DOI:
10.1039/B921203P
(Paper)
Green Chem., 2010,
12, 114-122
In vitro biocompatibility of nanoscale zerovalent iron particles (NZVI) synthesized using tea polyphenols
Received 7th April 2009, Accepted 9th October 2009
First published on 4th November 2009
Abstract
A “green” protocol was used for the rapid generation of nanoscale zerovalent iron (NZVI) particles using tea polyphenols. The NZVI particles were subsequently examined for in vitro biocompatibility using the human keratinocyte cell (HaCaT) line as a representative skin exposure model. The cells were exposed to NZVI for time periods of 24 and 48 h. Biocompatibility was assessed using the methyl tetrazolium, or MTS, (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)) and lactate dehydrogenase (LDH) assays to determine in vitro cytotoxicity. The evaluation of mitochondrial function (MTS) and membrane integrity (LDH) in human keratinocytes showed that these “green” synthesized NZVI particles were nontoxic in the human keratinocytes exposed when compared with control samples synthesized using a borohydride protocol. In fact, in most cases, these “green” nanoparticles induced a prolific response in the cellular function even at the highest concentration (100μg ml−1).
Introduction
Nanoscale zerovalent iron (NZVI) particles have been found to be effective in degrading various chlorinated organic contaminant hydrocarbons such as trichloroethylene,1 and trichloroethene (TCE).2–4 Recent environmental applications include removal of nitrite by ultrasound dispersed NZVI,5 dechlorination of dibenzo-P-dioxins,6 reduction of chlorinated ethanes,7 adsorption of humic acid and its effect on arsenic removal8–9 and hexavalent chromium removal.10 However, field applications of NZVI have been limited to granular particles used in permeable reactive barriers (PRB).4,5 While PRBs are found to be effective for remediation of shallow aquifers, more cost-effective in situ technologies are lacking for rapid and complete destruction of chlorinated contaminants in deep aquifers and in source zones.6 Recently, there is great interest in testing an in situ remediation strategy where reactive NZVI particles are directly delivered into the contaminant source zones compared to passive technologies such as PRB. The in situ preparation of the NZVI particles offers unique advantages, such as (i) the applicability to source zones in deep aquifers or to areas where lands are occupied, (ii) a much shorter overall remediation time, (iii) transportation and chemical reactivity and (iv) it helps relieve surface oxidation problems. However, for this technology to be feasible, the NZVI particles must be small enough to be mobile in the targeted zones, and the transport behaviors (or size) of the nanoparticles in various soils must be controllable. Yet, there has been no technique available for preparing NZVI particles of controlled size and transport properties, and a method is lacking to extend the reactive lifetime of these relatively short-lived nanoparticles. There are several methods which have been employed to prepare NZVI particles including sodium borohydride (NaBH4),11–14 ethylene glycol15 and corbothermal synthesis.10 While NZVI particles have been reported to be attainable by these methods, it was recognized that NZVI particles tend to rapidly agglomerate to form larger aggregates due to Van der Waals and magnetic forces, rendering them undeliverable to the targeted contaminant locations.11,12To prevent aggregation of metallic nanoparticles, particle stabilization has been commonly practiced by attaching a stabilizer such as a soluble polymer or surfactant onto the nanoparticles.13 The stabilizer molecules are designed to provide strong inter-particle electrostatic and/or steric repulsions to outweigh the attractive Van der Waals and magnetic forces. To stabilize NZVI, two general strategies have been employed: (i) Use of stabilizers before the nanoparticles or aggregates are formed or (ii) to mechanically break down the formed nanoparticle agglomerates and add a stabilizer (post-agglomeration stabilization).
We have seen a renewed interest in using green chemistry principles in the preparation of nanomaterials and nanocomposites.14–27 There is a tremendous increase in biological applications of these nanostructures, as well as a continuous interest in using biodegradable polymers or surfactants to cap these nanoparticles that prevent them from aggregation. Most of these polymers or surfactants have a tendency to be water soluble and it is of great interest to know how good dispersion or capping can be obtained using these biodegradable polymers or surfactants. The tea extract (poly phenols) was chosen as a reducing agent and stabilizing agent because poly phenols are biodegradable (non toxic) and water soluble at room temperature, unlike other polymers. Second, poly phenols can form complexes with metal ions and thereafter can reduce the metals. Third, tea extract contains molecules bearing alcoholic functional groups which can be exploited for reduction as well as stabilization of the nanoparticles.
