A highly efficient Cu/La2O3 catalyst for transfer dehydrogenation of primary aliphatic alcohols
Ruijuan Shi, Fei Wang, Tana, Yong Li, Xiumin Huang and Wenjie Shen*
State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: shen98@dicp.ac.cn; Fax: +86-411-84694447; Tel: +86-411-84379085
First published on 27th October 2009
Abstract
La2O3-supported copper nanoparticles are sufficiently active for transfer dehydrogenation of primary aliphatic alcohols to aldehydes. When used for 1-octanol, the yield of 1-octanal can be as high as 63%. The catalyst is also highly effective for a wide variety of substrates, such as aromatic, aliphatic, and cyclic alcohols. The yield of the desired products approaches 100% and the turnover frequency is 86.5 h−1. The synergistic effect between the basicity of La2O3 and the hydrogen spillover of Cu particle contributes significantly to the extremely high activity of the Cu/La2O3 catalyst for transfer dehydrogenation of primary aliphatic alcohols.
Introduction
Catalytic transformation of alcohols to aldehydes or ketones is perhaps the most demanding reaction in organic and green chemistry.1 Selective oxidation of alcohols using molecular oxygen and/or hydrogen peroxide as green oxidants has been used to replace the conventional oxidation process using stoichiometric quantities of inorganic oxidants (typically, KMnO4 and K2Cr2O7) which produce enormous amounts of toxic waste.2,3 In this field, supported Au, Pd, Pt, and Ru nanoparticles exhibit very high activity for the oxidation of allylic and benzylic alcohols to aldehydes or ketones using molecular oxygen, but they are poorly active for the oxidation of primary aliphatic alcohols.3–6 Only Ru-based catalysts afford desirable performance in the oxidation of primary aliphatic alcohols, with the yield of aldehydes in the range 70–90%.6–10 However, the oxidation of alcohols using molecular oxygen often causes safety problems linked with the use of flammable organic solvents, particularly in large-scale production processes.4–6Transfer dehydrogenation of alcohols has been recently proposed to be a green and efficient reaction route, in which the alcohol is directly dehydrogenated to aldehyde or ketone and the readily available unsaturated organic molecule (as a hydrogen acceptor) removes the hydrogen.6 Unfortunately, only a few heterogeneous catalysts are known to be effective for this novel process, and they are active only for secondary and allylic/aromatic alcohols.11–18 Hayashi and co-workers11,12 reported that Pd/C catalyzed the dehydrogenation of allylic and aromatic alcohols with moderate yields (75–86%, 323 K, 2-4 days) using ethylene or nitrobenzene as hydrogen acceptor. Baiker and co-workers13 reported that transfer dehydrogenation of aromatic alcohols occurred over a Pd/Al2O3 catalyst using cyclohexene as hydrogen acceptor, but this catalyst was nearly inactive for aliphatic and alicyclic alcohols, and the yield of 1-octanal was only 1% in the case of 1-octanol. Ravasio and co-workers14,15 revealed that a Cu/Al2O3 catalyst was highly active for the transfer dehydrogenation of allylic and alicyclic alcohols, but it was less active for 1-octanol, the yield of 1-octanal being only 4%. Over a Ru/AlO(OH) catalyst at 383 K for 24 h under argon atmosphere, 1-octanol was converted into a complex mixture containing 23% of 1-octanal.16 More recently, Kaneda and co-workers17,18 found that Ag and Cu nanoparticles supported on a basic hydrotalcite (HT) showed excellent catalytic performance for acceptor-free dehydrogenation of benzylic and secondary aliphatic alcohols. However, they were not effective for primary aliphatic alcohols, and the conversion of 1-octanol was only 17%, although the selectivity of 1-octanal was 99% on the Ag/HT catalyst.17
Clearly, the majority of known catalysts are still poorly active for the dehydrogenation of primary aliphatic alcohols. Here we report that a Cu/La2O3 catalyst affords fairly high activity for the transfer dehydrogenation of primary aliphatic alcohols through a synergistic effect between the basicity of the support and the fast hydrogen spillover of Cu nanoparticles.
