Biomimetic synthesis of photoactive α-Fe2O3 templated by the hyperthermophilic ferritin from Pyrococus furiosus†
Michael T.
Klem
a,
Mark
Young
bc and
Trevor
Douglas
*c
aDepartment of Chemistry & Geochemistry, Montana Tech of the University of Montana, Butte, MT, USA
bDepartment of Plant Sciences, Center for BioInspired Nanomaterials, NASA Astrobiology Biogeocatalysis Center Montana State University, Bozeman, MT, USA
cDepartment of Chemistry and Biochemistry, Center for BioInspired Nanomaterials, NASA Astrobiology Biogeocatalysis Center Montana State University, Bozeman, MT, USA. E-mail: tdouglas@chemistry.montana.edu
First published on 9th November 2009
Abstract
Utilizing a biomimetic approach toward materials synthesis a ferritin protein cage, from the hyperthermophilic archeaon Pyrococcus furiosus, was utilized to first tempate the formation of a largely amorphous ferrihydrite iron oxide and then bring about its transformation to hematite (α-Fe2O3). This was achieved under boiling aqueous conditions by refluxing the ferritin protein cage/ferrihydrite composite. The resultant material showed diffraction indicative of hematite, a visible band gap semiconductor, and photocurrents were measured under visible illumination. The resultant protein/mineral composite was also studied via dynamic light scattering, transmission electron microscopy, and size exclusion chromatography.
In an effort to find alternatives to nonrenewable fossil fuels, researchers have focused on using sunlight to evolve hydrogen as a clean and renewable energy source. Fujishima and Honda using a TiO2 photoanode experimentally achieved direct light-induced water splitting.1 Hematite (α-Fe2O3) has also attracted interest as a photoanode because of its nearly ideal band gap, visible light absorption, nontoxicity, and excellent chemical stability under appropriate conditions.2 One of the difficulties in using hematite as a photoanode is its extremely short hole diffusion length of approximately 5 nm.3 This short diffusion length is the prevailing cause of the low efficiencies observed with iron-oxide photoanodes due to electron hole recombination.
It has been hypothesized that creation of nanoparticles of α-Fe2O3 with sizes commensurate with the hole diffusion length would exhibit improved efficiencies for photoanodic oxidation because of the hindrance of charge transport inside the nanoparticles. Currently, high temperature vapor phase methods have been employed in the fabrication of iron oxide photoanodes, but these methods have had limited success in producing crystalline iron oxides with particle sizes in the region of 5 nm.3–6 The use of nanostructured films7–9 and heteroatom dopants4,10–16 have been explored as pathways to improved performance. The incorporation of nanoparticles in thin films have also been investigated.17
Biomimetic approaches toward materials chemistry have provided new avenues for the synthesis and assembly of nanomaterials.18–22 Bio-inspired approaches in material synthesis have utilized evolved molecular interactions,23,24 macromolecular templates,25 and well defined protein cage architectures26–31 to exert synthetic control over crystal morphology, phase, and orientation. While biological macromolecules have been utilized in materials synthesis their use has often been limited by their relatively low thermal stability compared with other synthetic methods. Isolation of protein templates from thermophilic and hyperthermophilic organisms is one approach which might ease this limitation.32,33 The isolation and synthetic utilization of a number of thermally stable proteins such as ferritins,34 heat shock proteins,35 and DNA-binding proteins from starved cells32,36 from hyperthermophilic archaea have significantly expanded the temperature range for in vitro biomimetic synthesis.
Recently, a hyperthermophilic ferritin was isolated from the archaeon Pyrococcus furiosus, a marine anaerobe that lives in high temperature environments where temperatures can reach 120 °C.37 This protein cage has been cloned from its original host, and overexpressed in Escherichia coli.37 The recombinant Pyrococcus ferritin (Pf_Fn) was subjected to reaction temperatures of 120 °C and was shown to retain the iron sequestering behavior for over 30 min at these extreme temperatures.37 The ability of this protein to template magnetic iron oxides (i.e. Fe3O4) has also been demonstrated at elevated temperatures with magnetic properties different from syntheses using mesophillic ferritin as a template.34
Ferrihydrite was initially synthesized inside Pf_Fn with a chemical loading of 1000 Fe atoms/cage using previously established methods.38 Isolated Apo-Pf_Fn protein cages were diluted to 0.3 mg mL−1 in 100 mM MES (pH = 6.5) and 50 mM NaCl. Dearated solutions of (NH4)2FeSO4·6H2O were added to the Pf_Fn for a total chemical loading of 1000 Fe/cage over a period of 24 h. The resulting solution was allowed to air oxidize to form 7.0 nm ferrihydrite particles. The Pf_Fn/ferrihydrite composite was dialyzed into 50 mM HEPES (pH = 7.0) and 100 mM NaCl. The solution was then refluxed at 97 °C in the presence of trace amounts of sodium oxalate (<1 mM) for 1–5 days.
