Cyanogen isocyanate (NC–NCO) revisited: thermal and chemical reactivity of a hydrogen-free precursor to C–N–(O) polymers
Carsten Ludwig
Schmidt
and
Martin
Jansen
*
Max-Planck-Institut für Festkörperforschung, Heisenbergstrasse 1, 70569, Stuttgart, Deutschland. E-mail: M.Jansen@fkf.mpg.de; Fax: +49 711 689 1502; Tel: +49 711 689 1501
First published on 6th November 2009
Abstract
The first inorganic polyisocyanate, (NC–NCO)x, has been prepared and investigated. By polyaddition (polymerisation) of the reactive monomer cyanogen isocyanate (NC–NCO) at low temperatures, the moisture-sensitive polymer was obtained as a phase pure solid. Using several analytical techniques (UV, IR, NMR, MS, DSC, TGA), the formation and the principal connectivity patterns of the hydrogen-free amorphous inorganic network have been settled. From the experimental results, a close structural relation to the organic “nylons” was deduced. Upon thermal impact, this inorganic macromolecule behaves like a typical thermosetting material. Exposing polymeric C2N2O to a temperature between 400 and 500 °C leads to quantitative removal of oxygen in the form of CO2. Depolymerisation and fragmentation at higher temperatures resulted in a virtually complete mass loss due to release of gaseous species. Some preliminary results concerning the high-pressure transformation towards a crystalline “C2N2O” (carbon oxynitride) are also given.
Introduction
The use of molecular single-source precursors has proven a versatile tool in the synthesis of new materials. Besides the famous sol-gel process,1,2 the so-called “polymer route” has attracted growing attention.3 Both concepts are quite similar. Suitable molecular precursors are polymerized via polyaddition, or polycondensation reactions. Subsequently, the resulting inorganic polymers are transformed to the desired ceramics by thermolysis. Various systems have been studied using this approach. Especially binary or multinary compounds among the non-metallic elements of the second and third row have been in the focus and crystalline BN, Si3N4 and P3N54 have been prepared in such a way. The same is true for multinary amorphous networks in the system Si–B–N–(C).5 Regrettably, “C3N4” (carbon nitride) is still elusive or only available as a rather ill-defined material.6 Starting from different precursors, thermal treatment finally yielded (graphitic) carbon only. In some cases residual “impurities” of variable amounts were detected (N, H, O, Li, Cl etc.). Such samples are commonly addressed as “CNx” or “CNxHy”. Thermolysis of various C–N–H–(O) compounds seems to have yielded at the best amorphous “heterocarbons” (German: “Mischpolymerisate”7). Recently several papers claimed the synthesis of crystalline variants of aromatic C–N–H networks (polyheptazines).8 While most of the older claims have already been falsified,9 in some of the remaining cases it is rather difficult to properly assess the results. For example, in the cases of the crystalline variants of “melon-like” C–N–H phases, different structural solutions were presented although the reaction routes applied were virtually the same. This holds true even more for experiments where high pressure techniques have been used.10 According to an actual literature survey, we count claims of at least 18 different crystalline modifications, including “C2N2NH” (carbon nitride imide).11 Clearly, such a vast amount of polymorphs is rather unlikely to exist. Because of the complex decomposition pathways in the system C–N–H–(O),12 either under ambient or high pressure a countless number of different chemical species can form. In fact, besides polymeric hydrogenated carbon (C–H)x also other inorganic carbon polymers like “melon” (C–N–H)x,13 “azulmic acid” (C–N–H)x,14 paracyanogen (C–N)x15 and “cyamelide” (H–O–C–N)x16 are known. Most of these polymers have remained rather ill-defined.In summarizing, one can state that all the enormous efforts made in trying to synthesize pure and well-defined C3N4 starting from hydrogen-containing precursors have failed so far. We regard the strength of the C–H or N–H-bonds, the cleavage of which affords high thermal activation, and the planar shape of most of the molecular precursors, directing to the formation of graphite, obstructive to realizing three dimensional structures of C3N4, predicted to be stable.17 Moreover, besides the loss of N2 at higher temperatures (the main drawback in all efforts to prepare pure C3N4), using hydrogen-containing samples further loss of nitrogen is enabled by releasing volatile NH3, HCN or NC–NH2. Therefore, in another attempt to synthesize stoichiometric C3N4, we have resorted to hydrogen-free molecular or polymeric precursors. Molecular NC–NCO appeared a good candidate for us since it polymerizes and moreover has the ability to transform to C3N4 by release of CO2. The release of CO2 as a kind of polycondensation of reactive monomers has already been observed in inorganic polymer chemistry18 (see equation 1).