In this paper we have synthesized green NZVI which can ideally be utilized for toxicological studies. Control experiments were carried out using the standard NaBH4 reduction method and results were compared with the new green approach. Favorable conditions to make Fe nanoparticles were established with uniform size and shape.
Experimental procedure
Synthesis of “green” nanoscale zerovalent iron particles (NZVI)
Tea extract preparation: about 2 grams of tea powder (Red label from Tata, India Ltd. 99%) were extracted in 100 mL hot water and were then used to carry out the reaction with 0.1 N Fe(NO3)3. The different compositions were prepared at room temperature and are shown in Table 1. Control experiments were carried out using 0.1 N NaBH4 solutions at room temperature and the compositions are listed in Table 1.
Table 1 Preparation of nanoscale zerovalent iron (NZVI) particles using tea
| Sample Code No | Description |
|---|
| T1 | 10mL tea extract + 1mL 0.1 N Fe(NO3)3 solution |
| T2 | 5mL tea extract + 5mL 0.1 N Fe(NO3)3 solution |
| T3 | 1mL tea extract + 5mL 0.1 N Fe(NO3)3 solution |
| T4 | 1mL tea extract + 10mL 0.1 N Fe(NO3)3 solution |
| T5 | 5mL tea extract + 4mL 0.1 N Fe(NO3)3 solution |
| T6 | 5mL epicatechin (0.01 N) extract + 1mL 0.1 N Fe(NO3)3 |
| T7 | 4mL epicatechin (0.01 N) extract + 4mL 0.1 N Fe(NO3)3 |
| T8 | 5mL tea extract + 2mL 0.1 N Fe(NO3)3 solution |
| C1 | (Control) 2mL Fe(NO3)3 + 10mL NaBH4 |
| C2 | (Control) 1mL Fe(NO3)3 + 10mL NaBH4 |
| C3 | (Control) 1mL Fe(NO3)3 + 5mL NaBH4 |
Nanomaterial characterization
Transmission electron microscopy (TEM) characterization was performed to obtain nanoparticle size and morphology on a Hitachi H-7600 tungsten-tip instrument at an accelerating voltage of 100kV. Nanoparticles were examined after suspension in water and subsequent deposition of 5 μL of solution onto formvar/carbon-coated TEM grids. The AMT software for the digital TEM camera was calibrated for size measurements of the nanoparticles at each magnification level. Information on mean size and standard deviation was calculated from measuring over 100 nanoparticles in random fields of view in addition to images that show general morphology of the nanoparticles. A Panalytical X-pert diffractometer with a copper Kα source was used to identify crystalline phases of the lead precipitates. The tube was operated at 45 kV and 40 mA for the analyses. Scans were performed over a 2-theta ranging from 5 to 70° with a step of 0.02° and a one-second count time at each step. Pattern analysis was performed by following ASTM procedures using the computer software Jade (Versions 8, Materials Data, Inc.), with reference to the 1995-2002 ICDD PDF-2 data files (International Center for Diffraction Data, Newtown Square, PA). UV spectra were recorded using a Varian UV-visible spectrometer (Model Cary 50 Conc).HaCaT cells were donated generously by Dr. James F. Dillman III, of the U.S. Army Medical Research Institute of Chemical Defense.28 The HaCaT cells were cultured with RPMI-1640 media with 10% fetal bovine serum and 1% penicillin/streptomycin. The HaCaT line was incubated at 37 °C, 100% humidity, and 5% CO2. During exposure, the HaCaT line was cultured with RPMI-1640 media with 1% penicillin/streptomycin and no fetal bovine serum (HEM).Treatment protocol