Experimental
Catalyst preparation
The Cu/La2O3 catalyst was prepared by a deposition–precipitation method. 0.55 g copper acetate monohydrate (Cu(CH3COO)2·H2O) was dissolved in 200 ml water, and 1.8 g commercially available La2O3 powder was then dispersed into the solution with stirring at room temperature. Subsequently, 40 ml sodium carbonate aqueous solution (0.1 M) was added dropwise to the mixture to achieve a final pH value of 8. The precipitate was further aged in the mother liquid for 2 h. After being filtered and washed thoroughly with water, the resultant solid was dried at 383 K overnight and calcined at 723 K for 4 h in air, giving the CuO/La2O3 precursor. 200 mg CuO/La2O3 sample was then loaded in a fixed-bed reactor and reduced with pure hydrogen (40 ml min−1) at 573 K for 1 h, yielding the Cu/La2O3 catalyst.Catalyst characterization
The actual loading of copper was measured by inductively coupled plasma atomic emission spectroscopy (ICP-AES) on a Plasma-Spec-II instrument. 20 mg CuO/La2O3 was dissolved in concentrated hydrochloric acid and the mixture was diluted with water to meet the detection range of the instrument.The N2 adsorption–desorption isotherms were performed using a Micrometrics ASAP 2010 instrument at 77 K. Before the measurement, the sample was degassed at 573 K for 3 h. The surface area was calculated from a multipoint BET analysis of the adsorption isotherm.
The X-ray powder diffraction (XRD) patterns were recorded on a D/MAX 2500/PC powder diffractometer (Rigaku) operated at 40 kV and 200 mA, using Cu-Kα (1.5418 Å) radiation. In situ XRD measurement for the reduction of the CuO/La2O3 sample was performed in a high-temperature chamber installed in the diffractometer. The CuO/La2O3 sample was pressed into a flake and mounted in the chamber. After heating to 573 K in the flow of He, pure hydrogen was introduced into the chamber and kept at 573 K for 1 h and the XRD patterns were then recorded.
Transmission electron microscopy (TEM) images were recorded on a FEI Tecnai G2 Spirit microscope operated at 120 kV. The specimen was prepared by ultrasonically dispersing the sample powder in ethanol and drops of the suspension were deposited on a clean carbon-coated copper grid and dried in air.
Temperature-programmed reduction (H2-TPR) was conducted in a U-shape quartz reactor connected to a mass spectrometry (OmniStar 200). 100 mg CuO/La2O3 sample was heated to 673 K at a rate of 10 K min−1 under He flow (50 ml min−1) and maintained at this temperature for 1 h. After being cooled to room temperature, a 20 vol.% H2/He mixture (30 ml min−1) was introduced and the sample was heated to 723 K at a rate of 10 K min−1 and kept at this temperature for 1 h.
The dispersion of copper in the Cu/La2O3 catalyst was determined by N2O chemisorption.19 50 mg of the CuO/La2O3 sample was loaded in a U-shape quartz reactor and treated with a 20 vol.% O2/N2 mixture (30 ml min−1) at 673 K (10 K min−1) for 0.5 h. After being cooled to room temperature, a 5 vol.% H2/N2 mixture (30 ml min−1) was introduced and the sample was heated to 673 K at a rate of 5 K min−1. Then, the sample was cooled to 363 K under He flow and further purged for 30 min. Subsequently, the sample was exposed to a 10 vol.% N2O/He mixture for 30 min at 363 K and then cooled to room temperature under a He flow. A 5 vol.% H2/N2 mixture (30 ml min−1) was introduced and the sample was heated to 573 K at a rate of 5 K min−1. The dispersion of copper and the average size of copper particles were calculated by assuming 1.46 × 1019 copper atoms per m2 and a N2O/Cu ratio of 1:2.
The IR spectra of CO adsorption were recorded on a Bruker vector-22 FTIR spectrometer. The sample powder was pressed into a self-supporting wafer (20 mg) and mounted into the IR cell. The sample was first reduced with pure H2 (40 ml min−1) at 573 K for 1 h and then evacuated at 673 K (2 × 10−2 Pa) for 5 h. After acquisition of the background spectrum, the sample was exposed to CO with increasing pressure (5–500 Pa) at room temperature. Then the cell was evacuated and heated from room temperature to 373 K. The spectra of CO adsorption on the Cu/La2O3 catalyst were obtained by subtracting the background spectrum.
