Supramolecular replication of peptide and DNA patterned arrays†
Anna
Laromaine‡
ab,
Ozge
Akbulut‡
b,
Francesco
Stellacci
*b and
Molly M.
Stevens
*a
aDepartment of Materials and Institute of Biomedical Engineering, Imperial College, Exhibition Road, SW7 2AZ, London, UK. E-mail: m.stevens@imperial.ac.uk; Fax: +44(0)2075946757; Tel: +44(0)2075946804
bDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, 02139, Cambridge, MA, USA. E-mail: frstella@mit.edu
First published on 14th October 2009
Abstract
Here we present a novel approach to the parallel fabrication of protein and protein/DNA features on surfaces.
Recently, much effort has focused on the development of DNA or protein arrays. Applications span fields as diverse as genomics, proteomics and diagnostics with examples in drug development, biomarker detection, and elucidation of genomic and proteomic pathways and networks.1,2 Current methods for DNA array fabrication utilize spotting, ink-jet printing and in situ photochemical synthesis.3–6 Protein arrays can be produced by a subset of these methods.7,8 All current fabrication methods to produce biomolecular arrays are serial; they build arrays typically point-by-point (e.g. by spotting each biomolecule), or layer-by-layer (e.g. by in situ synthesis of biomolecules), a serious limitation that ultimately affects the quality and price of the final product. DNA array fabrication methods based on new approaches (mostly based on in situ synthesis) are emerging, whereas alternative approaches for the fabrication of protein arrays or mixed arrays are in their infancy.9,10
Parallel fabrication methods for DNA and proteins are derived from microcontact printing (µcp) whereby a patterned and DNA-11 or protein-inked12,13 elastomeric stamp is employed to create micropatterns on a substrate. In its basic form µcp has the drawback of transferring only one kind of molecule onto the target substrate. Supramolecular recognition approaches can improve on the efficiency of µcp for biomolecule printing. Affinity contact printing (αcp) introduces supramolecular interactions into the printing cycle. For instance, adding a pre-inking step to establish supramolecular interaction between the ink and the stamp leads to selectivity and more reliable printing.14,15
Crook's16 and our17 group independently developed a parallel soft-material stamping technique (supramolecular nanostamping (SuNS)) to replicate DNA features from one surface onto another (Fig. S1†). Recently, we modified this method using a liquid prepolymer as the secondary substrate to achieve better contact (liquid supramolecular nanostamping (LiSuNS)).18 Both SuNS and LiSuNS have the ability to produce a DNA microarray in a single stamping cycle, hence in a more efficient and less expensive way when compared to all presently available alternative fabrication approaches. LiSuNS has the ability to stamp an array within a microfluidic channel.19
LiSuNS harnesses the supramolecular interaction between complementary biomolecules in a three step process: 1) hybridization, 2) printing and 3) dehybridization. In the first step (hybridization), the master is hybridized with its complementary strand bearing a ‘sticky group’; that is a chemical group targeting a secondary surface. In the subsequent step (printing), the elastomeric prepolymer is poured onto the hybridized master and cured; thus allowing the ‘sticky groups’ to covalently attach to this substrate. In our experiments with DNA, we introduced acrylic phosphoramidite groups (sticky groups) which reacted covalently with the elastomeric prepolymer. For the printing of coiled-coil peptides, amine groups within the peptide were able to covalently react with the elastomeric prepolymer. In the last step (dehybridization), the secondary structure is broken by mechanical forces,16 effectively lifting off the cured polymer from the master, making the secondary surface a complementary replica of the master (Fig. 2a).18
To date SuNS and LiSuNS have solely been used to stamp DNA features.18 Yet, the possibility to print different types of biomolecules in just one cycle, for instance DNA and other biomolecules, is very attractive. Therefore, we venture here to assess the multiplexing capability of LiSuNS in terms of producing mixed DNA and coiled-coil peptide arrays. Coiled-coil peptides exhibit selective and reversible supramolecular recognition at surfaces20 and thus are ideal protein candidates to fulfil the requirements imposed by LiSuNS.