Cells were seeded to 60-80% confluency in 96 well plates within a growth period of 24-48 h. Upon reaching 80% confluency, typically 48 h, cells were treated with either 0μg mL−1, 5μg mL−1, 10μg mL−1, 25 μg mL−1, 50μg mL−1 or 100μg mL−1 concentrations of nanoparticles suspended in HEM. After a 24h or 48h exposure, the nanoparticle biocompatibility was assessed using cytotoxicity assays.Cytotoxicity assays
The HaCaT cells were exposed to the NZVI at concentrations of 0μg mL−1, 5μg mL−1, 10μg mL−1, 25μg mL−1, 50μg mL−1 or 100μg mL−1 in HEM. After either 24h or 48h incubation with the nanoparticles, cell proliferation was measured using the CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega), and membrane leakage was evaluated using the CytoTox 96® Non-Radioactive Cytotoxicity Assay (Promega). The mitochondrial function and membrane leakage were then assessed spectrophotometrically with a SpectraMAX Plus 190 microplate reader. The data are represented as an average of three independent trials ± the standard deviation.Statistical analysis
The data are represented as an average of three independent trials ± the standard deviation. The cytotoxicity assays were run and any group with a p value less than 0.05 was considered significant.Results and discussion
Typically, nanomaterials possess special properties - chemical, optical, magnetic, and biological, which make them desirable for commercial or medical applications. However, these same properties potentially may lead to a response in the human body that is different from and is not directly predicted by the constituent chemicals and compounds. Understanding properties of nanomaterials obtained through different routes is still a challenge. In this context, we found that the preparation of NZVI particles of controlled size and transport properties using benign tea polyphenols is very attractive. The reactive lifetime of these relatively short-lived nanoparticles is extended because the tea polyphenols not only reduce the iron salts but cap the ensuing nanoparticles. The reduction potential of caffeine is 0.3 V vs. SCE which is sufficient to reduce metals viz. Fe (reduction potential −0.44 V vs. SCE). The other polyphenols have reduction potential values29 ranging from 0.3 to 0.8 V (see Table 2 for reduction potential values).
The formation of Fe nanoparticles with caffeine/polyphenols took place via the following steps: (1) complexation with Fe salts, (2) simultaneous reduction of Fe, (3) capping with oxidized polyphenols/caffeine. The reduction of Fe was confirmed using UV spectra and is shown in Fig. 1. The blank extract has an absorption beginning at 500 nm which is similar to the control Fe(NO3)3 solutions. The reaction between Fe(NO3)3 and tea extract was instantaneous and the color of the reaction mixture changed from yellow to dark brown as shown in the inset of Fig. 1. After the reaction the UV spectra had broad absorption at a higher wavelength and there was no sharp absorption at lower wavelengths as shown in the controls.
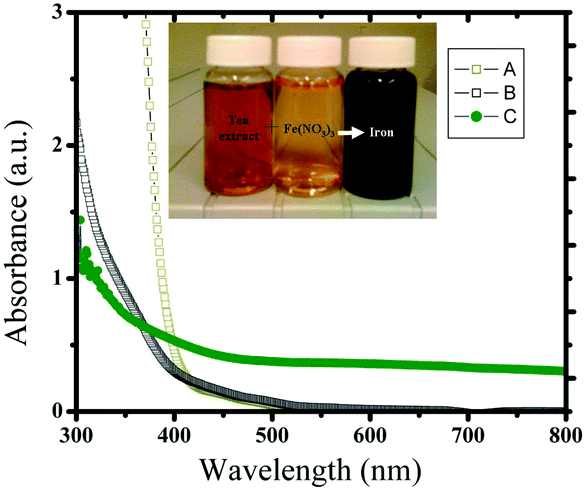 |
| | Fig. 1 UV spectra of (a) Fe(NO3)3 control, (b) control tea extract and (c) the reaction product (Fe nanoparticles) obtained from Fe(NO3)3 and tea extract. The inset shows the photographic image of the tea extract, control Fe(NO3)3 solution and after mixing them (from left to right vials). | |