The temperature-programmed desorption of CO2 (CO2-TPD) was conducted in a U-shape quartz reactor connected to a mass spectrometry (OmniStar 200). 100 mg CuO/La2O3 sample was heated at a rate of 10 K min−1 to 723 K under He flow (30 ml min−1) and maintained at this temperature for 1 h in order to remove the surface impurities. Then the sample was reduced with pure H2 at 573 K for 1 h. After being cooled to room temperature under He flow, the sample was exposed to a mixture of 30 vol.% CO2/He (20 ml min−1) for 0.5 h. Subsequently, the sample was purged with He (50 ml min−1) for 2 h and then heated to 1073 K at a rate of 10 K min−1. Desorption of CO2 was monitored by the mass spectrometry.
The X-ray photoelectron spectroscopy (XPS) and Auger electron spectroscopy (AES) were performed on an ESCALAB MK-II X-ray photoelectron spectrometer (VG Scientific Ltd, UK). The CuO/La2O3 sample was pressed into a thin disc and mounted on a sample rod placed in a pretreatment chamber, in which the sample was reduced by pure H2 at 573 K for 1 h. Then the sample was transferred into the analysis chamber where the spectra of Cu 2p, Cu L3VV levels were recorded. The charging effect was corrected by adjusting the binding energy of C 1 s to 284.6 eV.
Catalytic test
Transfer dehydrogenation of alcohols was conducted with a stainless steel autoclave (50 ml). Typically, the reaction mixture contained 2 mmol alcohol, 4 mmol styrene (hydrogen acceptor), 200 mg Cu/La2O3 catalyst and 8 ml mesitylene. The reaction was performed at 363–423 K for a certain period (0.5–24 h) under N2 atmosphere. The yield of products was analyzed by Agilent GC-7890T (HP-5 capillary column) using decane as an internal standard. The turnover frequency (TOF) was defined as the moles of product formed per mole of Cu per hour, and the dispersion of copper was determined by N2O chemisorption measurement.Results and discussion
Fig. 1 shows the XRD patterns of the samples. Bare La2O3 showed typical diffraction peaks of hexagonal structure (JCPDS 05-0602) and the average crystallite size was 94 nm. The Cu/La2O3 catalyst also exhibited the diffraction peaks of hexagonal La2O3, but lanthanum carbonate (La2O2CO3, JCPDS 37-0804) and oxyhydroxide (LaOOH, CPDS 19-0656) phases were also detected. This is caused by the interaction of lanthanum oxide with water and carbon oxide during copper loading and calcination process.20 The absence of diffraction lines of Cu species indicates that they were highly dispersed on the surface of La2O3, or they were too small to be detected. The Cu/La2O3 catalyst had a surface area of 22 m2 g−1, and the copper loading was 8.4 wt%.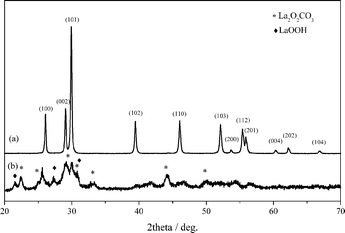 | ||
| Fig. 1 XRD patterns of La2O3 (a) and Cu/La2O3 (b). | ||
Fig. 2 shows the H2-TPR profiles of bulk CuO and CuO/La2O3. The reduction of bulk CuO occurred at 539 K, whereas the reduction of CuO in the CuO/La2O3 sample shifted to a much lower temperature region – the main reduction took place at 432 K with a small shoulder at 415 K. The minor reaction peak was ascribed to the reduction of finely dispersed CuO species, while the major peak corresponded to the reduction of CuO species strongly interacting with La2O3. The H2 consumption for the CuO/La2O3 sample was estimated as 1.25 mmol g−1, which is very close to the stoichiometric value (1.32 mmol g−1) for the total reduction of CuO species, indicating the sole presence of metallic copper in the Cu/La2O3 catalyst.
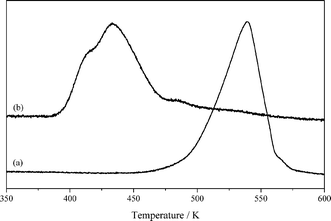 | ||
| Fig. 2 H2-TPR profiles of CuO (a) and CuO/La2O3 (b). | ||
Fig. 3 shows the TEM images of the Cu/La2O3 catalyst. The La2O3 support was composed of spherical nanoparticles with an average size of about 30 nm. The Cu particles could not be clearly observed, probably due to the small size and the poor contrast between Cu and La2O3. However, the N2O chemisorption measurement revealed that the mean size of copper particle was about 6 nm, giving a copper dispersion of about 17%.