The α-helical coiled-coil motif is an oligomerization domain present in many cytoskeletal proteins, transcription factors, motor proteins, or viral proteins, amongst others.21–23 Two or more amphipathic α-helical peptide sequences form a supra-coil stabilized by a hydrophobic interface. The peptide sequence consists of heptad repeats (a-b-c-d-e-f-g)n of amino acids whereby hydrophobic residues in positions (a/d) stabilize the motif and polar residues in positions (e/g) impart specificity through electrostatic interactions. A helical wheel representation of the peptides E and K utilized here is shown in Fig. 1a.24
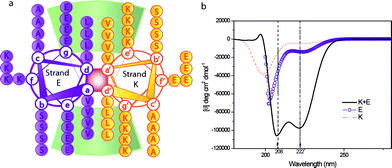 | ||
| Fig. 1 a) Helical wheel projection, looking down the axes of the helices of the dimerization domain of the coiled-coil peptides used in this work. Heptad positions are labelled a to g for strand E and a′ to g′ for strand K. Residues e/g′ and g/e′ (i, i′ + 5) participate in the interhelical electrostatic interactions, residues a/a′ and d/d′ contribute in the stabilization by hydrophobic interactions. b) Circular dichroism spectra of single stranded peptides K and E and the heterodimeric coiled coil peptide K/E. | ||
The coiled-coil motif, though relatively structurally simple, is highly versatile in design in terms of oligomerization state, rigidity, force of interaction between complementary strands and response to environmental conditions such as temperature and pH.25–27 Elucidation of sequence-structure relationships means, at least in protein terms, rational design of the motif is now somewhat analogous to design and use of DNA-based building blocks.28–37
The sequence of the heterodimeric coiled-coil used here was initially reported by Litowski and Hodges38 and has been slightly modified to introduce a terminal thiol group and a tri-glycine spacer to each of the complementary peptides E and K (Fig. 1a). E and K heterodimerisation was assessed using circular dichroism (CD), Fig. 1b. The solid line in the CD spectra shows an α-helical structure with characteristic minima at 208 and 222nm for K + E, whereas homopeptides (E or K) exhibit a random coil structure (dashed and circle line in Fig. 1b). Experimental information is provided in ESI.†
Here we demonstrate that LiSuNS can print coiled-coil peptides as outlined in Fig. 2a. First we determined that the functionality of the printed peptide is preserved and specific; namely, peptides only form coiled-coil structures with their complementary sequence. We utilised an array of 10µm × 10µm gold squares on a silicon wafer and immersed it in a solution of peptide K in phosphate buffered saline (PBS) to allow the peptides to bond on the gold features through their terminal thiol groups. After thorough washing in PBS and deionized (DI) water, this substrate was immersed in 6-mercapto-1-hexanol (MH) solution to minimize non-specific adsorption. Hereafter, the K-functionalised and passivated substrate is referred to as the ‘master’. For complementary peptide coiling, analogous to DNA-hybridization, the master was incubated in a solution of the complementary peptide E for 12 hours (Fig. 2a). We prepared a poly(dimethyl siloxane) (PDMS) prepolymer solution, a mixture of silicon elastomer and curing agent with a weight ratio of 10:1. The prepolymer mixture and the hybridized master were kept in an environmental chamber with 35% humidity to spontaneously remove any trapped air bubbles in the mixture.18 In our previous study,18 this humidity level was optimized to ensure that the level of condensation on the biomolecules does not affect printing onto PDMS. Thereafter, the prepolymer mixture was poured into the Petri dish containing the master and cured for 1.5 hours at 60 °C. The cured hardened PDMS (i.e. the secondary substrate) was separated from the master by mechanical force and immersed in a solution of fluorescent complementary printed peptide K sequence in PBS for 3 hours. After washing with DI water, fluorescence microscopy images of the substrate revealed that the peptide K array had been accurately replicated by LiSuNS, Fig. 2b. When the same experiment was carried out with a fluorescent non-complementary sequence, no pattern was observed (data not shown).
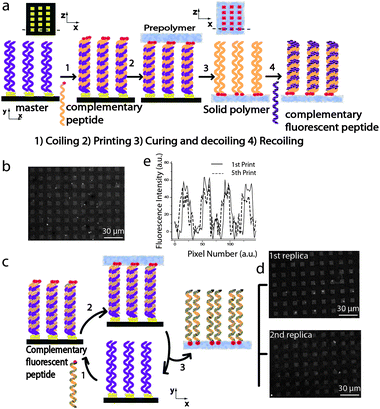 | ||
| Fig. 2 a) Schematic illustration of LiSuNS of coiled-coil peptides. b) Fluorescence micrograph of the PDMS secondary substrate with the peptide sequence K printed and labeled with the complementary fluorescent peptide E. c) Schematic illustration of repeat printing with LiSuNS. d) Fluorescence microscopy of the first replica and of the second replica. e) Comparison of fluorescence intensity of the printed patterns after the first and the fifth printing cycles. | ||
To ensure integrity of the master after printing, we repeated the printing cycle with the same master as depicted in Fig. 2c. The peptide K functionalized master was incubated with complementary fluorescent peptide E and this transferred to the PDMS secondary substrate enabling immediate assessment by fluorescence microscopy. Fluorescence microscopy of printed arrays generated from a first printing cycle and repeated printing cycles (where the same master was re-incubated in peptide E) is shown in Fig. 2d. We observed no significant loss in fluorescence intensity confirming the functional robustness of the master for multiple printing cycles (e.g. 5 print runs), Fig. 2e. We have also demonstrated that the initial sequence on the master does not transfer to the PDMS substrate during printing thus further enabling repeat printing applications and there is not any residual complementary peptide in the master after printing (data not shown).