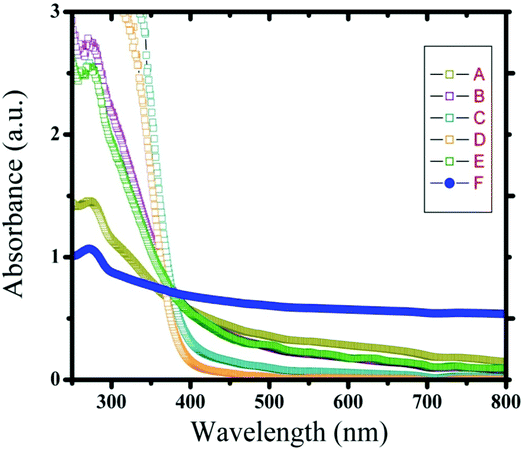
UV spectra of T1, T2, T3, T4, T5 and T8 (descriptive text codes are given in Table 1) are shown in Fig. 2. All the spectra looked similar in the visible region, but displayed slight differences in the UV region. T1 and T8 did not show strong absorption in the UV range when compared with T2 and T5. The spectra of T3 and T5 had sharp absorption in the UV range when compared with all the others plotted. X-ray diffraction experiments were conducted to confirm the phase formation. Fig. 3 shows the XRD patterns of T1, T2, T3, T4 and T8 samples respectively. Except sample T2, patterns (a) and (e) showed a small 100% intensity peak around 2θ 50° (patterns (c) and (d) were amorphous in nature) and could be indexed to hexagonal Fe (JCPDS No. 00-024-0072). The XRD pattern of T2 showed 100% intensity peak at 2θ, 33.115 which can be indexed to the rhombohedraphase of alpha Fe2O3 with plane (104). Fig. 4 shows TEM images of T1, T2, T3 and T4 samples respectively. Sample T1 yielded spherical particles ranging from 40 to 50 nm whereas T2 and T3 yielded ultra small nanoparticles which are difficult to visualize at this magnification. However, sample T4 showed different structures such as platelets and nanorods. We believe concentration of tea extract plays a key role in dictating the final structures and size of the iron nanoparticles. Sample T4 contained minimal amounts of caffeine/polyphenols hence the larger particle size. Caffeine/polyphenols are important for reducing and capping behavior.20 At higher caffeine/polyphenol concentrations in the reaction mixture we observed a decrease in particle size. From samples T5-T8, particle shapes were found to be spherical and larger in size when compared with T1 to T4 (Fig. 5). The control samples C1 and C2 formed irregular shapes with size varying from 50 nm (control C1) to nearly 500 nm (control C2) (Fig. 6).The MTS assay of all the tested samples at 24 and 48 h showed no decreases in cell proliferation similar to control samples (see Fig. 7 & 8). LDH leakage results after 24h didn't show any discernible difference from control samples except in samples T1, T2, T3, and T4, but after 48h results were significantly higher when compared with control samples indicating an increase in membrane leakage in HaCaT cells treated with NZVI (see Fig. 9 & 10). T1, T2, T3 and T4 showed increases in LDH leakage when the particle concentrations reached 50μg mL−1 and above. The increases in LDH leakage are likely due to the size of the particles created. Increases in LDH are due to stress on the cellular membrane, which could be caused by the larger particle size in this case. Larger particles are not able to enter the cells, but they apply pressure on the cellular membrane while trying to enter leading to cellular membrane stress. As the particles are created in the smaller nano-sizes the membrane leakage will likely decrease. Also we saw higher LDH leakage with higher concentrations of particles. The increase in LDH leakage with increasing particle concentration is due to the cell membrane being stressed by so many particles pushing on the surface and is therefore a result of concentration of the particles rather than the type of particles used.