 | ||
| Fig. 3 TEM images of the Cu/La2O3 catalyst. | ||
Fig. 4 shows the FTIR spectra of CO absorption on the Cu/La2O3 catalyst. Generally, the band of CO absorption on Cu2+ site is above 2140 cm−1; weakly bound CO on Cu0 shows a clear stretching frequency below 2110 cm−1; while CO adsorption on Cu+ is located between 2110 and 2135 cm−1.21 The Cu0–CO band is easily decomposed at 298–373 K, but the Cu+–CO band is relatively stable and can be present at 373 K.22 CO adsorption at room temperature on the Cu/La2O3 catalyst showed a band at 2098 cm−1, indicating the presence of a Cu0–CO band. This is similar to the previous observation of CO adsorption on Cu0 at 2100 cm−1 on a Cu/TiO2 catalyst.23 As the pressure of CO increased, the band slightly shifted to 2102 cm−1 and the intensity was enhanced significantly. Upon heating, the Cu0–CO band weakened, and it totally disappeared at 373 K. This further confirms that the copper species are present as Cu0 in the Cu/La2O3 catalyst.
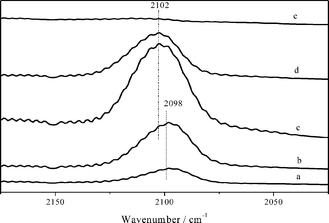 | ||
| Fig. 4 FTIR spectra of CO adsorbed on the Cu/La2O3 catalyst. CO pressure of 5 Pa (a), 20 Pa (b), and 100 Pa (c), followed by evacuation at 323 K (d) and 373 K (e). | ||
Fig. 5 shows the X-ray photoelectron spectra of Cu 2p and Cu L3VV Auger in the Cu/La2O3 catalyst. Obviously, the disappearance of the satellite peak and the binding energy of Cu 2p3/2 at 932.0 eV shows that the copper exists as Cu0 or Cu+ (Fig. 5A). The Cu0 and Cu+ species cannot be distinguished by the binding energy of Cu 2p3/2 because they are almost identical, but they can be easily distinguished from the Cu L3VV Auger lines, where Cu0 and Cu+ are separated by 2.0 eV.24 The kinetic energy of Cu L3VV at 918.3 eV confirmed that Cu0 is the main species in the Cu/La2O3 catalyst.25 These findings are in line with the H2-TPR and FTIR results, which showed the sole presence of metallic Cu after hydrogen reduction at 573 K.
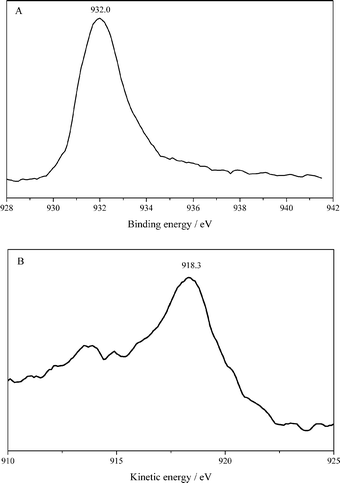 | ||
| Fig. 5 XPS spectra of Cu 2p (A) and Cu L3VV Auger (B) in the Cu/La2O3 catalyst. | ||
Fig. 6 shows the CO2-TPD profile of the Cu/La2O3 catalyst. Desorption of CO2 occurred at 354, 390 and 501 K, and the relative areas were 21, 41 and 38%, respectively. This indicates that there are three types of adsorption sites for CO2 with different basicity strengths. Previous CO2-TPD26,27 and FTIR26–28 studies have readily revealed that the hydroxy groups on the surface of La2O3 have a weakly basic character; CO2 adsorption on the medium strength basic sites (bidentate carbonates) gave CO2 desorption at intermediate temperature, and CO2 adsorption on the strong strength basic sites (unidentate carbonates) desorbed at high temperature. Therefore, the desorption peak at 354 K is ascribed to the weak basic sites (OH groups), while the desorption peak at 390 K and 501 K represents the moderately basic (La–O2− pairs) and strongly basic (O2− ions) sites on the Cu/La2O3 catalyst, respectively.