Using a biomimetic approach we have developed a synthetic technique that involves the conversion of a relatively amorphous hexagonal iron oxide (ferrihydrite) to ordered α-Fe2O3 within the Pf_Fn protein cage. Ferrihydrite was converted to hematite using processes previously described as a combination of catalytic dissolution/reprecipitation and topotactic solid-state transformation inside the Pf_Fn protein cage.39 The pre- and post- reaction products were characterized by TEM (Fig. 1). Unstained samples showed electron-dense cores whose sizes (6.9 ± 1.1 nm) were commensurate with the interior dimensions of the protein cage. When samples were negatively stained with uranyl acetate, the intact protein cages were clearly visible as a white “halo” surrounding the metal oxide cores on the interior surface. Electron diffraction was not observed in the ferrihydrite samples consistent with the relatively amorphous nature of ferrihydrite.40 Electron diffraction, with d-spacings corresponding to α-Fe2O3, was observed in the refluxed samples (ESI†). Lower temperature phase transformations (i.e. 65 °C) using Pf_Fn cages were unsuccessful at generating hematite nanoparticles from the ferrihydrite precursor. Control reactions using horse spleen ferritin were also unsuccessful at generating hematite nanoparticles from a ferrihydrite precursor due to precipitation of the protein at the high temperatures required.
 | ||
| Fig. 1 Transmission electron micrographs of (A) unstained, (B) stained refluxed Pf_Fn, and (C) unstained ferrihydrite in Pf_Fn. The dark electron dense cores in (A) have an electron diffraction pattern (insert) consistent with α-Fe2O3 while no diffraction was observed in (C) consistent with disordered ferrihydrite. | ||
The products were further characterized using dynamic light scattering and found to have the same size distribution as the original protein cage starting materials, 12.9 ± 0.5 nm vs. 13.0 ± 0.2 nm for unheated and heated, respectively (ESI†). This indicates the high degree of heat stability and spatial selectivity the protein cage exerts on the iron oxides. No colloidal material was detected exterior to the Pf_Fn protein cages. The protein absorbance at 280 nm was used to monitor elution of the composite protein/mineral by size exclusion chromatography (Fig. 2). The mineralized product had an elution profile with a retention time identical to that of empty Pf_Fn protein. This co-elution behavior points toward the composite nature of the material and suggests that the protein cage and mineral are intimately associated. Size exclusion chromatography is relatively sensitive to small changes in particle size, and thus these data suggest that the size of the protein cage has not been significantly perturbed by the reaction. It is also indicative of the mineral being encapsulated within the protein cage architecture demonstrating the spatial selectivity of the reaction.
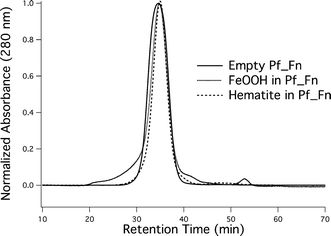 | ||
Fig. 2 Size exclusion chromatograph of empty (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ), FeOOH containing (⋯), and α-Fe2O3 ( ), FeOOH containing (⋯), and α-Fe2O3 (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) Pf_Fn protein cages. ) Pf_Fn protein cages. | ||
Photocurrents generated from the α-Fe2O3/Pf_Fn composite were collected at an ITO working electrode immersed in a suspension of α-Fe2O3/Pf_Fn (0.3 mg mL−1 α-Fe2O3/Pf_Fn, ethanol) using methyl viologen (MV2+, 2 mM) as an electron mediator.41,42 The photocurrents generated under illumination from a high intensity 420 nm LED are shown (Fig. 3). Conversely, control reactions involving the empty Pf_Fn cage and MV2+ exhibited photocurrents on the order of 0.0–0.5 μA. Photo-reactions performed with the α-Fe2O3/Pf_Fn composite gave rise to the photocurrents on the order of 5–37 μA cm−2 depending on concentration (Fig. 4). The maximum in the current density is most likely to be due to incomplete conversion of the ferrihydrite precursor to hematite. Further studies also indicate that the transformation is complete within 60 min of reaction. No further gain in photocurrent was observed after 60 min (Fig. 5).
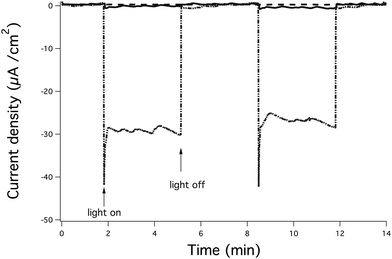 | ||
Fig. 3 Photocurrent generated upon exposure of ferrihydrite in Pf_Fn (), Methyl viologen (), and hematite Pf_Fn/Methyl viologen (![[dash dot, graph caption]](https://www.rsc.org/images/entities/char_e090.gif) ) to a high intensity LED light. ) to a high intensity LED light. | ||
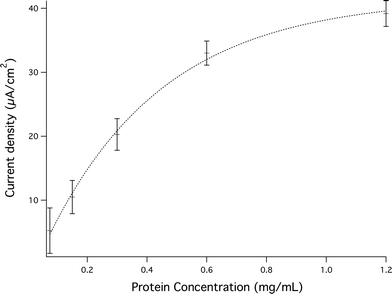 | ||
| Fig. 4 Photocurrents generated as a function of protein concentration. | ||
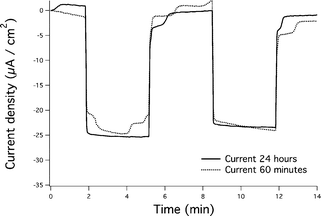 | ||
| Fig. 5 Photocurrents generated at 60 min vs. 24 h. The strong similarities in the currents observed suggest that the conversion of ferrihydrite to hematite is complete in one hour. | ||
Despite the favorable band gap for α-Fe2O3, the photocurrents observed are lower than those observed for sprayed α-Fe2O3 films.43 It is hypothesized that interactions between the protein cage and the photoexcited electron allow for electron/hole recombination to occur before the MV2+ can be reduced. This is most likely to be due to the mass transport limitation of MV into and out of the protein cage.
This work illustrates two important concepts. First, a ferritin protein cage isolated from a hyperthermophilic archea can be effectively used as a size constrained reaction vessel for the conversion of ferrihydrite to α-Fe2O3 under boiling aqueous conditions. Furthermore, the inherent temperature stability of this ferritin is a necessary requirement for a successful transformation. Second, the resultant hematite/protein composite acts as a visible band gap semiconductor for the photoreduction of methyl viologen using ethanol as a sacrificial reductant.
This research was supported in part by grants from the National Science Foundation (CBET-0709358), and the Department of Energy (DE-FG02-07ER46477).
Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CAS.
- R. van de Krol, Y. Q. Liang and J. Schoonman, J. Mater. Chem., 2008, 18, 2311 RSC.
- J. H. Kennedy and K. W. Frese, J. Electrochem. Soc., 1978, 125, 709 CAS.
- A. Duret and M. Gratzel, J. Phys. Chem. B, 2005, 109, 17184 CrossRef CAS.
- E. L. Miller, D. Paluselli, B. Marsen and R. E. Rocheleau, Sol. Energy Mater. Sol. Cells, 2005, 88, 131 CrossRef CAS.
- C. J. Sartoretti, B. D. Alexander, R. Solarska, W. A. Rutkowska, J. Augustynski and R. Cerny, J. Phys. Chem. B, 2005, 109, 13685 CrossRef CAS.
- A. G. Agrios, I. Cesar, P. Comte, M. K. Nazeeruddin and M. Gratzel, Chem. Mater., 2006, 18, 5395 CrossRef CAS.
- I. Cesar, A. Kay, J. A. G. Martinez and M. Gratzel, J. Am. Chem. Soc., 2006, 128, 4582 CrossRef CAS.
- A. Kay, I. Cesar and M. Gratzel, J. Am. Chem. Soc., 2006, 128, 15714 CrossRef CAS.
- T. Arai, Y. Konishi, Y. Iwasaki, H. Sugihara and K. Sayama, J. Comb. Chem., 2007, 9, 574 CrossRef CAS.
- V. M. Aroutiounian, V. M. Arakelyan, G. E. Shahnazaryan, G. M. Stepanyan, E. A. Khachaturyan, H. Wang and J. A. Turner, Sol. Energy, 2006, 80, 1098 CrossRef CAS.
- U. Bjorksten, J. Moser and M. Gratzel, Chem. Mater., 1994, 6, 858 CrossRef.
- M. M. Khader, G. H. Vurens, I. K. Kim, M. Salmeron and G. A. Somorjai, J. Am. Chem. Soc., 1987, 109, 3581 CrossRef CAS.
- C. Leygraf, M. Hendewerk and G. A. Somorjai, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 5739 CrossRef CAS.
- S. Mohanty and J. Ghose, J. Phys. Chem. Solids, 1992, 53, 81 CrossRef CAS.
- A. B. Murphy, P. R. F. Barnes, L. K. Randeniya, I. C. Plumb, I. E. Grey, M. D. Horne and J. A. Glasscock, Int. J. Hydrogen Energy, 2006, 31, 1999 CrossRef CAS.
- P. H. Borse, H. Jun, S. H. Choi, S. J. Hong and J. S. Lee, Appl. Phys. Lett., 2008, 93, 173103 CrossRef.
- S. W. Lee, C. B. Mao, C. E. Flynn and A. M. Belcher, Science, 2002, 296, 892 CrossRef CAS.
- S. Mann, Biomimetic Materials Chemistry, VCH, New York, 1996 Search PubMed.
- S. Mann, D. D. Archibald, J. M. Didymus, T. Douglas, B. R. Heywood, F. C. Meldrum and N. J. Reeves, Science, 1993, 261, 1286 CAS.
- R. R. Naik, S. J. Stringer, G. Agarwal, S. E. Jones and M. O. Stone, Nat. Mater., 2002, 1, 169 CrossRef CAS.
- M. Uchida, M. T. Klem, M. Allen, P. Suci, M. Flenniken, E. Gillitzer, Z. Varpness, L. O. Liepold, M. Young and T. Douglas, Adv. Mater., 2007, 19, 1025 CrossRef CAS.
- M. Allen, D. Willits, J. Mosolf, M. Young and T. Douglas, Adv. Mater., 2002, 14, 1562 CrossRef CAS.
- A. Sidorenko, T. Krupenkin, A. Taylor, P. Fratzl and J. Aizenberg, Science, 2007, 315, 487 CrossRef CAS.
- W. Shenton, T. Douglas, M. Young, G. Stubbs and S. Mann, Adv. Mater., 1999, 11, 253 CrossRef CAS.
- M. Allen, D. Willits, M. Young and T. Douglas, Inorg. Chem., 2003, 42, 6300 CrossRef CAS.
- A. S. Blum, C. M. Soto, C. D. Wilson, T. L. Brower, S. K. Pollack, T. L. Schull, A. Chatterji, T. W. Lin, J. E. Johnson, C. Amsinck, P. Franzon, R. Shashidhar and B. R. Ratna, Small, 2005, 1, 702 CrossRef CAS.
- T. Douglas, D. P. E. Dickson, S. Betteridge, J. Charnock, C. D. Garner and S. Mann, Science, 1995, 269, 54 CAS.
- T. Douglas and M. Young, Nature, 1998, 393, 152 CrossRef CAS.
- S. Mann and G. A. Ozin, Nature, 1996, 382, 313 CrossRef CAS.
- F. C. Meldrum, B. R. Heywood and S. Mann, Science, 1992, 257, 522 CAS.
- B. Wiedenheft, J. Mosolf, D. Willits, M. Yeager, K. A. Dryden, M. Young and T. Douglas, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10551 CrossRef CAS.
- B. Wiedenheft, M. L. Flenniken, M. A. Allen, M. Young and T. Douglas, Soft Matter, 2007, 3, 1091 RSC.
- M. J. Parker, M. A. Allen, B. Ramsay, M. T. Klem, M. Young and T. Douglas, Chem. Mater., 2008, 20, 1541 CrossRef CAS.
- M. T. Klem, D. Willits, D. J. Solis, A. M. Belcher, M. Young and T. Douglas, Adv. Funct. Mater., 2005, 15, 1489 CrossRef CAS.
- B. Ramsay, B. Wiedenheft, M. Allen, G. H. Gauss, C. M. Lawrence, M. Young and T. Douglas, J. Inorg. Biochem., 2006, 100, 1061 CrossRef CAS.
- J. Tatur, P. L. Hagedoorn, M. L. Overeijnder and W. R. Hagen, Extremophiles, 2006, 10, 139 CrossRef CAS.
- I. G. Macara, P. M. Harrison and T. G. Hoy, Biochem. J., 1972, 126, 151 CAS.
- Z. Pu, M. Cao, J. Yang, K. Huang and C. Hu, Nanotechnology, 2006, 17, 799 CrossRef CAS.
- F. M. Michel, L. Ehm, S. M. Antao, P. L. Lee, P. J. Chupas, G. Liu, D. R. Strongin, M. A. A. Schoonen, B. L. Phillips and J. B. Parise, Science, 2007, 316, 1726 CrossRef CAS.
- H. Park, W. Choi and M. R. Hoffmann, J. Mater. Chem., 2008, 18, 2379 RSC.
- H. W. Park, J. S. Lee and W. Y. Choi, Catal. Today, 2006, 111, 259 CrossRef CAS.
- J. D. Desai, H. M. Pathan, S. K. Min, K. D. Jung and O. S. Joo, Appl. Surf. Sci., 2005, 252, 1870 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Pf_Fn purification from E.coli and characterization details. See DOI: 10.1039/b918620d |
| This journal is © The Royal Society of Chemistry 2010 |