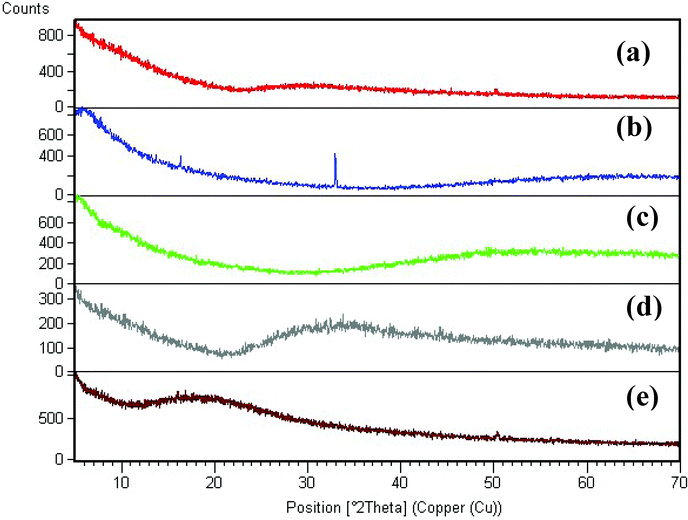 |
| | Fig. 3 XRD pattern of iron nanoparticles obtained using different ratios of tea extract and polyphenols; (a) T1, (b) T2, (c) T3, (d) T4 and (e) T8 samples. | |
 |
| | Fig. 4 TEM images of (a) T1, (b) T2, (c) T3 and (d) T4 samples. | |
 |
| | Fig. 5 TEM images of (a) T5, (b) T6, (c) T7 and (d) T8 samples. | |
 |
| | Fig. 6 TEM images of (a) C1 and (b) C2 control samples. | |
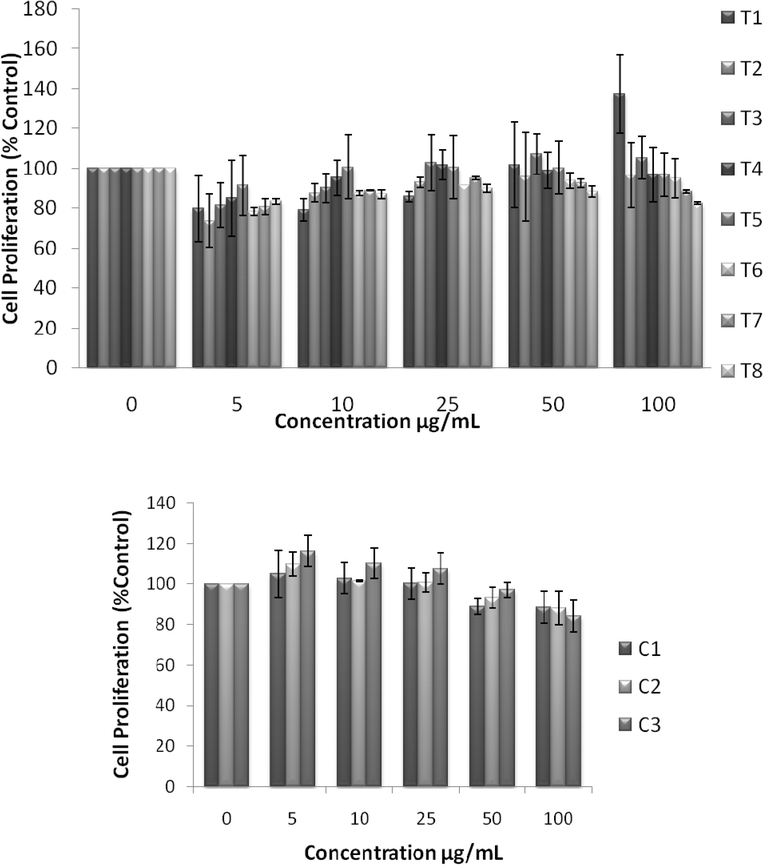 |
| | Fig. 7 Effect of NZVI on mitochondrial function after 24h exposure. Mitochondrial function was determined by the MTS assay as described in the Experimental procedure section. Each experimental point is a composite of three independent experiments, with n≥ 3 for each point. Top: MTS showing cell proliferation after 24h exposure to various concentrations of NZVI. The figure shows no significant reductions in cell proliferation, but instead increased cell proliferation after 24h. Bottom: MTS showing changes in cell proliferation after 24h exposure to various concentrations of control particles made with sodium borohydride. These particles produced no significant decrease in cell proliferation after 24h. | |
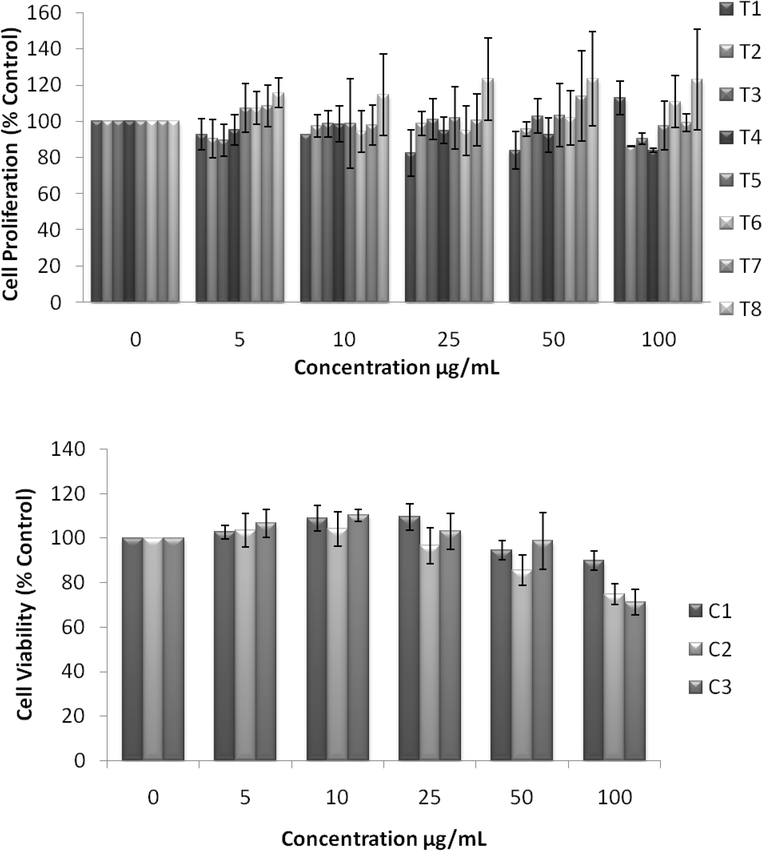 |
| | Fig. 8 Effect of NZVI on mitochondrial function after 48h exposure. Mitochondrial function was determined by the MTS assay as described in the Experimental procedure section. Each experimental point is a composite of three independent experiments, with n≥ 3 for each point. Top: MTS showing cell proliferation after 48h exposure to various concentrations of NZVI. The figure shows no significant reductions in cell proliferation, but instead increased cell proliferation after 48h. Bottom: MTS showing changes in cell proliferation after a 48h exposure to various concentrations of control particles made with sodium borohydride. These particles produced no significant decrease in cell proliferation after 48h. | |
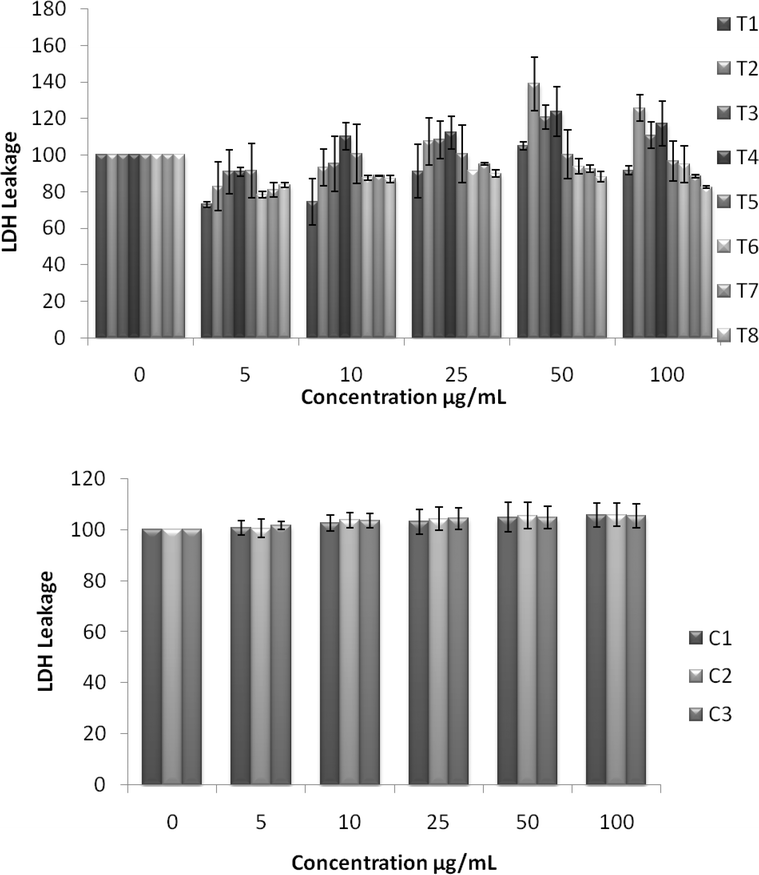 |
| | Fig. 9 Effect of NZVI on cellular membrane leakage after 24h exposure. Cellular membrane leakage was determined by the lactate dehydrogenase (LDH) assay as described in the Experimental procedure section. Each experimental point is a composite of three independent experiments, with n≥ 3 for each point. Top: LDH leakage after 24h exposure to various concentrations of NZVI. Bottom: LDH leakage after a 24h exposure to various concentrations of control particles made with sodium borohydride. | |
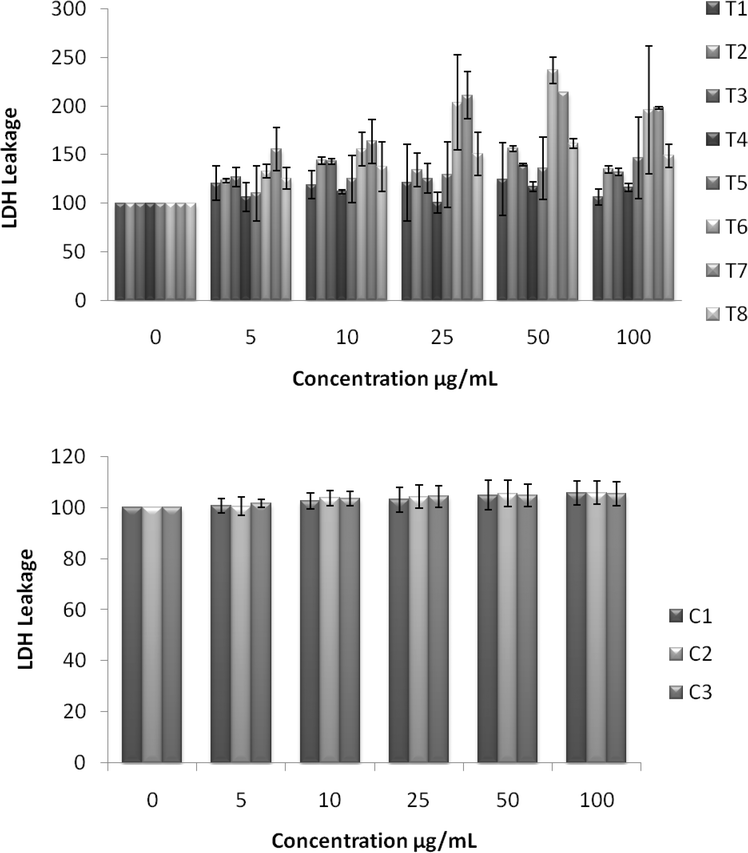 |
| | Fig. 10 Effect of NZVI on cellular membrane leakage after 48h exposure. Cellular membrane leakage was determined by the lactate dehydrogenase (LDH) assay as described in the Experimental procedure section. Each experimental point is a composite of three independent experiments, with n≥ 3 for each point. Top: LDH leakage after 48h exposure to various concentrations of NZVI. Bottom: LDH leakage after 48h exposure to various concentrations of control particles made with sodium borohydride. | |
Conclusions
A “green” one-step approach has been developed for the synthesis of nanoscale zerovalent iron particles at room temperature. The method does not use a typical noxious chemical reducing agent or a surfactant and can be scaled up to bulk level. The “green” synthesized NZVI samples were assessed with MTS and LDH toxicological assays and some of them were found to be nontoxic when compared with control samples prepared using the conventional borohydride reduction protocols. This simple method offers spherical zerovalent iron nanoparticles with unique size and shape when compared with control samples. We anticipate the development of various technological and environmental remediation applications for these stable particles including dechlorination of dibenzo-P-dioxins, reduction of chlorinated ethanes, arsenic and hexavalent chromium removal to name a few.
References
- K.-C. Huang and S. H. Ehrman, Langmuir, 2007, 23, 1419 CrossRef CAS.
- Q. Huang, X. Shi, R. A. Pinto, E. J. Petersen and W. J. Weber Jr., Environ. Sci. Technol., 2008, 42, 8884 CrossRef CAS.
- L. Guo, Q.-J. Huang, X.-Y. Li and S. Yang, Langmuir, 2006, 22, 7867 CrossRef CAS.