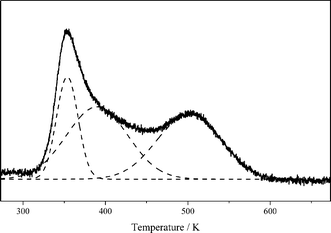 | ||
| Fig. 6 CO2-TPD profile of the Cu/La2O3 catalyst. | ||
Table 1 summarizes the reaction results of transfer dehydrogenation of alcohols on the Cu/La2O3 catalyst. 1-Octanol, which is a representative of aliphatic primary alcohols, was readily and selectively converted into 1-octanal with a yield of about 10% even at 363 K (entry 1). The yield of 1-octanal rapidly increased to 35% at 403 K for 10 h and 63% at 423 K for 2 h (entries 2 and 3). The TOF increased rapidly with temperature, and it reached 13.7 h−1 at 423 K, clearly demonstrating the high activity of the Cu/La2O3 catalyst. Here, the TOF is as high as 0.2 h−1 at 363 K, which is almost 20 times larger than that obtained over a Cu/Al2O3 catalyst (TOF = 0.01 h−1) at the same reaction temperature.14,15 The TOF of 1.5 h−1 at 403 K is about 10 times larger than that obtained over the most active Ru/AlO(OH) (TOF = 0.16 h−1)16 and Cu/HT (TOF = 0.1 h−1 for heptanol)18 catalysts. Moreover, the Cu/La2O3 catalyst was also highly active for the dehydrogenation of other aliphatic primary alcohols. 1-Hexanol and 1-decanol were selectively converted to 1-hexanal (yield 48%) and 1-decanal (yield 45%) at 423 K (entries 4 and 5). This extremely high activity of the Cu/La2O3 catalyst may rely on the active Cu particles and the basic nature of La2O3, which favors the effective and selective dehydrogenation of alcohols.
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Product | T/K | t/h | Conv./% | Sel./% | TOF/h−1 |
| a Reaction conditions: alcohol (2 mmol), styrene (4 mmol), Cu/La2O3 (200 mg), mesitylene (8 ml), N2 atmosphere. | |||||||
| 1 | 1-Octanol | 1-Octanal | 363 | 24 | 12 | 97 | 0.2 |
| 2 | 1-Octanol | 1-Octanal | 403 | 10 | 35 | 100 | 1.5 |
| 3 | 1-Octanol | 1-Octanal | 423 | 2 | 63 | 100 | 13.7 |
| 4 | 1-Hexanol | 1-Hexanal | 423 | 8 | 48 | 100 | 2.6 |
| 5 | 1-Decanol | 1-Decanal | 423 | 10 | 45 | 100 | 2.0 |
| 6 | Benzyl alcohol | Benzaldehyde | 423 | 3 | 99 | 100 | 14.3 |
| 7 | 1-Phenylethanol | Acetophenone | 423 | 0.5 | 99.5 | 100 | 86.5 |
| 8 | 2-Octanol | 2-Octanone | 423 | 1 | 99.5 | 100 | 43.3 |
| 9 | Cyclohexanol | Cyclohexanone | 423 | 1 | 77 | 100 | 33.5 |
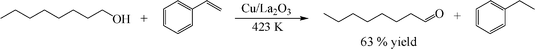
This methodology can also be applied to the conversion of benzylic and secondary aliphatic alcohols. As expected, the Cu/La2O3 catalyst was quite active for aromatic alcohols. Benzyl alcohol and 1-phenylethanol were converted to the corresponding aldehydes and ketones quantitatively and the TOFs were much higher than those for primary aliphatic alcohols (entries 6 and 7). When used for benzyl alcohol, 99% yield of benzaldehyde was obtained within 3 h with a TOF of 14.3 h−1. Previously, the reaction of benzyl alcohol using of Pd/C–ethylene system only gave a 43% yield of benzyl aldehyde in 4 days at 323 K,11 and the Pd/Al2O3–cyclohexene system gave an 18% yield of benzyl aldehyde within 15 h at 353 K.13 Clearly, the reaction rate of Cu/La2O3 catalyst is at least 5 times larger than the previous catalysts. In the case of 1-phenylethanol, the yield of acetophenone was 99.5% and the TOF reached 86.5 h−1 (entry 7). This value is also much higher than those reported for Cu catalysts with or without a hydrogen acceptor: Cu/Al2O3 (TOF = 8.7 h−1)14,15 and Cu/HT (TOF = 4.4 h−1).18 In addition, the Cu/La2O3 catalyst was also highly effective for the dehydrogenation of aliphatic secondary alcohols. 2-Octanol was quantitatively converted to 2-octanone within 1 h (TOF = 43.3 h−1, entry 8). However, this reaction took 15 h at 383 K with a Ru/AlO(OH) catalyst (TOF = 1.5 h−1),16 4 h at 353 K with a Cu/Al2O3 catalyst (TOF = 1.5 h−1),14,15 and 6 h at 403 K with a Cu/HT catalyst (TOF = 2.2 h−1).18 More importantly, cyclohexanol, which is poorly reactive over noble metals (Pd and Ru) even under aerobic conditions,5,6 was converted to cyclohexanone with 77% yield within 1 h (TOF = 33.5 h−1, entry 9). In contrast, the Pd/Al2O3–cyclohexene system only gave 1% yield of cyclohexanone within 5 h at 353 K,13 and it took 12 h at 353 K over a Ru/AlO(OH) catalyst to achieve total conversion to cyclohexanone.16
It is well known that the acid–base properties of the catalyst play a central role in alcohol dehydrogenation.27,29 In order to facilitate the dehydrogenation of alcohols, organic or inorganic bases are often added to the reaction media as co-catalysts. In particular, strong bases like KOH or NaOH or sodium alkoxides are frequently used as promoters in H-transfer reaction of alcohols for promoting the reaction rates. Meanwhile, the basic component also inhibits over-oxidation of primary alcohols to carboxylic acids under aerobic conditions, improving the selectivity. This can be alternatively achieved by using basic solid oxides, such as MgO, Mg–Al, and Cu–Mg–Al hydrotalcite.27,30 Pillai et al.31 has reported that a palladium catalyst supported on hydrotalcite (a basic MgO-like material) showed comparable performance to that of Pd/MgO, whereas Pd catalysts supported on acidic materials (silica, alumina and zeolite-β) were not active for alcohol oxidation. Probably, the La2O3 support here acts as a strong base with a large number of basic sites,32 which favors proton abstraction from the substrate alcohol under anaerobic conditions.
However, a blank test with La2O3 alone only gave 1.5% conversion of 1-octanol, implying that La2O3 favors the dehydrogenation of alcohol but the finely dispersed Cu particles are also needed to transfer hydrogen species. A mechanistic study on a Cu/Al2O3 catalyst for alcohol dehydrogenation revealed that the rate-determining step is the formation of a carbocation-type intermediate by abstracting the OH group in the substrate alcohol.15 Because the primary carbocation is highly unstable and the removal of the second hydrogen atom from the α carbon normally does not occur, the primary aliphatic alcohols are almost inactive over the Cu/Al2O3 catalyst. Concerning the current Cu/La2O3 catalyst, however, the reaction might follow an alternative route. La2O3 provides available basic sites for alcohol adsorption and hydrogen abstraction from the substrate alcohol. It is likely that the basic sites of La2O3 can display nucleophilic reactivity and abstract a proton from the alcohol to form a reactive negatively charged intermediate (alkoxide). Simultaneously, the Cu particles catalyze the hydrogenation of styrene based on the fast hydrogen spillover and the facile release of hydrogen from its surface. Unlike noble metals, which tend to adsorb hydrogen strongly with the formation of stable metal hydride species,13,17 Cu adsorbs hydrogen only weakly and thus favors a fast spillover.
Therefore, it seems reasonable that transfer dehydrogenation of alcohols occurs at the Cu–La2O3 boundaries, following a bifunctional process (Scheme 1). The substrate alcohol dehydrogenates to alkoxide on the basic sites of La2O3, and then the dissociated hydrogen atom migrates to the nearby Cu. Subsequently, the extraction of α-hydrogen from the alkoxide intermediate occurs on a Cu particle or Cu–La2O3 interface, forming the aldehyde or ketone. Finally, hydrogenation of styrene takes place on Cu, yielding ethylbenzene. In summary, the basic sites of La2O3 are mainly responsible for proton abstraction from alcohol, while the Cu particles promote the hydrogenation of styrene.