LiSuNS has the ability to print spatial as well as chemical information: that is this technique not only transfers the shape, size and the position of the features on the master (spatial information), but also their chemical nature (chemical information). The potential use in the transfer of multiplexed nucleic acid and protein based chemical information in one cycle is a tantalizing goal.39 Here we explored such multiplexing capability to print a substrate containing DNA and peptide features in one cycle. We created a master using a microfluidic approach and immobilized peptide K and a single stranded thiolated DNA on a gold slide (ESI†). The master was passivated with MH and was treated with its complementary peptide E and complementary DNA sequence terminated with acridine. The secondary substrate was cured and mechanically separated from the master. The printed secondary substrate was then incubated in complementary fluorescent DNA and NHS-fluorescein, which is a marker for primary amines, and imaged with fluorescence microscopy. Fig. 3 shows a successful replication of the original patterns of DNA and peptide. When the hybridization step was carried out with a fluorescent non-complementary DNA sequence, no pattern was observed.
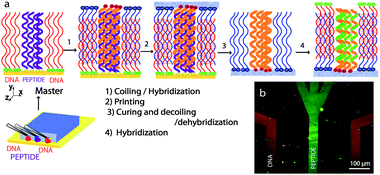 | ||
| Fig. 3 a) Multiplexed LiSuNS of peptide and DNA patterned array. a) Schematic illustration of the microfluidic channels used for master preparation and the steps involved in the process. b) Fluorescence micrograph of printed peptide and DNA lines with false colour overlay. | ||
In conclusion, we have utilized LiSuNS to generate arrays of peptides over large areas and demonstrated that the approach retains the key features of DNA-LiSuNS. We also advance the multiplexing capability of LiSuNS by demonstrating (to the best of our knowledge) the first example of DNA/peptide printing in a single step. We believe that this work lays the foundation for the use of SuNS and LiSuNS with complex biomolecules leading to the fabrication of surfaces and devices that go beyond microarrays. Future work will include the hierarchical fabrication of biomolecular features stemming from patterns printed in the way illustrated in this report. Additionally we point out that this work opens up new avenues in the facile generation of arrays of multiplexed chemical information.
Acknowledgements
We thank Dr M. Murugesan for his help in the peptide synthesis. This work was supported by the Packard Foundation (FS), and the EPSRC and FP7 ERC grant Naturale (MMS). AL thanks Beatriu de Pinós, Generalitat de Catalunya for funding.Notes and references
- S. F. Kingsmore, Nat. Rev. Drug Discovery, 2006, 5, 310–320 CrossRef CAS.
- C. Wingren and C. A. K. Borrebaeck, Drug Discovery Today, 2007, 12, 813–819 CrossRef CAS.
- S. K. Kufer, E. M. Puchner, H. Gumpp, T. Liedl and H. E. Gaub, Science, 2008, 319, 594–596 CrossRef CAS.
- A. B. Braunschweig, F. W. Huo and C. A. Mirkin, Nat. Chem., 2009, 1, 353–358 Search PubMed.
- F. W. Huo, Z. J. Zheng, G. F. Zheng, L. R. Giam, H. Zhang and C. A. Mirkin, Science, 2008, 321, 1658–1660 CrossRef CAS.
- M. J. Heller, Annu. Rev. Biomed. Eng., 2002, 4, 129–143 CrossRef CAS.
- F. G. Zaugg and P. Wagner, MRS Bulletin, 2003, 28, 837–842 CAS.
- P. S. Petrou, M. Chatzichristidi, A. M. Douvas, P. Argitis, K. Misiakos and S. E. Kakabakos, Biosens. Bioelectron., 2007, 22, 1994–2002 CrossRef CAS.
- M. He, O. Stoevesandt, E. A. Palmer, F. Khan, O. Ericsson and M. J. Taussig, Nat. Methods, 2008, 5, 175–177 CrossRef CAS.
- B. Spurrier, P. Honkanen, A. Holway, K. Kumamoto, M. Terashima, S. Takenoshita, G. Wakabayashi, J. Austin and S. Nishizuka, Biotechnol. Adv., 2008, 26, 361–369 CrossRef CAS.
- S. A. Lange, V. Benes, D. P. Kern, J. K. H. Hrber and A. Bernard, Anal. Chem., 2004, 76, 1641–1647 CrossRef CAS.
- A. Bernard, J. P. Renault, B. Michel, H. R. Bosshard and E. Delamarche, Adv. Mater., 2000, 12, 1067–1070 CrossRef CAS.
- J. P. Renault, A. Bernard, A. Bietsch, B. Michel, H. R. Bosshard, E. Delamarche, M. Kreiter, B. Hecht and U. P. Wild, J. Phys. Chem. B, 2003, 107, 703–711 CrossRef CAS.
- A. Bernard, D. Fitzli, P. Sonderegger, E. Delamarche, B. Michel, H. R. Bosshard and H. Biebuyck, Nat. Biotechnol., 2001, 19, 866–869 CrossRef CAS.
- J. P. Renault, A. Bernard, D. Juncker, B. Michel, H. R. Bosshard and E. Delamarche, Angew. Chem., Int. Ed., 2002, 41, 2320–2323 CrossRef CAS.
- H. Lin, L. Sun and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 11210–11211 CrossRef CAS.
- A. A. Yu, T. A. Savas, G. S. Taylor, A. Guiseppe-Elie, H. I. Smith and F. Stellacci, Nano Lett., 2005, 5, 1061–1064 CrossRef CAS.
- A. A. Yu and F. Stellacci, Adv. Mater., 2007, 19, 4338–4342 CrossRef CAS.
- A. A. Yu and F. Stellacci, in preparation.
- M. M. Stevens, N. T. Flynn, C. Wang, D. A. Tirrell and R. Langer, Adv. Mater., 2004, 16, 915–918 CrossRef CAS.
- P. Burkhard, J. Stetefeld and S. V. Strelkov, Trends Cell Biol., 2001, 11, 82–88 CrossRef CAS.
- J. R. Beasley and M. H. Hecht, J. Biol. Chem., 1997, 272, 2031–2034 CrossRef CAS.
- D. A. D. Parry, R. D. B. Fraser and J. M. Squire, J. Struct. Biol., 2008, 163, 258–269 CrossRef CAS.
- B. Tripet, L. Yu, D. L. Bautista, W. Y. Wong, R. T. Irvin and R. S. Hodges, Protein Eng., Des. Sel., 1996, 9, 1029–1042 CrossRef CAS.
- M. M. Stevens, S. Allen, M. C. Davies, C. J. Roberts, J. K. Sakata, S. J. B. Tendler, D. A. Tirrell and P. M. Williams, Biomacromolecules, 2005, 6, 1266–1271 CrossRef CAS.
- Y. Tsatskis, S. C. Kwok, E. Becker, C. Gill, M. N. Smith, R. A. B. Keates, R. S. Hodges and J. M. Wood, Biochemistry, 2008, 47, 60–72 CrossRef CAS.
- B. Koksch, K. Pagel, S. C. Wagner and J. Scheike, J. Pep. Sci., 2006, 12, 198 Search PubMed.
- A. Lupas, Trends Biochem. Sci., 1996, 21, 375–382 CrossRef CAS.
- K. M. Muller, K. M. Arndt and T. Alber, in Applications of Chimeric Genes and Hybrid Proteins, Pt C, Academic Press, San Diego, CA, 2000, pp. 261–282 Search PubMed.
- W. D. Kohn and R. S. Hodges, Trends Biotechnol., 1998, 16, 379–389 CrossRef CAS.
- H. Dietz, T. Bornschlogl, R. Heym, F. Konig and M. Rief, New J. Phys., 2007, 9, 424 CrossRef.
- E. H. C. Bromley, R. B. Sessions, A. R. Thomson and D. N. Woolfson, J. Am. Chem. Soc., 2009, 131, 928 CrossRef CAS.
- R. Y. Sweeney, E. Y. Park, B. L. Iverson and G. Georgiou, Biotechnol. Bioeng., 2006, 95, 539–545 CrossRef CAS.
- J. M. Mason, K. M. Muller and K. M. Arndt, Biochemistry, 2007, 46, 4804–4814 CrossRef CAS.
- M. O. Steinmetz, I. Jelesarov, W. M. Matousek, S. Honnappa, W. Jahnke, J. H. Missimer, S. Frank, A. T. Alexandrescu and R. A. Kammerer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7062–7067 CrossRef CAS.
- M. Sales, J. J. Plecs, J. M. Holton and T. Alber, Protein Sci., 2007, 16, 2224–2232 CrossRef CAS.
- G. Grigoryan and A. E. Keating, Curr. Opin. Struct. Biol., 2008, 18, 477–483 CrossRef CAS.
- J. R. Litowski and R. S. Hodges, J. Pept. Res., 2001, 58, 477–492 CrossRef CAS.
- A. Perrin, D. Duracher, M. Perret, P. Cleuziat and B. Mandranda, Anal. Biochem., 2003, 322, 148–155 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and methods. See DOI: 10.1039/b915803k |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2010 |