A multi-layer microfluidic device for efficient culture and analysis of renal tubular cells†
Kyung-Jin
Jang
ab and
Kahp-Yang
Suh
*ab
aInterdisciplinary Program in Nano-Science and Technology, Seoul, 151-747, Korea
bSchool of Mechanical and Aerospace Engineering, Seoul National University, Seoul, 151-747, Korea. E-mail: sky4u@snu.ac.kr
First published on 26th August 2009
Abstract
We have developed a simple multi-layer microfluidic device by integrating a polydimethyl siloxane (PDMS) microfluidic channel and a porous membrane substrate to culture and analyze the renal tubular cells. As a model cell type, primary rat inner medullary collecting duct (IMCD) cells were cultured inside the channel. To generate in vivo-like tubular environments for the cells, a fluidic shear stress of 1 dyn/cm2 was applied for 5 hours, allowing for optimal fluidic conditions for the cultured cells, as verified by enhanced cell polarization, cytoskeletal reorganization, and molecular transport by hormonal stimulations. These results suggest that the microfluidic device presented here is useful for resembling an in vivo renal tubule system and has potential applications in drug screening and advanced tissue engineering.
1. Introduction
The kidney is a complex osmoregulatory organ, where the reabsorption of water and molecules is controlled by endocrine mechanisms. To maintain homeostasis, the renal transporters and water channels are localized in the apical and basolateral membranes of tubular epithelial cells which are subjected to a urinary flow along the tubule.1–3 Although the kidney tubular cells are exposed to fluidic environments, the effect of fluidic shear stress on the tubular cells is much less understood, as compared to that on the vascular endothelial cells.4 Like endothelial cells lining the vasculature, renal tubular epithelial cells experience mechanical forces in response to variations in urinary flow rate, i.e., fluid flow-induced shear forces.5–7 The flow rate within the renal tubular system is estimated in the range of 0.2∼20 dyn/cm2, which is ∼10% of the endothelial cell. This fluid flow-induced shear stress may play an important role in the regulation of ions and water balance.Recently, some fluidic studies have been carried out to clarify unrevealed functions and phenomena of renal tubular cells. For example, Duan et al.4 and Essig et al.8 showed that shear stress can cause cytoskeletal reorganization and junctional reformation in renal proximal tubular epithelial cells. This result is opposed to what has been observed for the vascular endothelial cells and may be associated with optimum exchange of transcellular molecules. In inner medullary collecting duct (IMCD) cells, Cai et al.6 reported that nitric oxide production was modulated by shear stress, and Liu et al.7 demonstrated that an acute increase in tubular fluid flow rate led to an increase in intracellular concentration of Ca2+. The shear stress-induced cytoskeletal reorganization and the coordinated remodeling of junctional complexes, however, have not been reported for the renal IMCD cells.
In addition to the control of fluidic environments, it is important for the IMCD cells to form polarized membrane traffic and cell polarity that can be obtained in cultures on a porous membrane support. Tubular cells grown on the porous membrane can be provided nutrients from the basolateral region and become highly differentiated and polarized with enhanced cell junction and increased cell height.9 Despite these needs, there have been a handful of trials to culture tubular cells with the polarization because of the convenience of using traditional cell culture substrates. It is noted that such cell cultures may generate extracellular microenvironments that are quite different from those in vivo and thus a truly biomimetic microfluidic platform incorporating the effects of shear stress and porous membrane support is potentially of great benefit (Supplementary Fig. 1†).10–18
Here, we introduce a simple multi-layer microfluidic device (MMD) to culture and analyze the renal tubule cells. As a model cell type, primary cultured rat IMCD cells (Fig. 1) were used and cultured inside the channel. The device was formed by constructing two compartments (one flow chamber + one static chamber) inside the microfluidic device divided by a porous membrane.19,20 Accordingly, IMCD cells were exposed to a defined laminar shear stress within the channel, and immunofluorescence staining on the major proteins of the cytoskeleton and cell junctions was performed. Then, the generation of cell polarity was analyzed with apical and basolateral marker proteins. Furthermore, it was observed that the transport of water and ions through renal cell bodies could be measured directly by collecting the fluid stream and subsequent analysis, when triggered by hormonal stimulations. These results demonstrate that the combination of shear stress and porous membrane support is appropriate for creating biological tubule environments for various applications.
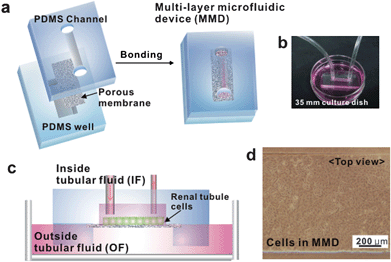 | ||
| Fig. 1 Fabrication and operation of a multi-layer microfluidic device (MMD). (a) The MMD is a sandwich assembly of polydimethyl siloxane (PDMS) channel, polyester membrane, and PDMS reservoir bonded with plasma treatment. (b) Photograph showing the operation of MMD that is connected to a syringe pump with silicone tubes. The MMD is placed on a culture dish containing an outside tubular fluid (OF). (c) Schematic of the device on a culture dish. (d) Microscope image of IMCD cells within the MMD. Cells are grown confluently after seeding 3 days. This image shows complete isolation of cells in the channel without any leakage. | ||
2. Materials and methods
Materials and reagents
Materials for the device fabrication were obtained from the following suppliers: polydimethyl siloxane (PDMS) (Sylgard 184 Silicone elastomer kit, Dow Corning, Midland, MI), polyester porous membrane (pore size; 0.4 µm, transwell insert, Corning Inc, Acton, MA). Reagents for the cell culture: Dulbecco's phosphate-buffered saline (D-PBS). Dulbecco's modified Eagle's medium: Ham's nutrient mixture F-12, 1:1 mix (DMEM/F-12), fetal bovine serum, penicillin-streptomycin were obtained from GIBCO BRL (Grand Island, NY). Collagenase B was purchased from Roche (Mannheim, Germany), chamber slides (Lab-Tek Chamber Slides System) and Petri dish were purchased form NUNC (Roskilde, Denmark), and cytotoxicity assay kit was purchased from Molecular Probes Inc. (Eugene, Oregon). Antibodies were obtained from the following suppliers: tetramethylrhodamine isothiocyanate (TRITC)-phalloidin (Sigma, St. Louis, MO), mouse anti-vinculin (Sigma), mouse anti-paxillin (Chemicon, Temecula, CA), rabbit anti-β-catenin (Sigma), mouse anti-E-cadherin (Chemicon), goat anti-aquaporin 2 (Santa Cruz Biotechnology, Inc., CA), mouse anti-Na-K-pump (Chemicon), and secondary antibodies: goat anti-mouse IgG-FITC, donkey anti-goat IgG-FITC, donkey anti-rabbit IgG-FITC, and donkey anti-mouse IgG-TRITC (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Lipofectamine reagent was purchased from Invitrogen (San Diego, CA). All other reagents were purchased from Sigma (St. Louis, MO).Device fabrication
PDMS microfluidic moulds (1 mm width, 1 cm length, and 100 µm height) were fabricated by curing PDMS pre-polymer on silicon masters with the impression of microfluidic channels (features sticking out) that had been prepared by photolithography. To cure the PDMS pre-polymer, a mixture containing the silicone elastomer and the curing agent (10:1 weight ratio) was poured onto the master and baked at 70 °C for 1 h. After peeling off from the silicon masters, the PDMS replicas were cut into narrow strips and holes were punched for inlets and outlets of the microfluidic devices. Square wells as a mass reservoir were formed on the bottom PDMS surface by cutting out a block (∼2 mm width and ∼0.6 cm long) with a sharp blade. For the confocal microscopy analysis, the thickness of PDMS well was controlled between 1 and 2 mm. The polyester membrane (pore size; 0.4 µm, 10 µm thickness) was prepared by cutting (∼6 mm in width and ∼1.6 cm in long) with scissors.To generate a MMD, a sandwiched assembly of PDMS microfluidic channel, porous membrane, and PDMS well was bonded after oxygen plasma treatment (60 W, PDC-32G, Harrick Scientific, Ossining, NY) for 1 min. As a control, a microfluidic PDMS channel was boned on glass substrate after both surfaces were treated by oxygen plasma.21,22
Primary culture of rat kidney IMCD cells and flow experiments
Primary IMCD cell culture was performed as described previously.3 After anesthetization by diethyl ether, both kidneys were removed from male Sprague-Dawley rats (7 weeks old, 200 g body wt, Samtako, Osan, Korea). The inner medulla was quickly dissected from the kidney and then cut into small pieces (∼1 mm cubes) with a surgical blade in cold D-PBS (pH 7.4, supplemented with 80 mM urea, and 130 mM NaCl, 640 mOsm/kgH2O). The tissues were digested in enzyme solution (1 ml of DMEM/F-12, 2 mg of collagenase B, 0.7 mg of hyaluronidase, 80 mM urea, and 130 mM NaCl) and incubated at 37 °C in a humidified 5% CO2 incubator for 60∼90 min with occasional mixing. After observing the tubule fragments, the digestion of tissue was stopped by adding a hypertonic culture medium (DMEM/F-12, 80 mM urea, 130 mM NaCl, 10 mM HEPES, 2 mM L-glutamine, penicillin-streptomycin (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U/ml) and 10% FBS (pH 7.4, 640 mOsm/kgH2O)] and centrifuged at 160 g for 1 min. After repeating the washing step three times, the IMCD cells were seeded on the fibronectin-coated (10 µg/ml for 1 h) device and chamber slides (glass control). The PDMS well was filled with medium and the MMD was plated on the Petri dish (30-mm) with sufficient medium.