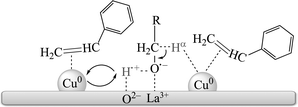 | ||
| Scheme 1 A possible reaction pathway for transfer dehydrogenation of alcohols on the Cu/La2O3 catalyst. | ||
Conclusions
Basic La2O3-supported Cu nanoparticles are highly effective for the transfer dehydrogenation of various alcohols, especially aliphatic primary alcohols. When used for 1-octanol, the yield of 1-octanal was as high as 63%. This unique performance is attributed to the synergistic effect between the basicity of La2O3 and the efficient spillover effect of Cu particles. The basic sites of La2O3 abstract protons from the substrate alcohol, while the Cu particles accelerate the extraction of α-hydrogen from the intermediate and simultaneously catalyze the hydrogenation of styrene.References
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
- R. A. Sheldon, I. W. C. E. Arends and A. Dijksman, Catal. Today, 2000, 57, 157–166 CrossRef CAS.
- B. Z. Zhan and A. Thompson, Tetrahedron, 2004, 60, 2917–2935 CrossRef CAS.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS.
- T. Matsumoto, M. Ueno, N. Wang and S. Kobayashi, Chem. Asian J., 2008, 3, 196–214 CrossRef CAS.
- R. Anderson, K. Griffin, P. Johnston and P. L. Alsters, Adv. Synth. Catal., 2003, 345, 517–523 CrossRef CAS.
- B. Z. Zhan, M. A. White, T. K. Sham, J. A. Pincock, R. J. Doucet, K. V. R. Rao, K. N. Robertson and T. S. Cameron, J. Am. Chem. Soc., 2003, 125, 2195–2199 CrossRef CAS.
- K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2000, 122, 7144–7145 CrossRef CAS.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538–4542 CrossRef CAS.
- M. Hayashi, K. Yamada, S. Nakayama, H. Hayashi and S. Yamazaki, Green Chem., 2000, 2, 257–260 RSC.
- T. Tanaka, H. Kawabata and M. Hayashi, Tetrahedron Lett., 2005, 46, 4989–4991 CrossRef CAS.
- C. Keresszegi, T. Mallat and A. Baiker, New J. Chem., 2001, 25, 1163–1167 RSC.
- F. Zaccheria, N. Ravasio, R. Psaro and A. Fusi, Chem. Commun., 2005, 253–255 RSC.
- F. Zaccheria, N. Ravasio, R. Psaro and A. Fusi, Chem. Eur. J, 2006, 12, 6426–6431 CrossRef CAS.
- W.-H. Kim, I. S. Park and J. Park, Org. Lett., 2006, 8, 2543–2545 CrossRef CAS.
- T. Mitsudome, Y. Mikami, H. Funai, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem. Int. Ed., 2008, 47, 138–141 CrossRef CAS.
- T. Mitsudome, Y. Mikami, K. Ebata, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2008, 4804–4806 RSC.
- S. Sato, R. Takahashi, T. Sodesawa, K. Yuma and Y. Obata, J. Catal., 2000, 196, 195–199 CrossRef CAS.
- B. Klingenberg and M. A. Vannice, Chem. Mater., 1996, 8, 2755–2768 CrossRef CAS.
- A. A. Davydov, Infrared Spectroscopy of Adsorbed Species on the Surface of Transition Metal Oxides, New York, 1990 Search PubMed.
- V. Z. Fridman and A. A. Davydov, J. Catal., 2000, 195, 20–30 CrossRef CAS.
- F. Boccuzzi, S. Coluccia, G. Martra and N. Ravasio, J. Catal., 1999, 184, 316–326 CrossRef CAS.
- G. Moretti, G. Fierro, M. L. Jacono and P. Porta, Surf. Interface Anal., 1989, 14, 325–336.
- M. Vijayaraj and C. S. Gopinath, J. Catal., 2006, 241, 83–95 CrossRef CAS.
- S. C. Shen, X. Chen and S. Kawi, Langmuir, 2004, 20, 9130–9137 CrossRef CAS.
- J. I. Di Cosimo, V. K. Díez, M. Xu, E. Iglesia and C. R. Apesteguía, J. Catal., 1998, 178, 499–510 CrossRef CAS.
- G. Colón, J. A. Navío, R. Monaci and I. Ferino, Phys. Chem. Chem. Phys., 2000, 2, 4453–4459 RSC.
- W. C. Conner and J. L. Falconer, Chem. Rev., 1995, 95, 759–788 CrossRef CAS.
- P. Haider and A. Baiker, J. Catal., 2007, 248, 175–187 CrossRef CAS.
- U. R. Pillai and E. S. Demessie, Green Chem., 2004, 6, 161–165 RSC.
- A. Auroux and A. Gervasini, J. Phys. Chem., 1990, 94, 6371–6379 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |