used, the latter valve can be more efficiently operated with lower pressures due to the preferential adhesion between PDMS and glass. However, fabrication of these valves is time and labor-intensive, and requires etching of glass which can create an unfavorable environment for cell culture. The valve system developed in this study takes advantage of the adhesiveness of PDMS to glass, while still being able to support eukaryotic cell culture. The valve was also designed such that the center of the valve is fixed and the entire valve layer does not move (see close-up of valve operation in Fig. 1). Keeping the center fixed and moving the valve walls alone enables generation of larger islands and reduces the volume of liquid that is replaced when the valve is opened or closed (∼50 nL with movement of the valve wall compared to ∼130 nL with movement of the entire valve layer). This feature is especially important as the bacterial signaling molecules present in the island microenvironment can be potentially lost with large volume changes.
Since the trapped bacteria and surrounding epithelial cells need to be cultured separately without any bacteria escaping the island and contaminating the epithelial cell regions, it was important to ensure the fidelity of the cell trapping scheme and demonstrate the ability to sequester different cell types in specific locations for long periods of time. This capability is demonstrated in Fig. 2C using color dyes. Positive pressure was applied through the pneumatic channel to lower the PDMS wall and form the bacterial islands. Purple dye was injected into the islands while yellow dye was injected into the surrounding areas. The microdevice was imaged after 48 h. The data show that the purple dye in the island is distinct from the surrounding yellow dye after 48 h. When the PDMS wall is raised, the yellow dye rapidly fills the island; thereby, demonstrating the effectiveness of the pneumatic trapping system in sequestering contents of the island from the surroundings.
3.2 Co-culture of commensal bacteria and epithelial cells
The pneumatically-controlled islands were formed by applying positive pressure to lower the PDMS wall. Exponential-phase commensal E. coli BW25113 expressing GFP was introduced into the islands while HeLa cells were seeded outside the islands. This culture scheme resulted in commensal bacterial islands surrounded by a monolayer of HeLa cells after 48 h. Both E. coli BW25113 and HeLa cells were allowed to grow in their respective culture regions for 48 h (i.e., until a commensal E. coli biofilm was established and HeLa cells reached confluence). Fig. 3A and B show representative images of HeLa cells and GFP-expressing commensal bacteria in the device, respectively. Fig. 3C and D show superimposed images of HeLa cells and E. coli after 48 h culture in the microfluidic device. A monolayer of HeLa cells is observed and more importantly, minimal GFP expressing bacteria are detected outside the island. These results clearly demonstrate that the co-culture model promotes the development of an epithelial cell monolayer and a bacterial biofilm; thereby, creating a system that enables investigation of the local microenvironment on pathogen colonization.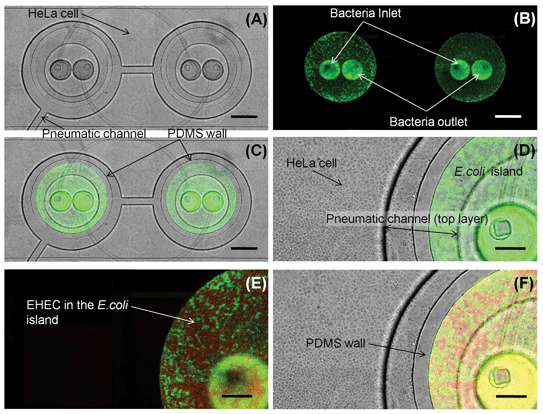 | ||
| Fig. 3 Co-culture of HeLa cells and bacteria. (A) Transmitted light image of HeLa cell monolayer. (B) Fluorescence image of GFP-expressing E. coli BW25113 localized in the bacterial islands. (C) Overlay of transmitted and green fluorescence images showing co-culture of HeLa cells and E. coli BW25113 for 48 h. (D) Close-up view of HeLa cells and E. coli BW25113 in bacterial-island after 48 h. (E) Fluorescence image of RFP-expressing EHEC and GFP-expressing E. coli BW25113 in island. (F) Overlay of transmitted, green, and red fluorescence images in the device. Scale bar represents 500 µm in panels (A)–(C) and 200 µm in panels (D)–(F). | ||
The commensal bacteria in the island region were not loosely attached but formed a biofilm. Confocal microscopy and three-dimensional reconstruction of the biofilm using IMARIS (Fig. 4) show that the commensal E. coli (green) uniformly colonized the island and grew to a thickness of ∼30 µm after 48 h, which is consistent with prior reports on E. coli BW25113 biofilm formation in flow cells.34 The organization of the biofilm indicated minimal void space (or polysaccharides that can occupy the acellular region) as the bacteria were densely packed and present at all depths and locations of the biofilm.
 | ||
| Fig. 4 Localization of EHEC in E. coli BW25113 biofilms. IMARIS visualization of EHEC (red) in E. coli BW25113 biofilms (green) developed on glass inside the bacterial island. The average E. coli BW25113 biofilm thickness was 30 µm. Red and green renditions were overlayed to obtain the distribution of EHEC in the commensal biofilm. | ||
3.3 EHEC colonization of commensal E. coli biofilm
In the GI tract, infections occur when a pathogen navigates the commensal bacterial layer present in the GI tract lumen and migrates to the epithelial cell surface. During the process of navigation, the pathogen is exposed to the signals present in the commensal microenvironment. The sequence of events in GI tract infections was mimicked by exposing EHEC to the commensal E. coli BW25113 biofilm prior to infection of HeLa cells. EHEC expressing RFP was introduced into the isolated bacterial island and allowed to navigate the commensal layer (and thereby, get exposed to the signals present in the commensal bacterial microenvironment) for 6 h. Fig. 3E shows colonization of the commensal biofilm (green) island by EHEC (red) and Fig. 3F shows the juxtaposition of epithelial cells and commensal E. coli with EHEC in the bacterial island. As was observed with the commensal bacteria, the RFP expressing EHEC are also localized to the island when the PDMS wall is lowered, and illustrates that the bacterial island can be isolated from epithelial cells.Conventional assays for pathogen attachment and colonization utilize a monolayer of eukaryotic cells in tissue culture plates into which pathogens are added. These models are not physiologically-relevant as they do not incorporate a commensal bacterial biofilm developed on eukaryotic cells. Simple addition of a pre-grown bacterial culture to eukaryotic cells is unlikely to lead to this conformation as biofilms are highly organized structures that develop over time, and it is extremely difficult, if not virtually impossible, to culture eukaryotic cells in the presence of bacteria for extended periods of time without significant loss in viability. Since pathogens do not navigate through a commensal biofilm in these models to attach to epithelial cells, these models do not accurately mimic the organization of epithelial cells and commensal bacteria in the GI tract. Moreover, in order to investigate the effect of different signals on pathogen colonization, molecules are added exogenously to the eukaryotic cells such that their concentration is uniform throughout the culture. This is also different from what pathogens encounter in a biofilm, as the heterogeneity and spatial organization of bacteria in biofilms3 result in highly localized zones of signals with varying concentrations (i.e., a gradient). The microfluidic co-culture model developed here addresses these two issues by enabling localization of commensal bacteria and epithelial cells, as well as pre-exposing pathogens to commensal bacteria prior to encountering epithelial cells; thereby, presenting a more physiologically-relevant environment during colonization.
The distribution of EHEC within the commensal biofilm is an important determinant of its attachment and infectivity as it needs to navigate through the commensal film to initiate attachment to epithelial cells. For attachment to proceed effectively, EHEC needs to be present near the bottom of the commensal biofilm. Fig. 4 shows that EHEC (red) is incorporated in all depths of the biofilm, including the bottom. The uniform distribution of EHEC throughout the commensal biofilm also suggests it is likely to have been exposed to the signals present in the commensal bacterial biofilm microenvironment.
3.4 Investigating the role of commensal molecules in EHEC colonization
The microfluidic co-culture model was used to investigate the effect of molecules present in the commensal microenvironment on EHEC ability to colonize and infect epithelial cells. A co-culture of HeLa cells, commensal E. coli BW25113, and EHEC was established as described above. After 6 h of EHEC exposure to the commensal microenvironment, the PDMS walls were raised by applying a negative pressure through the pneumatic channel. Raising of the PDMS walls resulted in EHEC migrating out of the bacterial islands and infecting the HeLa cells surrounding the island, leading to cell death.7 EHEC infection was allowed to proceed for 6 h without any flow (i.e., under its own ability to swim, migrate, and attach to HeLa cells). The extent of cell death in the HeLa cell regions surrounding the island was determined using a Live/Dead assay. In order to minimize interference with the Live/Dead assay, EHEC without RFP was used in these experiments. Minimal cell death was observed in the absence of EHEC (not shown) and the presence of dead cells surrounding the bacterial islands (Fig. 5A) clearly demonstrates that EHEC infects HeLa cells; thereby, validating the co-culture model.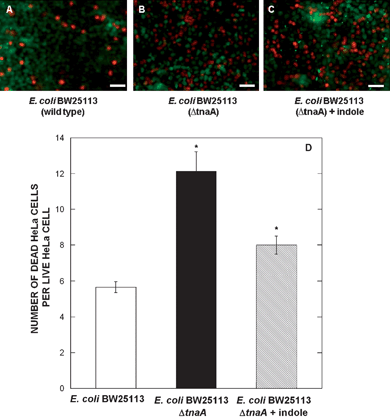 | ||
| Fig. 5 EHEC infection in co-culture device. Live/Dead staining of EHEC infection in bacterial islands containing (A) wild-type E. coli BW25113, (B) E. coli BW25113 ΔtnaA, and (C) E. coli BW25113 ΔtnaA with indole pre-treatment. Infection was performed at a multiplicity of infection of 200 : 1 (EHEC : HeLa cells). Images shown are from one representative location from five locations and two independent experiments. Scale bar represents 50 µm. (D) Quantification of the percentage of dead cells per live cell. Data shown are averaged from five images in two independent experiments (total of 10 locations). | ||
The effect of the commensal biofilm microenvironment (i.e., extracellular signals present in the biofilm) on EHEC attachment and infection was also investigated using the co-culture model. A commensal biofilm lacking the bacterial signal indole was developed by forming a commensal E. coli BW25113 ΔtnaA (isogenic mutant strain lacking the tnaA gene) biofilm, and EHEC was introduced into the biofilm. While prior work from our lab has shown that externally-added indole at a concentration of 500 µM inhibits EHEC attachment to HeLa cells,8 the effect of in situ produced indole (i.e., by E. coli in a biofilm) on EHEC colonization has not been studied. Our data (Fig. 5B and D) show that EHEC exposed to a commensal biofilm that lacks indole demonstrates a 2-fold increase in infectivity (as determined by the ratio of dead to live HeLa cells) compared to the wild-type strain, and is comparable to our previous data in standard tissue culture formats.8 In order to establish whether local indole exposure is required for attenuating EHEC infectivity, we pre-treated EHEC with 500 µM indole for 6 h prior to its introduction into an E. coli ΔtnaA biofilm, followed by infection of HeLa cells. Our data (Fig. 5C and D) show that indole pre-treatment decreases the extent of HeLa cell death but not to the levels observed with the wild-type strain. This suggests that local exposure to signals is more effective than pre-treatment, presumably because local concentrations of indole (not measured in this study) could be higher than the uniform concentration in the liquid phase. However, this observation does not preclude role(s) for other bacterial signals could in attenuating EHEC infection. Current work in our laboratory focuses on investigating the effect of different commensal signals on EHEC colonization of epithelial cells.
These results are especially significant as they suggest a spatial bias to colonization and the initiation of infection. Since the commensal microflora in the GI tract is heterogeneous and not uniform throughout,3 it is reasonable to expect that the distribution of signals is also heterogeneous. That is, bacteria that produce certain signals may be located only in certain niches and the ability of EHEC to colonize those niches should be different from other locations where no favorable or antagonistic signals are present. Since E. coli makes up ∼1% of the GI tract microflora,14 it is tempting to speculate that EHEC infections are minimal at locations where commensal E. coli is present. Current work in our laboratory focuses on testing this hypothesis.
It should be noted while we utilized a ∼30 µm commensal biofilm in our studies, the thickness of the commensal layer in vivo (and hence, the time taken to navigate the commensal layer) is not known. However, based on the large number of commensal bacteria (∼1014) present in the GI tract, the commensal layer is expected to be at least of comparable thickness, and a colonizing pathogen would be expected to be exposed to signals for a comparable duration. While we did not vary the thickness of the commensal layer in our experiments, further studies are required to fully investigate the relationship between the commensal biofilm thickness (i.e., time of exposure to commensal signals) and the extent of pathogen colonization. A second area of improvement of the model system presented here is on using polarized intestinal epithelial cells on-chip. While the co-culture model described here is based on non-polarized cells, epithelial cells in the GI tract are polarized and only the apical side of the cells is exposed to the commensal biofilm and pathogen. Hence, using polarized epithelial cells would more accurately mimic the GI tract organization.
The microfluidic co-culture model of bacteria and epithelial cells described here can be used for mechanistic studies on the role of different signals in infections to screening as well as for identifying probiotic molecules for combating bacterial infections. Our observation that local exposure may be more effective than pre-treatment is especially significant, as it strongly suggests the potential utility of the co-culture model for the identification and screening putative probiotic strains. Since the GI tract microflora is extremely diverse with more than 500 species, the ability to rapidly screen commensal bacteria for countering pathogen colonization in a physiologically-relevant model could lead to the identification of potential probiotic strains. In this regard, the co-culture model is advantageous as the isolated island facilitates the use of culture conditions (e.g., growth media, microaerophilic environment) optimal for each strain being tested.
4.0 Summary
In summary, we have developed a microfluidic device that uses pneumatically-controlled trapping for co-culture of eukaryotic cells and bacteria. Using HeLa cells as the model eukaryotic cell line and EHEC as the model pathogen, we show that the co-culture device can keep the island region isolated as well as support the cultivation and development of a HeLa cell monolayer and a commensal E. coli biofilm. In addition, we demonstrate that the co-culture model can be a useful tool for fundamental studies focused on investigating the role of specific signals on EHEC infectivity as well as for applications such as screening potential probiotic strains.Acknowledgements
This work was supported by funds from the National Science Foundation (CBET 0846453) and the Texas Engineering Experiment Station to AJ. JYK was partially supported by the South Korea Research Foundation Grant funded by the Korean Government (KRF-2008-357-D00318).References
- W. L. Hao and Y. K. Le, Methods Mol. Biol., 2004, 268, 491–502.
- L. S. Collier-Hyams and A. S. Neish, Cell. Mol. Life Sci., 2005, 62, 1339–1348 CrossRef CAS.
- S. Macfarlane and J. F. Dillon, J. Appl. Microbiol., 2007, 102, 1187–1196 CrossRef CAS.
- R. D. Berg, Trends Microbiol., 1996, 4, 430–435 CrossRef CAS.
- M. B. Clarke and V. Sperandio, Am. J. Physiol.: Gastrointest. Liver Physiol., 2005, 288, G1105–1109 CrossRef CAS.
- J. J. LeBlanc, Crit. Rev. Microbiol., 2003, 29, 277–296 CAS.
- J. B. Kaper, J. P. Nataro and H. L. T. Mobley, Nat. Rev. Microbiol., 2004, 2, 123–139 CrossRef CAS.
- T. Bansal, D. Englert, J. Lee, M. Hegde, T. K. Wood and A. Jayaraman, Infect. Immun., 2007, 75, 4597–4607 CrossRef CAS.
- T. Bansal, P. Jesudhasan, S. Pillai, T. K. Wood and A. Jayaraman, Appl. Microbiol. Biotechnol., 2008, 78, 811–819 CrossRef CAS.
- M. Hegde, T. K. Wood and A. Jayaraman, Appl. Microbiol. Biotechnol., 2009, 84, 763–776 CrossRef CAS.
- V. Sperandio, A. G. Torres, B. Jarvis, J. P. Nataro and J. B. Kaper, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8951–8956 CrossRef CAS.
- J. B. Kaper and V. Sperandio, Infect. Immun., 2005, 73, 3197–3209 CrossRef CAS.
- J. Lee, A. Jayaraman and T. K. Wood, BMC Microbiol., 2007, 7, 42 CrossRef.
- D. L. Hartl and D. E. Dykhuizen, Annu. Rev. Genet., 1984, 18, 31–68 CrossRef CAS.
- D. A. Karlin, A. J. Mastromarino, R. D. Jones, J. R. Stroehlein and O. Lorentz, J. Cancer Res. Clin. Oncol., 1985, 109, 135–141 CrossRef CAS.
- D. L. Englert, M. D. Manson and A. Jayaraman, Appl. Environ. Microbiol., 2009, 75, 4557–4564 CrossRef CAS.
- S. N. Bhatia, U. J. Balis, M. L. Yarmush and M. Toner, FASEB J., 1999, 13, 1883–1900 CAS.
- G. Stybayeva, H. Zhu, E. Ramanculov, S. Dandekar, M. George and A. Revzin, Biochem. Biophys. Res. Commun., 2009, 380, 575–580 CrossRef CAS.
- M. C. Hansen, R. J. J. Palmer, C. Udsen, D. C. White and S. Molin, Microbiology, 2001, 147, 1383–1391 CAS.
- R. L. Rodriguez and R. C. Tait, Recombinant DNA techniques: An Introduction, Benjamin/Cummings Publishing, Menlo Park, CA, 1983 Search PubMed.
- V. Sperandio, A. G. Torres and J. B. Kaper, Mol. Microbiol., 2002, 43, 809–821 CrossRef CAS.
- J. C. McDonald, M. L. Chabinyc, S. J. Metallo, J. R. Anderson, A. D. Stroock and G. M. Whitesides, Anal. Chem., 2002, 74, 1537–1545 CrossRef CAS.
- N. L. Jeon, D. T. Chiu, C. J. Wargo, H. Wu, I. S. Choi, J. R. Anderson and G. M. Whitesides, Biomed. Microdevices, 2002, 4, 117–121 CrossRef.
- J. Lee, T. Bansal, A. Jayaraman, W. E. Bentley and T. K. Wood, Appl. Environ. Microbiol., 2007, 73, 4100–4109 CrossRef CAS.
- V. Janakiraman, D. L. Englert, A. Jayaraman and H. Baskaran, Ann. Biomed. Eng., 2009, 37, 1206–1216 CrossRef.
- C. H. Hsu and A. Folch, Appl. Phys. Lett., 2005, 86, 023508 CrossRef.
- E. E. Hui and S. N. Bhatia, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5722–5726 CrossRef CAS.
- J. Y. Lee, S. S. Shah, J. Yan, M. C. Howland, A. N. Parikh, T. Pan and A. Revzin, Langmuir, 2009, 25, 3880–3886 CrossRef CAS.
- E. Leclerc, K. El Kirat and L. Griscom, Biomed. Microdevices, 2008, 10, 169–177 CrossRef.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS.
- J. Y. Baek, J. Y. Park, J. I. Ju, T. S. Lee and S. H. Lee, J. Micromech. Microeng., 2005, 15, 1015–1020 CrossRef.
- W. H. Grover, R. H. Ivester, E. C. Jensen and R. A. Mathies, Lab Chip, 2006, 6, 623–631 RSC.
- W. H. Grover, A. M. Skelley, C. N. Liu, E. T. Lagally and R. A. Mathies, Sens. Actuators, B, 2003, 89, 315–323 CrossRef.
- T. K. Wood, A. F. Gonzalez Barrios, M. Herzberg and J. Lee, Appl. Microbiol. Biotechnol., 2006, 72, 361–367 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures S1 and S2. See DOI: 10.1039/b911367c |
| This journal is © The Royal Society of Chemistry 2010 |