- S. R. Kanel, R. R. Goswami, T. P. Clement, M. O. Barnett and D. Zhao, Environ. Sci. Technol., 2008, 42, 896 CrossRef CAS.
- F. He and D. Zhao, Environ. Sci. Technol., 2007, 41, 6216 CrossRef CAS.
- L. B. Hoch, E. J. Mack, B. W. Hydutsky, J. M. Hershman, J. M. Skluzacek and T. E. Mallouk, Environ. Sci. Technol., 2008, 42, 2600 CrossRef CAS.
- Y. Liu and G. V. Lowry, Environ. Sci. Technol., 2006, 40, 6085 CrossRef CAS.
- S. R. Kanel, J.-M. Grenche and H. Choi, Environ. Sci. Technol., 2006, 40, 2045 CrossRef CAS.
- Y. Liu, T. Phenrat and G. V. Lowry, Environ. Sci. Technol., 2007, 41, 7881 CrossRef CAS.
- A. B. M. Giasuddin, S. R. Kanel and H. Choi, Environ. Sci. Technol., 2007, 41, 2022 CrossRef CAS.
- H. Song and E. R. Carraway, Environ. Sci. Technol., 2005, 39(16), 6237 CrossRef CAS.
- J.-H. Kim, P. G. Tratnyek and Y.-S. Chang, Environ. Sci. Technol., 2008, 42, 4106 CrossRef CAS.
- V. Sarathy, P. G. Tratnyek, J. T. Nurmi, D. R. Baer, J. E. Amonette, C. L. Chun, R. L. Penn and E. J. Reardon, J. Phys. Chem. C, 2008, 112, 2286 CrossRef CAS.
- M. N. Nadagouda and R. S. Varma, Green Chem., 2006, 8, 516 RSC.
- P. Raveendran, J. Fu and S. L. Wallen, J. Am. Chem. Soc., 2003, 125, 13940 CrossRef CAS.
- M. N. Nadagouda and R. S. Varma, Green Chem., 2007, 9, 632 RSC.
- R. R. Naik, S. J. Stringer, G. Agarwal, S. E. Jones and M. O. Stone, Nat. Mater., 2002, 1, 169 CrossRef CAS.
- M. N. Nadagouda and R. S. Varma, J. Nanomaterials, 2008 DOI:1155/2008/782358.
- P. Mukherjee, A. Ahmad, D. Mandal, S. Senapati, S. R. Sainkar, M. I. Khan, R. Parishcha, P. V. Ajaykumar, M. Alam, R. Kumar and M. Sastry, Nano Lett., 2001, 1, 515 CrossRef CAS.
- M. N. Nadagouda and R. S. Varma, Green Chem., 2008, 10, 859 RSC.
- P. T. Anastas, J. C. Warner, Green, Chemistry: Theory and Practice, Oxford University Press, Inc.New York, 1998 Search PubMed.
- M. N. Nadagouda and R. S. Varma, Cryst. Growth Des., 2008, 8, 291 CrossRef CAS.
- M. N. Nadagouda and R. S. Varma, Cryst. Growth Des., 2007, 4, 686.
- M. N. Nadagouda and R. S. Varma, Biomacromolecules, 2007, 8, 2762 CrossRef CAS.
- M. N. Nadagouda and R. S. Varma, Macromol. Rapid Commun., 2007, 28, 465 CrossRef CAS.
- M. N. Nadagouda, V. Polshettiwar and R. S. Varma, J. Mater. Chem., 2009, 19, 2026 RSC.
- V. Polshettiwar, B. Baruwati and R. S. Varma, Green Chem., 2009, 11, 127 RSC.
- P. Boukamp, R. T. Petrussevska, D. Breitkreutz, J. Hornung, A. Markham and N. E. Fusenig, J. Cell Biol., 1988, 106, 761 CrossRef CAS.
- Polyphenols, Wine and Health Edited by C. Chèze, J. Vercauteren and R. Verpoorte, France, 14–16 April 1999 (Proceedings of the Phytochemical Society of Europe, Volume 48) Springer, Dordrecht, The Netherlands, 2001 Search PubMed.
Footnote |
| † Present address: Pegasus Technical Services, 46 E Hollister Street, Cincinnati, Ohio 45219. |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.