| 2 C2N2O → C3N4 + CO2 | (1) |
As a first step in this direction, we have investigated the polymerisation of molecular NC–NCO and have characterized the polymer C2N2O in more detail. The results obtained are presented in this article. The analysis of polymeric C3N4 prepared in such a way (equation 1) will be presented in a subsequent publication. However, solid C2N2O might be an interesting material in itself because of possible analogies with Si2N2O, as they have been indicated by some theoretical studies.19
Experimental details
Synthesis
30 g of freshly prepared AgNCO (silver isocyanate) were dried under dynamic vacuum (p = 10−2 mbar) at T = 80 °C for several days. The absence of moisture was checked by infrared spectroscopy. The dried solid was decomposed at T = 750 °C and p = 10−2 mbar, and the gaseous products of decomposition were collected into a cold trap. The setup used is described in detail in a recent publication.20 The frozen solids were slowly warmed up to RT, while the gaseous by-products (N2, CO2) escaped. A yellow solid (between 0.5 and 1 g) was recovered which was highly moisture-sensitive and had to be handled under inert conditions.X-Ray powder diffractometry
For the XRD analysis, carefully ground samples were filled into a 0.3 mm lithium borate glass capillary (Hilgenberg, glass No 14) and measured in transmission geometry. Ambient and high temperature powder X-ray diffraction studies were performed on a STOE STADI P instrument with a linear PSD detector and with Cu-Kα1 radiation (λ = 1.5406 Å) from a curved Johansson-type monochromator using Ge (111).Thermal analysis
Simultaneous DTA/TG/MS (STA 409, Netzsch) was performed on a sample placed in a corundum crucible under flowing argon (100 ml/min) at a heating rate of 10 K/min. For the DSC (DSC 404, Netzsch), the sample was heated in a corundum crucible with a heating rate of 10 K/min under argon, the cooling rate was 2 K/min.Spectroscopy
Infrared spectra were recorded on a FT-IR spectrometer (IFS 113v, Bruker) from 400 cm−1 to 4000 cm−1 with KBr as the solid matrix. 1–2 mg substance was mixed with 300 mg of KBr and the powder pressed to pellets (diameter 1 cm), applying 300 MPa. For the Raman experiments the sample was filled in a 0.3 mm lithium borate glass capillary (Hilgenberg, glass No 14). The spectra were recorded with an excitation line of 632.817 nm (Labram 010, single grating) at acquisition times between 5 to 300 sec with a laser power between 0.4 and 0.004 mW. UV-vis spectroscopy was performed on a Perkin Elmer Lamda 9 spectrometer. For solutions, quartz cuvettes of 0.5 cm path length were used.He Pycnometry
The measurement of the density (Accupyc 1330 GB, micromeritics) was performed under flowing helium after calibrating with a given defined standard.NMR spectroscopy
Liquid NMR spectra of the dissolved samples were recorded using a Bruker Avance DPX-300 SB operating at 7.05 T. MAS-NMR studies were performed using a Bruker DSX-400 spectrometer operating at 9.4 T. The sample was filled in a 7 mm ZrO2 rotor and the magic-angle spinning frequency used was 79.49 MHz.Mass spectroscopy
MALDI-TOF mass spectra were obtained in reflection mode on a Bruker Daltonics (Bremen) Reflex VI (337 nm nitrogen laser). The DCTB served as a matrix for analysis of oligomers in the positive mode. The samples were prepared by mixing DMSO solutions of the oligomer and the matrix in the ratio 1:50. For GC-MS the setup TSQ 700 (Fa. Finnigan MAT, Bremen) was used.Elemental analysis
Elemental analysis was performed at the PML, Max Planck Institute for Metals Research, Stuttgart, using various apparatus (ELEMENTAR, Vario EL CHN-Determinator; ELTRA, CS 800, C/S Determinator; LECO, TC-436, N/O Determinator).Theory
Atomic charge calculations were performed applying the program Gaussian,21 using DFT with the hybrid functional B3LYP/6-31G(d). The geometrical parameters used were obtained from recent structural studies.20Experimental results
The molecular NC–NCO was prepared by decomposing AgNCO.20 Polymeric C2N2O was obtained in two different ways. Along the first, the “dry” monomer was warmed up to ambient conditions, with a yellow-coloured “glassy” solid resulting. In the second approach, a solvent was added at T = −76 °C and the solution was warmed up to RT. The choice of an appropriate solvent has proven to be crucial: while using toluene or pentane, a solid precipitate formed very fast; with ether and even more with acetonitrile, no phase separation occurred. In the latter cases, the initially yellow coloured solution turned orange and even at RT no solid was formed. However, upon storage at RT for several hours, the solution turned viscous and finally gave an orange-coloured gel. Taking all phenomena together, oligomeric/polymeric C2N2O behaves like a lyophilic colloid, as typically formed by macromolecules.22 This specific behaviour enables the colloidal solution to be applied by a sol-gel coating process.23 For further studies reported here, only the isolated resin-like polymer was used. The amorphous solid is not stable against moisture. Exposed to humid air at RT it transforms into cyanamide as monitored by powder X-ray diffraction studies, and during decomposition the evolution of a gas was noticed (equation 2):| NC–NCO + H2O → NC–NH2 + CO2 | (2) |
According to an elemental analysis of the yellow polymer (found [calculated for C2N2O]: C: 35.47 wt% [35.31], N: 41.51 wt% [41.18], O: 19.30 wt% [23.52]) the experimental sum formula is C2N2O0.9. No direct analysis of the hydrogen content has been performed. Due to the general abundance of hydrogen we have focused on potent indirect methods of high sensitivity like GC-MS, TGA-MS and MALDI-ToF (see below).