000 U/ml) and 10% FBS (pH 7.4, 640 mOsm/kgH2O)] and centrifuged at 160 g for 1 min. After repeating the washing step three times, the IMCD cells were seeded on the fibronectin-coated (10 µg/ml for 1 h) device and chamber slides (glass control). The PDMS well was filled with medium and the MMD was plated on the Petri dish (30-mm) with sufficient medium.
IMCD cells were cultured for 3 days in hypertonic culture medium at 37 °C in humidified 5% CO2 incubator. Two PDMS layers were strongly bonded without any leakage. The cells did not penetrate the border between the channel and the membrane and were grown confluently after 3 days (Fig. 1d). After the confluency, cells were exposed to a fluid shear stress of 1 dyn/cm2 for 5 h with hypertonic flow medium (DMEM/F12 with 1% FBS, 640 mOsm/kgH2O) using syringe pump (KD Scientific, Holliston, MA) at 37 °C. This flow rate was calculated using the following equation: τ = 6µQ/bh2, where µ is the medium viscosity (gm/cm/s), Q is the volumetric flow rate (cm3/s), b is the channel width, and h is the channel height.4 Each set of experiments was repeated five times.
Immunocytochemistry
After flow experiments, cell-attached membrane substrate was taken apart from the MMD. Cells were quickly washed with cold PBS and fixed with paraformaldehyde solution (3.7% in PBS) for 15 min. After washing three times with PBS, the cells were incubated with blocking and permeabilization solution (1% BSA and 0.3% Triton-X 100 in PBS) for 15 min. The cells were then stained with the following antibodies: TRITC-phalloidin (1:200 in PBS) for filamentous actins (F-actin), anti-vinculin (1:100 in PBS), anti-paxillin (1:100 in PBS), anti-β-catenin (1:2000 in PBS), anti-E-cadherin (1:100 in PBS), anti-aquaporin 2 (1:100 in PBS), and anti-Na-K-pump (1:500 in PBS) for overnight at 4 °C, washed with PBS three times, and incubated with fluorescein-conjugated secondary antibody for 1 h at room temperature. After washing three times with PBS, the cells were covered with mounting medium containing antifade reagent. Z-sectioned fluorescent images were captured under a confocal laser microscope (LSM510, Carl Zeiss, Jena, Germany).Cell viability analysis
After 5 h of shear stress, 1 mM of hydrogen peroxide (H2O2) solution for the oxidative stress condition was treated to the inner tubular fluid (IF) and outer tubular fluid (OF) regions. As a control, cells on the glass substrate were immersed in H2O2 solution. Before the oxidative stress condition, cell nuclei were labeled with 10 µM Hoechst for 30 min. The live/dead cell count was determined by a cytotoxicity assay kit and examined using an inverted fluorescence microscope (OLYMPUS IX71, Olympus Optical Co. Ltd., Tokyo, Japan). Initial cell number was measured using Hoechst staining. Cells were incubated with Eth-1 (4 mM in H2O2 solution, red fluorescence) for 20 min to detect dead cells during 0–4 h of experimental time points and incubated with H2O2 solution again. After 12 h, final cell viability was determined using calcein-AM (2 mM, green fluorescence) to detect live cells and Eth-1 solution for 20 min. The cells were washed in PBS and immediately viewed with a fluorescence microscope.23AQP2-transfected MDCK cell
Mouse AQP2 cDNA construct was inserted into the pEGFP-C1 vector using standard cloning techniques.3 MDCK (Madin-Darby canine kidney) cells were seeded in a culture dish in DMEM medium supplemented with 10% fetal bovine serum at 37 °C in 5% CO2 and then transiently transfected using lipofectamine 2000 (Invitrogen). Next, AQP2-GFP transfected cells were seeded in the MMD and maintained until confluence. For the arginine vasopressin (AVP) treatment, cells were exposed to a fluid shear stress of 1 dyn/cm2 for 5 h and then 10−9 M AVP was added in the PDMS well for the stimulation to the basal region of cell. The live imaging of AQP2-GFP was performed using confocal microscopy.3. Results and discussion
Device fabrication and operation
Fig. 1a shows a schematic procedure for the fabrication of a multi-layer microfluidic channel system for resembling an in vivo tubule-like environment. The top PDMS channel provides a lumen area, to which the apical membrane region of cells is exposed with an inner tubular fluid (IF, precursor fluid of urine). The bottom PDMS channel provides an interstitial area, which is in contact with the basolateral membrane region of cells with an outer tubular fluid (OF, blood stream) for the stimulation and exchange. To perform microfluidic experiment, the PDMS channel was connected to a syringe pump for applying a fluid shear to the cultured cells. Also, the PDMS well in the form of a square reservoir was filled with the medium and placed on the culture dish containing an OF to provide sufficient air and medium circulation and hormonal stimulation (Fig. 1b–c). Two PDMS layers and porous membrane were bonded strongly after oxygen plasma treatment without any leakage. IMCD cells were cultured for 3 days on the fibronectin coated membrane in hypertonic medium (640 mOsm/kgH2O) supplemented with 10% FBS at 37 °C in 5% CO2 and then exposed to 1 dyn/cm2 of shear stress for 5 h. The cells did not penetrate the border between the channel and the membrane and were grown confluently after 3 days (Fig. 1d).Cellular conformational rearrangement
We first analyzed the intracellular conformational rearrangement and functional changes under two conditions (i.e., MMD and glass control) to determine whether cell height is affected by shear stress and porous substrate. IMCD cells, when cultured on the porous membrane within the channel, are exposed to an IF and thus are provided nutrients and stimulus by an OF contacting with their basal region. For glass control, the cells are plated on the solid substrate without having an apical basal region (Fig. 2a). Cellular conformational change and cell height were measured by confocal microscope images of AQP2 (apical marker protein) and Na-K-pump (basolateral marker protein) with x–z optical sectioned slices. As shown in Fig. 2b, AQP2 was localized at the intracellular vesicle regions, while Na-K-pump was localized at the basolateral membrane, suggesting that the MMD provides an optimum condition for well-organized cell polarization. In contrast, AQP2 protein and Na-K-pump were not distributed separately in the cells when cultured on glass control. The cell height was also measured to investigate how cell polarization is affected by substrate and fluidic environments. Quantitative analysis of cell height demonstrated that cells cultured inside the channel with 5 h of fluidic stimulation showed 13.6 ± 1.6 µm height, while the control showed 4.6 ± 0.3 µm height (Fig. 2c). These results indicate that IMCD cells can recognize their apical and basolateral surfaces within the MMD and the fluid shear stress can cause a synergistic effect on the cell height, which was even thicker than that on the static transwell system (7.1 ± 0.4 µm, data not shown).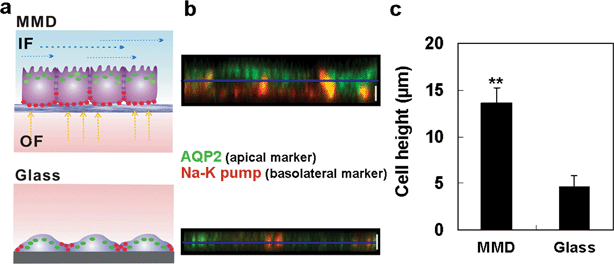 | ||
| Fig. 2 Cellular conformational rearrangement of IMCD cells cultured in MMD and glass substrate. (a) Schematic for the comparison of MMD and conventional culture systems. (b) x–z optical sectioned confocal microscope images of AQP2 (apical marker protein) and Na-K-pump (basolateral marker protein) after 5 h of fluid shear stress with MMD and glass control. Scale bar, 5 µm. (c) Quantitative analysis of cell height. Cells cultured in MMD with 5 h of fluidic stimulation show 13.6 ± 1.6 µm height and on the glass control show 4.6 ± 0.3 µm height. Error bar, s.d. of the mean. (n = 50) (**, p < 0.001). | ||
Cell viability under oxidative stress condition
To compare cell viability after inducing an oxidative stress, the cells in the MMD with 5 h of shear stress and glass substrate with static condition were treated with 1 mM of hydrogen peroxide (H2O2) solution. H2O2 is known as a major reactive oxygen species and related to apoptotic or necrotic cell death.24 To measure the cell viability rate after oxidative stress, initial cell number was counted after staining with Hoechst for the nuclei count. Both for MMD and glass substrate, IMCD cells were treated with H2O2, and the cell viability was determined at various time points (0–12 h). To elucidate the time course damage of oxidative stress for 0–4 h, Eth-1 solution was used to detect dead cell in red fluorescence. After 12 h, the number of live and dead cells was counted using an inverted fluorescence microscope after staining with calcein-AM (green fluorescence) and Eth-1 solution (Fig 3a). Quantitative data of cell viability rate was calculated with initial cell number and dead cell number at varying time points.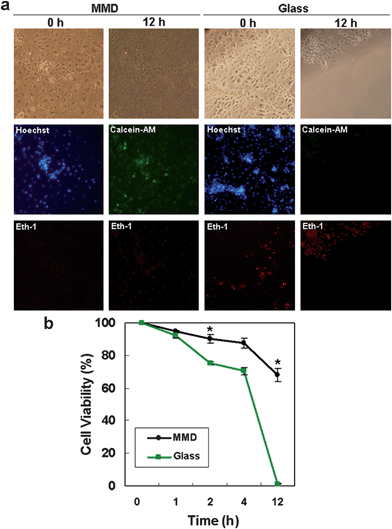 | ||
| Fig. 3 Cell viability under an oxidative stress condition in MMD and glass substrate. (a) Microscope images of IMCD cells in MMD after 5 h of shear stress and on glass control after 0 h and 12 h of exposure to hydrogen peroxide (H2O2) solution (1 mM). Hoechst was used to label cell nuclei to count the initial cell number. Eth-1 (red) for dead cell and Calcein-AM (green) for live cell were used to measure the cell viability. Images were taken under an inverted fluorescence microscope. (b) Quantitative analysis of cell viability. (*, p < 0.05). | ||
As shown in Fig. 3b, H2O2 induced higher cell death for the glass control after 2 h treatment of H2O2 solution (MMD 90.2 ± 2.7% vs glass 76.2 ± 1.1%) and showed a significant difference after 12 h treatment (MMD 68 ± 4.1% vs glass 1.2 ± 0.3%). The cell viability was much higher for the cells under fluidic and porous membrane environments, suggesting that the current microfluidic device can allow for robust cell cultures by inducing necessary structural changes and cell polarity.
Reorganization of cytoskeleton, focal adhesions, and cell junction
We next compared the cytoskeletal and cell junctional changes within the MMD as well as using three controls: transwell membrane (static), glass substrate (static), and PDMS channel + glass substrate (fluidic). To determine the influence of shear stress and porous substrate on the cytoskeleton and cell junction, the cells cultured under different conditions were stained for F-actin, vinculin, β-catenin and E-cadherin and imaged with confocal microscopy (LSM510, Carl Zeiss, Jena, Germany) (Fig. 4).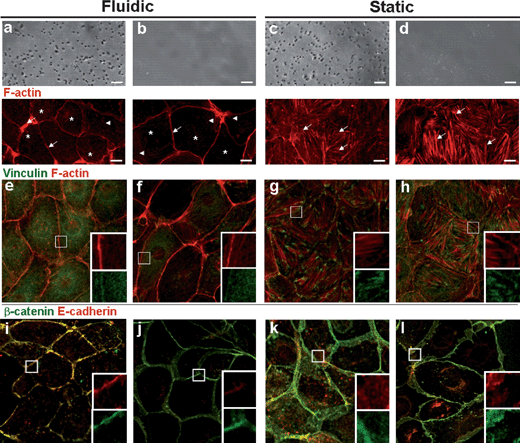 | ||
| Fig. 4 Reorganization of cytoskeleton, focal adhesions, and cell junction of IMCD cells under four different conditions: MMD (fluidic) (a, e, i), PDMS channel + glass substrate (fluidic) (b, f, j), transwell (static) (c, g, k), and glass substrate (static) (d, h, l). In fluidic condition, cells are subjected to 1 dyn/cm2 of fluid shear stress for 5 h. Optical (top panel) and confocal microscopy images of IMCD cells stained with antibodies for F-actin (red) (a–d, bottom, e–h), vinculin (green) (e–h), β-catenin (green) and E-cadherin (red) (i–l). The inset shows a magnified view of the boxed region (10 × 10 µm). Scale bar, 10 µm. | ||
As shown, the shear-induced mechanical stimulation gives rise to depolymerization of F-actin in IMCD cells within 5 h under fluidic conditions (Fig. 4a,b and Supplementary Fig. 2†). Similar to proximal tubule cells,4 the shear stress induced remarkable reorganization of F-actin to both IMCD cells cultured on the membrane and glass surfaces. The estimated flow sensor in IMCD cell is a primary cilium in the apical membrane.7 It can further be seen from the figure that depolymerized and dot-like F-actin patterns (asterisks) and thin actin fibers (arrowheads) were present at the cell interior, whereas thick actin fibers (arrows) were observed at the cell periphery for the IMCD cells cultured under fluidic conditions (Fig. 4a,b). Comparing cell cultures on the membrane and glass substrates, one can find more scattered, dot-like F-actin patterns for the cells on the porous membrane (Fig. 4a, asterisks). In contrast, F-actins under static conditions assembled into long bundles and networks throughout the cell body without a distinct formation of cell periphery (Fig. 4c, d). F-actins were clustered thick and abundant with a more organized parallel pattern throughout the cell interior on the glass substrate (Fig. 4d, arrows) than on the membrane support (Fig. 4c). These data indicate that F-actins act as a submembranous barrier to vesicular fusion under static conditions and the fluidic shear stress induces actin depolymerization at the cell interior and actin reinforcement at the cell periphery.
In parallel, vinculin, E-cadherin and β-catenin were analyzed under four different conditions. In fluidic conditions, vinculin, which is a cell–cell and cell–matrix junction associated protein, was diffused weakly throughout the cell body as a result of F-actin depolymerization, and localized at the cell membrane (Fig. 4e, f). In static conditions, vinculin was localized at the cell membrane and cell interior and co-localized with F-actin strikingly at the end of actin fibers with dense and short fiber shapes (Fig. 4g, h). In fluidic conditions, E-cadherin and β-catenin, which are adherens junction proteins related to epithelial polarity and tissue architecture,25,26 were co-localized at narrow and reinforced cell junctional sites with much reinforced junction in MMD (Fig. 4i, j). In static conditions, E-cadherin and β-catenin were densely and broadly localized at adherens junctional sites as well as at the cell interior (Fig. 4o, p).
Analysis of molecular transport by AQP2 trafficking and drug screening
As shown in Fig. 2 and 4, the kidney tubular cells cultured within the MMD exhibit several striking features. First, the cells demonstrate the distinct, well-organized separation of apical and basal regions on the porous membrane support (Fig. 2), which is direct evidence of cell polarization and can further be used as a microfluidic platform to test AQP2 trafficking from intracellular vesicle to apical cell membrane. In addition, the biomechanical response of the cells such as reorganization of cytoskeleton, focal adhesions, and cell junction can actively be controlled by a simple application of the shear stress (Fig. 4), which can be used to examine transcellular and paracellular molecular transport for the cultured cells. It is noted that other sophisticated techniques are available for measurements at the single cell level such as electrophysiological measurement and measurement of cell volume kinetics.27–30 For these techniques, freshly isolated tubule fragments in microperfusion, cell monolayers in a transwell system or radioactive isotopes have been used. As these methods are typically complex and indirect, and require different physiological conditions, it is of great benefit to improve compatibility and simplicity of the measurement using a simple multi-channel microfluidic system under in-vivo like tubule environments.In order to evaluate molecular transport in renal tubule cells, we measured water and Na uptake after hormonal stimulations of vasopressin and aldosterone using the IMCD cells (Fig. 5a–b). It is known that vasopressin (anti-diuretic hormone) induces water uptake by stimulating AQP2 trafficking, while aldosterone induces Na uptake by activating Na-K pump in the tubular cells (Fig 5c). In the experiments, the IMCD cells were cultured within the MMD for 3 days and then exposed to a shear stress for 5 h on their apical surface with fluid medium, having the stimulation of 10−8 M arginine vasopressin (AVP+) in a complete cell culture medium or 10−7 M aldosterone (Aldo+) in a complete cell culture medium on their basal region. The measurement of osmolarity and Na ion was performed using an osmometer and ICP (inductively coupled plasma) emission spectrometer with 1 h of fluidic samples. As shown in Fig. 5a–b, the osmolarity of apical fluidic sample was changed to 681 ± 13.9 mOsm/kgH2O by vasopressin (vehicle means a complete medium, 649 ± 2.8 mOsm/kgH2O) and Na transport was changed to 69.8 ± 8.3 mM by aldosterone (vehicle, 85 ± 3.8 mM). These results reveal that water and ion transports are strictly regulated by hormonal stimulations. Shown in Fig. 5c are immunofluorescence images of F-actin, AQP2, and Na-K-pump of the IMCD cells in response to hormonal stimulations of AVP+ and Aldo+ along with those of the vehicle as a control. As shown, the stimulation of AVP+ and Aldo+ at the basal region induced dynamic changes in F-actin reorganization. Also, AQP2 labeling was increased to the plasma membrane in response to AVP+ with the inset showing a magnified view of the boxed region (10 × 10 µm).
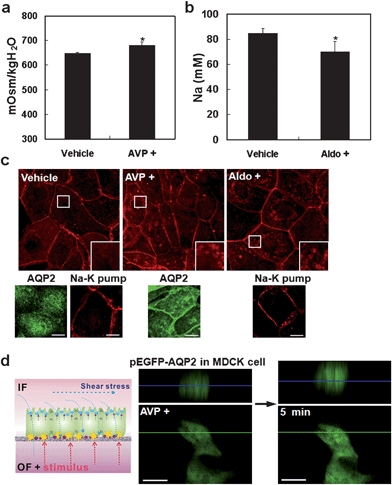 | ||
| Fig. 5 Analysis of molecular transport by AQP2 trafficking and drug screening. The hormone is treated to the basal region of the cells (PDMS well) while applying a fluid shear stress to the cells on the apical region. (a) Quantitative analysis of osmolarity with an apical fluid for 1 h in the presence of 10−8 M arginine vasopressin (AVP) or in the absence of hormone (vehicle) at the basal region. (b) Quantitative analysis of Na transport on vehicle and 10−7 M aldosterone (Aldo+). Error bar, s.d. of the mean. (n = 5) (*, p < 0.05). (c) Immunofluorescence images of F-actin, AQP2, and Na-K-pump in response to hormonal stimulations (AVP+ and Aldo+). The images for vehicle are also shown as a control. Each inset shows a magnified view of the boxed region (10 × 10 µm). (d) Schematic of drug screening and test of the real-time AQP2 trafficking using AQP2-GFP transfected MDCK cells. After stimulation of AVP+ for 5 min, AQP2-GFP translocated to the plasma membrane. Upper images show the x–z optical sectioned confocal microscope images into the green line. The blue line indicates the middle plane of the cell. Scale bar, 10 µm. | ||
To further demonstrate the potential as a drug screening platform towards the regulation of water balance, we tested the real-time translocation of AQP2 in response to a hormonal stimulation in the MMD using AQP2-transfected MDCK cells.31 This transfected cell was established independently to observe the movement of fluorescein-labeled protein (e.g., GFP) over time (see Experimental section). Prior to the AVP treatment, the cells were cultured within the MMD confluently, and a fluid shear stress of 1 dyn/cm2 was applied for the time period of 5 h. As shown in Fig. 5d, AQP2-GFP gradually translocated to the plasma membrane after 5 min while 10−9 M AVP was being supplied to the basal region of the cells under a confocal microscope. This finding indicates that one can perform simple screening of unknown drugs at the basal region of the cells under in-vivo like tubule environments. Together with the ability to treat a specific stimulus at the apical region, the current device would be useful in a wide range of applications involving drug screening and tissue engineering of kidney cells.
4. Summary
The simple multi-layer microfluidic device presented in this work provides a truly biomimetic, microfluidic platform for creating in vivo-like extracellular environments for efficient culture and analysis of the renal tubule cells. The fluidic shear stress of ∼1 dyn/cm2 for the time period of 5 h on the porous membrane support was found to be sufficient for the IMCD cells to enhance cell polarization and rearrange cytoskeleton and cell junctions. The IMCD cells, when cultured under in-vivo like tubular environments within the channel performed their inherent role of regulating water and ion balance via molecular transport by hormonal stimulations. The simple multi-layer microfluidic device presented here would find uses in a variety of applications including a disease model system such as nephrogenic diabetes insipidus (NDI) or edema, drug screening, and a model system for studying renal physiology.Acknowledgements
We thank Prof. Tae-Hwan Kwon for helpful discussions and technical support (osmolarity measurement) and Se-Yon Hwang and Do-Hyun Kang for animal experiments. This work was supported by the WCU (World Class University) program on Multiscale Design (R31-2008-000-10083-0), the Korea Research Foundation Grant funded by the Korean Government (MOEHRD) (Grant KRF-J03003), and the Nano Systems Institute-National Core Research Center (NSI-NCRC).References
- C. Palfrey and A. Cossins, Nature, 1994, 371, 377–378 CrossRef CAS.
- S. Nielsen, J. Frokiaer, D. Marples, T. H. Kwon, P. Agre and M. A. Knepper, Physiol. Rev., 2002, 82, 205–244 CAS.
- Y. J. Lee, I. K. Song, K. J. Jang, J. Nielsen, J. Frokiaer, S. Nielsen and T. H. Kwon, Am. J. Physiol.: Renal Physiol., 2007, 292, F340–F350 CAS.
- Y. Duan, N. Gotoh, Q. S. Yan, Z. P. Du, A. M. Weinstein, T. Wang and S. Weinbaum, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11418–11423 CrossRef CAS.
- Z. P. Du, Y. Duan, Q. S. Yan, A. M. Weinstein, S. Weinbaum and T. Wang, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13068–13073 CrossRef CAS.
- Z. Q. Cai, J. D. Xin, D. M. Pollock and J. S. Pollock, Am. J. Physiol.: Renal Physiol., 2000, 279, F270–F274 CAS.
- W. Liu, S. Y. Xu, C. Woda, P. Kim, S. Weinbaum and L. M. Satlin, Am. J. Physiol.: Renal Physiol., 2003, 285, F998–F1012 CAS.
- M. Essig, F. Terzi, F. M. Burtin and G. Friedlander, Am. J. Physiol.: Renal Physiol., 2001, 281, F751–F762 CAS.
- M. M. P. Zegers, L. E. O'Brien, W. Yu, A. Datta and K. E. Mostov, Trends Cell Biol., 2003, 13, 169–176 CrossRef CAS.
- S. M. Kim, S. H. Lee and K. Y. Suh, Lab Chip, 2008, 8, 1015–1023 RSC.
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS.
- A. Khademhosseini, R. Langer, J. Borenstein and J. P. Vacanti, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2480–2487 CrossRef CAS.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–U1210 CrossRef CAS.
- A. M. Taylor, M. Blurton-Jones, S. W. Rhee, D. H. Cribbs, C. W. Cotman and N. L. Jeon, Nat. Methods, 2005, 2, 599–605 CrossRef CAS.
- A. Groisman, C. Lobo, H. J. Cho, J. K. Campbell, Y. S. Dufour, A. M. Stevens and A. Levchenko, Nat. Methods, 2005, 2, 685–689 CrossRef CAS.
- C. J. Wang, X. Li, B. Lin, S. Shim, G. L. Ming and A. Levchenko, Lab Chip, 2008, 8, 227–237 RSC.
- D. Lim, Y. Kamotani, B. Cho, J. Mazumder and S. Takayama, Lab Chip, 2003, 3, 318–323 RSC.
- K. R. King, S. Wang, A. Jayaraman, M. L. Yarmush and M. Toner, Lab Chip, 2008, 8, 107–116 RSC.
- D. Huh, H. Fujioka, Y. C. Tung, N. Futai, R. Paine, J. B. Grotberg and S. Takayama, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18886–18891 CrossRef CAS.
- Y. S. Torisawa, B. H. Chueh, D. Huh, P. Ramamurthy, T. M. Roth, K. F. Barald and S. Takayama, Lab Chip, 2007, 7, 770–776 RSC.
- K. W. Kwon, S. S. Choi, S. H. Lee, B. Kim, S. N. Lee, M. C. Park, P. Kim, S. Y. Hwang and K. Y. Suh, Lab Chip, 2007, 7, 1461–1468 RSC.
- P. Kim, K. W. Kwon, M. C. Park, S. H. Lee, S. M. Kim and K. Y. Suh, Biochip. J., 2008, 2, 1–11 Search PubMed.
- K. J. Jang and J. M. Nam, Small, 2008, 4, 1930–1935 CrossRef CAS.
- I. Arany, J. K. Megyesi, H. Kaneto, S. Tanaka and R. L. Safirstein, Kidney Int., 2004, 65, 1231–1239 CrossRef CAS.
- B. M. Gumbiner, Nat. Rev. Mol. Cell Biol., 2005, 6, 622–634 CrossRef CAS.
- D. M. Bryant and J. L. Stow, Trends Cell Biol., 2004, 14, 427–434 CrossRef CAS.
- K. Maric, B. Wiesner, D. Lorenz, E. Klussmann, T. Betz and W. Rosenthal, Biophys. J., 2001, 80, 1783–1790 CrossRef CAS.
- D. M. Cohen, Clin. Exp. Pharmacol. Physiol., 1999, 26, 69–73 CrossRef CAS.
- Y. Fujii, F. Takemoto and A. I. Katz, Am. J. Physiol.: Renal Physiol., 1990, 259, F40–F45 Search PubMed.
- Y. F. Guan, C. M. Hao, D. R. Cha, R. Rao, W. D. Lu, D. E. Kohan, M. A. Magnuson, R. Redha, Y. H. Zhang and M. D. Breyer, Nat. Med., 2005, 11, 861–866 CrossRef CAS.
- F. de Mattia, P. J. M. Savelkoul, E. J. Kamsteeg, I. B. M. Konings, P. van der Sluijs, R. Mallmann, A. Oksche and P. M. T. Deen, J. Am. Soc. Nephrol., 2005, 16, 2872–2880 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and Fig. S2. See DOI: 10.1039/b907515a |
| This journal is © The Royal Society of Chemistry 2010 |