The density of polymeric C2N2O was found to be 1.74 g/cm3 at RT. For comparison, the calculated X-ray density of molecular NC–NCO at T = −100 °C is 1.51 g/cm3.20
A UV-Vis spectrum recorded on the slightly yellow solution of redispersed polymeric NC–NCO (Fig. 1) consists of two bands. A strong band was found at 230 nm and a weaker one at 274 nm. For comparison, HNCO (isocyanic acid) absorbs at 280 nm,24 (CN)2 (cyanogen) at 220 nm and 272 nm25 and NC–NCS at 267 nm.26 Therefore, the UV-Vis spectrum provides evidence that both functional groups, CN and NCO, are present in our polymer.
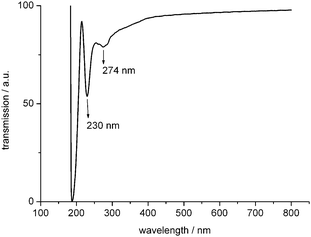 | ||
| Fig. 1 UV-Vis spectrum of NC–NCO oligomers in acetonitrile. | ||
No liquid 13C-NMR spectrum could be recorded using the same dispersion, and instead, a solid-state 13C-MAS experiment was performed. One sharp resonance (plus sidebands) was detected at 139 ppm (Fig. 2). This shift is between the known values for an isocyanate (–NCO) group (160 ppm) and a carbodiimide/cyanamide group (116–125 ppm).27 Although the NC–NCO sample investigated was polymeric, the presence of only one rather sharp signal indicates a rather well developed and defined short range structure. This is quite common for polyisocyanates and polynitriles.28 For comparison, in polymeric paracyanogen (CN)x, one 13C signal was found at 153 ppm.29
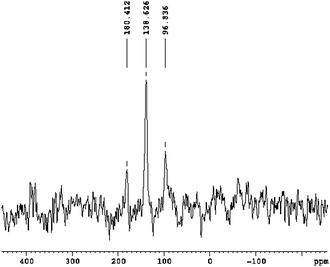 | ||
| Fig. 2 13C-MAS-NMR spectrum of polymeric NC–NCO. | ||
The IR spectrum of the polymer is shown in Fig. 3. A strong absorption at 2288 cm−1 and a weak one at 2342 cm−1 match the characteristic group frequencies of nitriles (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), or isocyanates (–NCO). A very intense band found at 1793 cm−1 is indicative for a carbonyl group (–C
N), or isocyanates (–NCO). A very intense band found at 1793 cm−1 is indicative for a carbonyl group (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), while the absorption at 1382 cm−1 is in the typical region for di-substituted amide functions.30 At lower wavenumbers, three additional bands were clearly identified at 1178 cm−1, 995 cm−1 and a strong one at 720 cm−1, which however, are difficult to assign unambiguously. A very weak and rather broad band was found between 3000–3250 cm−1, most likely resulting from an undesired hydrolysis of this highly sensitive compound, taking place during the preparation of the pellets. However, as further shown below, no indications for the presence of hydrogen in significant amounts have been obtained by other experimental methods. In addition, comparing this IR data with an IR spectrum of “melon” (containing only 1.8% of hydrogen8) may give further support that our sample can be classified as being hydrogen-free. Raman spectroscopy on the yellow polymer failed due to strong photoluminescence.
O), while the absorption at 1382 cm−1 is in the typical region for di-substituted amide functions.30 At lower wavenumbers, three additional bands were clearly identified at 1178 cm−1, 995 cm−1 and a strong one at 720 cm−1, which however, are difficult to assign unambiguously. A very weak and rather broad band was found between 3000–3250 cm−1, most likely resulting from an undesired hydrolysis of this highly sensitive compound, taking place during the preparation of the pellets. However, as further shown below, no indications for the presence of hydrogen in significant amounts have been obtained by other experimental methods. In addition, comparing this IR data with an IR spectrum of “melon” (containing only 1.8% of hydrogen8) may give further support that our sample can be classified as being hydrogen-free. Raman spectroscopy on the yellow polymer failed due to strong photoluminescence.
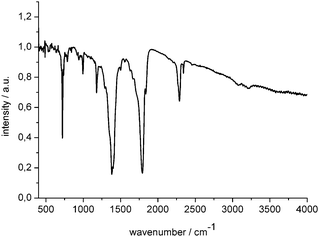 | ||
| Fig. 3 IR spectrum (in KBr) of solid NC–NCO polymer obtained by solid-state polymerisation. | ||
In laser-ionsation-ToF-MS studies on polymeric NC–NCO, fragmentation of the thermally sensitive compound occurred, and with laser intensities of 10%, 30% and 65%, quite different ToF spectra were obtained. Without a matrix and with an intensity of 10%, a group of signals centred at 544 m/z was detected (Fig. 4), a mass that corresponds to a volatile oligomer (NC–NCO)8 (544 m/z). The observed pattern is typical for a highly cross-linked polymer fragmenting into various oligomers. With a higher thermal impact (65% laser intensities), only the primary fragments –NCO and –CN were seen.20 No indications for the presence of hydrogen could be found. For example HCN, HOCN and NH3 were absent. In order to circumvent the massive thermal degradation (local heating) induced by the laser beam, the NCNCO polymer was independently analyzed using GC-MS. The spectrum obtained is shown in Fig. 5. As volatile products, CO2 (mass 44) and molecular NCNCO (mass 68) were clearly observed. No higher mass, e.g. 204 m/z that would indicate the presence of trimers of NCNCO, could be detected. In addition, also with this method, the presence of hydrogen-containing species could be ruled out. In the purely thermal activation process, the signal for CO2 was always observed with higher intensity than the NC–NCO molecular peak. Therefore, it was concluded that at these conditions, the polymer decomposed rather than de-polymerized. Finally, a solution of NC–NCO in DMSO was analysed via MALDI-ToF (see Fig. 6). Here, the formation of the NC–NCO dimer (m/z: 136) as well as of the trimer (m/z: 204) could be proven. This result is similar to that obtained for HOCN.24 Summarizing the results of MS spectroscopy, bulk NC–NCO consists of several oligomers of rather moderate size. Upon energetic impact the oligomers depolymerise and finally decompose forming the volatile monomer NC–NCO and CO2 and other non-volatile products of decomposition.
 | ||
| Fig. 4 Laser-ionsation-ToF mass spectrum (without matrix) of polymeric (C2N2O)x. | ||
 | ||
| Fig. 5 GC-MS spectrum (EI mode) of polymeric C2N2O. The signal at m/z: 68 corresponds to the molar mass of monomeric NC–NCO. | ||
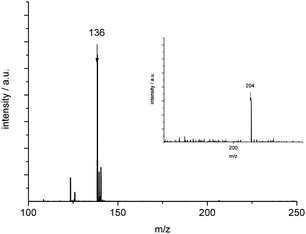 | ||
| Fig. 6 MALDI-ToF-MS data on polymeric NC–NCO. Intense signals for the dimer (NC–NCO)2 (m/z: 136) and a weak signal for the trimer (NC–NCO)3 (m/z: 204) (enlarged insert) have been recorded. | ||
According to TGA-MS studies of the amorphous polymer (heating rate 10 °C/min, see Fig. 7), decomposition starts around T = 170 °C with CO2 as the only detectable gaseous species evolving. Other volatile molecules (especially hydrogen-containing ones like NH3, HCN, HOCN or NC–NH2) were not detected. The mass loss as a function of time in this step (up to T = 618 °C: 37%) follows an exponential law. Mathematical analysis of the data from T = 190 °C up to T = 560 °C yields a good fitting function (y = 116.79e(−x/162.43) + 61.60; r = 0.99899). The observed exponential decay is commonly noticed for cross-linking reactions in the solid state.
 | ||
| Fig. 7 TGA-MS trace of polymeric C2N2O. TGA trace in black, fitted curve in cyan, MS for CO2 in green, (CN)2 in red and N2 in blue. | ||
Concerning inorganic polymers, it has been studied in great detail for siloxanes and silicones.31 The polycondensation will slow down more and more as the as formed network becomes more and more stiff and inflexible. At temperatures beyond T = 560 °C the experimental data deviate from the fit, pointing towards a growing contribution of another mechanism. Since at this temperature no further masses could be detected in the MS, it was concluded that molecular or oligomeric NCNCO was released which polymerized or condensed again at colder parts of the DTA setup. This assumption is in agreement with the results of the GC-MS study (see Fig 5 and equation 3).
| (NCNCO)x(s) → CO2(g) + NCNCO(g) + C3N4(s) | (3) |
The ideal reaction (no formation of molecular NCNCO) would result in an overall mass loss of 32%. Applying the fitting function of the TG data, this mass loss would be reached at T = 467 °C. In fact, in closed vessels phase pure C3N4 can be prepared by thermal treatment between 400 and 500 °C. Formation of gaseous NC–NCO within these closed vessels has been proven. Upon heating only a certain part of the ampoule up to 400 °C, NC–NCO sublimed to the colder parts of the ampoule. The full analysis of this hydrogen-free variant of C3N4 will be presented in a subsequent publication. The second step of decomposition is clearly seen around T = 620 °C. N2, (CN)2 as well as additional CO2 was detected in the MS. At this point bond breaking reactions set in. At the end of this step of fast decomposition (T = 780 °C) no significant amounts of a solid residue were left, nearly complete decomposition had occurred. The walls of the crucible were covered by a very thin black film, however, it was not possible to harvest significant amounts of that material for further characterisation.
DSC measurements (data not shown) have indicated that the evolution of CO2 starting at T = 170 °C is an exothermic process (broad weak signal) while the decomposition at T > 600 °C is strongly endothermic.
Discussion
Molecular NC–NCO (cyanogen isocyanate)20 has been shown to be sensitive to moisture and to be a highly reactive species. Without any further initiation, it polymerises upon warming from T = −76 °C to RT, forming a yellow inorganic amorphous material. On warming up molecular NC–NCO in a solvent, polymerisation results in either resin-like solids (toluene) or highly viscous gels (acetonitrile).Even nowadays it is still a challenge to elucidate the structure of amorphous materials. Applying UV-Vis, IR and 13C-NMR spectroscopy, CN, CO and N–CN/NCN functions have been identified to be the main building units of amorphous NC–NCO. Remarkably, only one signal has been detected in 13C-MAS NMR spectra, indicating a rather homogeneous and uniform surrounding of the carbon atoms. Backed by DFT/B3LYP/6-31G(d) calculations, we identify three possible reaction routes for the oligomerisation of the monomer (Fig. 8): A, trimerisation of the CN groups, forming a substituted triazine (as known from HCN); B, trimerisation of the OCN groups may be possible forming a cyanurate core (as known from HNCO); and finally C, under certain conditions, an extended linear polymer could be formed (also known to occur with HNCO “cyamelide”). The presence of both functional groups (NCO and CN) in the polymer was validated by UV-Vis, and IR spectroscopy, which would not comply with trimers formed along routes A or B. Analysing the Mulliken charges on molecular NC–NCO provided further clues concerning possible intermolecular reactions towards the final polymer (Fig. 9).
 | ||
| Fig. 8 Possible reaction paths for the oligomerisation/polymerisation of molecular NC–NCO. | ||
 | ||
| Fig. 9 Mulliken charge populations for molecular NC–NCO. | ||
It turned out that the highest charges are localized on the CN double bond of the isocyanate group (N: −0.42, C: +0.65). Therefore, we expect the polyaddition reaction to occur most likely at this specific point of high polarity. This kind of regioselective reactivity is well known and commonly observed in organic isocyanate chemistry.32 For example, in their reviews, Tiger28 and Bur30 discuss several examples of this dominant regioselective reaction resulting in polymers with substituted amide functions. For this reason, we assign an uretdione-type of constitution to the observed dimer (Fig. 6) 32 and we regard the trimer (Fig. 6) as a molecular cyanurate (Fig. 10).
 | ||
| Fig. 10 The oligomerisation/polymerisation of molecular NC–NCO. | ||
Therefore, if trimerisation is a significant reaction path at all, it would most likely occur according to route B (Fig. 8). In fact, the IR data did not give any evidence for the presence of the triazine core.
The dimer and trimer were identified as unstable intermediates only in the case of MALDI-ToF experiments. Studying the bulk material with several techniques, non-destructive as well as destructive ones, did not give any indication for the presence of these molecules in significant amounts. Thus, we conclude that the formation of molecular species is not favoured at all and instead, like in the cases of HNCO24 or RNCO,32 polymerisation (Fig. 8, reaction C) takes place. The absence of stable trimers is especially noteworthy, since other RNCO polymers (organic polyisocyanates) are known to form monomers as well as trimers upon thermal depolymerisation.32 In the case of NC–NCO, the polymer depolymerises mainly to the monomer. Although commonly observed for other polymers, in this case no indications for a ring-chain equilibrium were noticeable.33 Another hint supporting mechanism C might be deduced from the gel-like behaviour upon self-polymerisation in a polar solvent like acetonitrile. The effect that during the development of an extended inorganic polymer a gel-like or visco-elastic state can be recovered is not uncommon.34
From the DSC data, we conclude that our inorganic polymer behaves like a highly-crosslinked inorganic network. This again is in full agreement with general concepts of polymer chemistry.34 Similar thermal effects have been previously observed for various polyisocyanates.32 In general, this behaviour is rather typical for “non-graphitizing” highly cross-linked polymers (“Duroplaste” or “thermosets”).35 Evolution of CO2 was seen to be a favoured process which is interesting with respect to the formation of a pure C–N network.
Several decomposition mechanisms seem to be responsible for the mass losses observed: massive CO2 abstraction starting at T = 170 °C, additional decomposition reactions noticeable at T > 560 °C and finally nearly complete vaporization starting at T = 620 °C. This latter temperature of decomposition as observed with the thermoanalytical methods applied (see experimental section) seems to be quite common. Also for hydrogen-containing C–N–O–H samples of various kinds, a decomposition temperature of T > 600 °C has been reported.8 In our case, the observed steps of decomposition are not well separated and at higher temperatures take place simultaneously. Therefore, in order to remove CO2 efficiently without any side reactions, the temperature has to be kept below 560 °C. From the fitting function to the experimental TGA trace it was suggested that complete CO2 abstraction could be even possible at T < 470 °C.
It is important to note that replacing the nitrile group in the (NC–NCO)x polymer either by H or by an organic function R would yield the well known polymers “cyamelide”16 or “nylon-1”.36 Especially the latter (organic) polymers have been studied in depth. Comparing the analytical data available, all of them seem to have the same polymeric structure (Fig. 8, C). Also the conditions of synthesis are quite similar to the case of (C2N2O)x. At low temperatures, polyaddition proceeds through the reactive isocyanate group.36
Polymeric C2N2O, which according to our findings is an inorganic polyisocyanate, can be seen as a new candidate for establishing extended non-molecular crystalline “low-Z solids”37 like N2, C2N2, CO and CO2. All these latter initial hydrogen-free molecular compounds have been polymerized and crystallized under extremely high pressure conditions using diamond anvil cells. Following the same path, also a crystalline “carbon oxynitride” should be accessible. In fact, during preliminary experiments applying pressures up to p = 20 GPa and temperatures up to T = 600 °C, the (C2N2O)x polymer transformed into an amorphous black-coloured carbon phase (93 wt% carbon) only containing carbon nanotubes. Obviously, the pressure conditions applied were not sufficient to prevent loss of nitrogen (and oxygen). Maybe the commonly observed escape of N2 from C–N–(O) networks at elevated temperatures can be prevented at even higher pressures. For the transformation of molecular CO2 to extended tridymite-like solid structures, pressures and temperatures up to 40 GPa and 1800 K were needed.38 We expect similar extreme conditions to be necessary for achieving analogous high pressure transformations (crystallisation) for (C2N2O)x.
Conclusion
The all inorganic random network (C2N2O)x was obtained by polymerisation (polyaddition) of unstable molecular NC–NCO (cyanogen isocyanate). Using several overlapping and complementary probes, insights into its structure and thermal behaviour have been deduced. Striking analogies between (C2N2O)x and both cyamelide as well as nylon-1 have been elaborated. Therefore, we classify C2N2O as an inorganic hydrogen-free polyisocyanate. From the thermal studies it has followed that the polymer is thermally stable up to T = 170 °C. Above this threshold, mainly CO2 is evolving until at T > 600 °C complete decomposition sets in, releasing (CN)2, CO2 and N2. Upon subjecting (C2N2O)x to elevated pressure and temperature conditions of 20 GPa and 600 °C, respectively, no evidence for crystallisation was obtained. For the realisation of a crystalline variant of C2N2O, further studies at even higher pressures appear necessary. Polymerisation of molecular NC–NCO in suitable solvents gave colloidal dispersions of this inorganic macromolecule enabling sol-gel processing of thin films.Acknowledgements
The authors would like to thank Jasmin Jarczak for experimental support. C.L.S. also thanks the “Studienstiftung des Deutschen Volkes” as well as the “Fonds der chemischen Industrie” (Kekule scholarship) for scholarships.References
- H. Schmidt, J. Non-Cryst. Solids, 1988, 100, 51 CAS.
- J. Livage, Prog. Solid State Chem., 1988, 18, 259 CrossRef CAS.
- K. J. Wynne and R. R. Rice, Annu. Rev. Mater. Sci., 1984, 14, 297 CAS; M. Birot, J.-P. Pillot and J. Dunogues, Chem. Rev., 1995, 95, 1443 CrossRef CAS; H.-P. Baldus and M. Jansen, Angew. Chem., 1997, 109, 338; R. J. P. Corriu, Angew. Chem., 1990, 102, 818 CrossRef.
- R. T. Paine and C. K. Narula, Chem. Rev., 1990, 90, 73 CrossRef CAS; C. Boberski, R. Hamminger, M. Peukert, F. Aldinger, R. Dillinger, J. Heinrich and J. Huber, Adv. Mater., 1989, 101, 1592 CAS; W. Schnick, J. Lücke and F. Krumeich, Chem. Mater., 1996, 8, 281 CrossRef CAS.
- M. Jansen, B. Jäschke and Th. Jäschke, Struct. Bonding, 2002, 101, 137 CAS.
- J. V. Badding, Adv. Mater., 1997, 9, 877 CrossRef CAS; M. J. Yacaman, J. M. Gil, F. J. M. Gil, M. Sarikaya and M. X. Quian, Mater. Chem. Phys., 1997, 47, 109 CrossRef CAS; E. G. Wang, Prog. Mater. Sci., 1997, 41, 241 CrossRef CAS; A. K. Sharma and J. Narayan, Int. Mater. Rev., 1997, 42, 137 Search PubMed; S. Muhl and J. M. Mendez, Diamond Relat. Mater., 1999, 8, 1809 CrossRef CAS; I. Widlow and Y. W. Chung, Int. Mater. Rev., 2002, 47, 153 Search PubMed; J. C. Sung, New Diamond Front. Carbon Tech., 2002, 12, 47 Search PubMed; E. Kroke and M. Schwarz, Coord. Chem. Rev., 2004, 248, 493 CrossRef CAS; G. Goglio, D. Foy and G. Demazeau, Mater. Sci. Eng., R, 2008, 58, 195 CrossRef.
- H. Krebs, Angew. Chem., 1953, 65, 293 CrossRef CAS; H. Krebs, Angew. Chem., 1958, 70, 615 CrossRef CAS.
- Z. Zang, K. Leinenweber, M. Bauer, L. A. J. Garvie, P. F. McMillan and G. H. Wolf, J. Am. Chem. Soc., 2001, 123, 7788 CrossRef CAS; M. J. Bojdys, J.-O. Müller, M. Antonietti and A. Thomas, Chem.–Eur. J., 2008, 14, 8177 CrossRef CAS; B. V. Lotsch, M. Döblinger, J. Sehnert, L. Seyfarth, J. Senker, O. Oeckler and W. Schnick, Chem.–Eur. J., 2007, 13(17), 4969 CrossRef CAS; M. Döblinger, B. V. Lotsch, J. Wack, J. Thun, J. Senker and W. Schnick, Chem. Commun., 2009, 1541 RSC.
- T. Malkow, Mater. Sci. Eng., A, 2001, 302, 311 CrossRef.
- E. Horvath-Bordon, R. Riedel, A. Zerr, P. F. McMillan, G. Auffermann, Y. Prots, W. Bronger, R. Kniep and P. Kroll, Chem. Soc. Rev., 2006, 35, 987 RSC.
- E. Horvath-Bordon, R. Riedel, P. F. McMillan, P. Kroll, G. Miehle, P. A. van Arken, A. Zerr, P. Hoppe, O. Shebanova, I. McLaren, S. Lauterbach, E. Kroke and R. Boehler, Angew. Chem., 2007, 119, 1498 CrossRef; E. Horvath-Bordon, PhD thesis, TU Darmstadt, 2004.
- B. Bann and S. A. Miller, Chem. Rev., 1958, 58, 131 CrossRef CAS; F. Kurzer, Chem. Rev., 1956, 56, 95 CrossRef CAS.
- E. C. Franklin, J. Am. Chem. Soc., 1922, 44, 486 CrossRef CAS (but also many other references of this author).
- Th. Völker, Angew. Chem., 1960, 72, 379 CrossRef.
- L. W. Jenneskens, J. W. G. Mahy, E. J. Vlietstra, S. J. Goede and F. Bickelhaupt, J. Chem. Soc., Faraday Trans., 1994, 90, 327 RSC.
- W. Kern, H. Paul and W. Mehren, Makromol. Chem., 1954, 14, 146 CrossRef CAS.
- A. Y. Liu and M. L. Cohen, Science, 1989, 245, 841 CAS.
- E. Mayer, Monatsh. Chem., 1970, 101, 834 CAS.
- A. L. Ivanovskii, N. I. Medvedeva and G. P. Shveikin, Mendeleev Commun., 1999, 9(1), 34 CrossRef; A. L. Ivanovskii, N. I. Medvedeva, O. Yu. Kontsevoi and G. P. Shveikin, Phys. Status Solidi B, 2000, 221, 647 CrossRef CAS.
- C. L. Schmidt, R. Dinnebier and M. Jansen, Solid State Sci., 2009, 11, 1107 CrossRef CAS; C. L. Schmidt, D. Fischer, H.-J. Himmel, R. Köppe, H. Schnöckel and M. Jansen, Z. Anorg. Allg. Chem., 2009, 635, 1172 CrossRef CAS.
- Gaussian 03, Revision D.02, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- H. Staudinger, Organische Kolloidchemie, 1950, Vieweg Verlag, Braunschweig, Deutschland Search PubMed.
- C. L. Schmidt, M. Jansen, Patent application, 2009.
- D. J. Belson and A. N. Strachan, Chem. Soc. Rev., 1999, 45, 41 Search PubMed.
- F. Cataldo, Eur. Polym. J., 1999, 35, 571 CrossRef CAS.
- P. Li, K. Wong and W. F. Fan, Chem. Phys. Lett., 2003, 380, 117 CrossRef CAS.
- D. Rovnyak, M. Baldus, B. A. Itin, M. Bennati, A. Stevens and R. G. Griffin, J. Phys. Chem. B, 2000, 104, 9817 CrossRef CAS.
- R. P. Tiger, L. I. Sarynina and S. G. Entelis, Russ. Chem. Rev., 1972, 41, 774 Search PubMed.
- L. Maya, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2595 CAS.
- A. J. Bur and L. J. Fetters, Chem. Rev., 1976, 76, 727 CrossRef CAS.
- W. Noll, Chemie und Technologie der Silicone, 1968, Verlag Chemie, Weinheim, Deutschland Search PubMed.
- H. J. Laas, R. Halpaap and J. Pedain, J. Prakt. Chem., 1994, 336, 185 CrossRef CAS; R. G. Arnold, J. A. Nelson and J. J. Verbanc, Chem. Rev., 1957, 57, 47 CrossRef CAS; J. H. Saunders and R. J. Slocombe, Chem. Rev., 1948, 43, 203 CrossRef CAS; S. Ozaki, Chem. Rev., 1972, 72, 457 CrossRef; Y. Iwakura, K. Uno and N. Kobayashi, J. Polym. Sci., Part A-1, 1968, 6, 2611 CrossRef CAS.
- J. B. Carmichael, J. Macromol. Chem., 1966, 1, 207 Search PubMed; J. R. van Wazer, J. Macromol. Sci., Part A: Pure Appl. Chem., 1967, 1, 29 Search PubMed; H. R. Allcock, J. Macromol. Sci.-Rev. Macromol. Chem. C, 1970, 4, 149 Search PubMed.
- L. Z. Rogovina and G. L. Slonimskii, Russ. Chem. Rev., 1974, 43, 503 Search PubMed; L. Z. Rogovina and V. G. Vasil'ev, Macromol. Symp., 1996, 106, 299 CAS.
- B. Lersmacher and H. Lydtin, Chem. Ing. Tech., 1970, 42, 659 CrossRef CAS.
- V. E. Shashoua, W. Sweeny and R. F. Tietz, J. Am. Chem. Soc., 1960, 82, 866 CrossRef CAS.
- V. V. Brazhkin and A. G. Lyapin, Nat. Mater., 2004, 3, 497 CrossRef CAS.
- V. Iota and C. S. Yoo, Phys. Status Solidi B, 2001, 223, 427 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |