A quantum-chemical-based guide to analyze/quantify the cytotoxicity of ionic liquids†
J. S. Torrecillaa, J. Palomar*b, J. Lemusb and F. Rodrígueza
aDepartamento de Ingeniería Química, Universidad Complutense de Madrid, 28040, Madrid, Spain
bSección de Ingeniería Química, Universidad Autónoma de Madrid, Cantoblanco, 28049, Madrid, Spain. E-mail: pepe.palomar@uam.es; Fax: +34 91 497 35 6
First published on 28th October 2009
Abstract
A COSMO-RS descriptor (Sσ-profile) has been used in quantitative structure–activity relationship studies (QSARs) based on a neural network for the prediction of the toxicological effect of ionic liquids (ILs) on a leukemia rat cell line (Log![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) EC50 IPC-81) for a wide variety of compounds including imidazolium, pyridinium, ammonium, phosphonium, pyrrolidinium and quinolinium ILs. Sσ-profile is a two-dimensional quantum-chemical parameter capable of characterising the electronic structure and molecular size of cations and anions. By using a COSMO-RS descriptor for a training set of 105 compounds (96 ILs and 9 closely related salts) with known biological activities (experimental LogEC50 IPC-81 values), a reliable neural network was designed for the systematic analysis of the influence of structural IL elements (cation side chain, head group, anion type and the presence of functional groups) on the cytotoxicity of ∼450 IL compounds. The Quantitative Structure–Activity Map (QSAM), a new concept developed here, was proposed as a valuable tool for (i) the molecular understanding of IL toxicity, by relating Log EC50 IPC-81 parameters to the electronic structure of compounds given by quantum-chemical calculations; and (ii) the sustainable design of IL products with low toxicity, by linking the chemical structure of counterions to the predictions of IL cytotoxicity in handy contour plots. As a principal contribution, quantum-chemical-based QSAM guides allow the analysis/quantification of the non-linear mixture effects of the toxicophores constituting the IL structures. Based on these favorable results, the QSAR model was applied to estimate IL cytotoxicities in order to screen commercially available compounds with comparatively low toxicities.
EC50 IPC-81) for a wide variety of compounds including imidazolium, pyridinium, ammonium, phosphonium, pyrrolidinium and quinolinium ILs. Sσ-profile is a two-dimensional quantum-chemical parameter capable of characterising the electronic structure and molecular size of cations and anions. By using a COSMO-RS descriptor for a training set of 105 compounds (96 ILs and 9 closely related salts) with known biological activities (experimental LogEC50 IPC-81 values), a reliable neural network was designed for the systematic analysis of the influence of structural IL elements (cation side chain, head group, anion type and the presence of functional groups) on the cytotoxicity of ∼450 IL compounds. The Quantitative Structure–Activity Map (QSAM), a new concept developed here, was proposed as a valuable tool for (i) the molecular understanding of IL toxicity, by relating Log EC50 IPC-81 parameters to the electronic structure of compounds given by quantum-chemical calculations; and (ii) the sustainable design of IL products with low toxicity, by linking the chemical structure of counterions to the predictions of IL cytotoxicity in handy contour plots. As a principal contribution, quantum-chemical-based QSAM guides allow the analysis/quantification of the non-linear mixture effects of the toxicophores constituting the IL structures. Based on these favorable results, the QSAR model was applied to estimate IL cytotoxicities in order to screen commercially available compounds with comparatively low toxicities.
1. Introduction
Room-temperature ionic liquids (ILs) possess an array of properties that make them attractive for academic studies and industry: extremely low vapour pressure, high thermal and chemical stabilities, non-flammability and high solvent capacity.1–3 Thus, ILs have been extensively examined as an alternative to conventional volatile organic solvents in reaction and separation processes.4,5 The number of commercial applications of ILs is rapidly growing, and the rate of appearance of patents on IL technology has been rising exponentially for years. Examples of these ILs applications in industry have been reported recently.6 ILs have often been frequently considered as an environmentally benign alternative to volatile organic solvents, mainly based on their negligible vapour pressure. However, recently it has been well documented that they can exhibit environmental toxicity (over a wide range) and persistency.7–13 Regarding the development of IL risk assessment, Prof. Ranke's group (UFT Centre for Environmental Research and Sustainable Technology) has performed the most extensive analysis, carried out in single laboratory tests, of the (eco)toxicological hazard potentials of ILs, following the T-SAR (thinking in terms of structure–activity relationships) strategy.14,15 By applying the T-SAR approach, a large sample of test IL compounds was selected to evaluate their inhibitory effects on a flexible (eco)toxicological test battery (enzymes, cells, microorganisms and organisms).16–23 In order to improve the understanding of toxicity, ILs were subdivided into the cationic size chain,17,18 head group22,23 and the corresponding anion,19,20 to handle the huge structural variability of these compounds. In addition, the test-kits included functionalized side chains and head groups to identify toxicophore elements in ILs.22,23 The systematic analysis of structural insights performed by Ranke et al. provided very relevant information for the sustainable design of ILs with reduced toxicological hazard potential. However, there are an unmanageable number of possible ILs (>106 simple ion combinations and near-endless potential IL mixtures, >1018)24 and the experimental quantification of the toxicity of each IL is not possible. As consequence, an important challenge in IL research is the development of reliable models to relate the physicochemical and structural properties of ILs with their biological effects.A few quantitative structure–activity relationships (QSARs) to predict IL toxicity have been reported in literature. Ranke et al. found a linear correlation between the toxicity of 74 ILs (based on imidazolium, pyrrolidinium, pyridinium, quinolinium, quaternary phosphonium and quaternary ammonium cations) and a empirical HPLC-derived lipophilicity parameter (correlation coefficient of estimated vs. real values, R2 = 0.78).21 A different approach by Luis et al.25 designed an algorithm based on group contribution methods to estimate the aquatic toxicity of 43 imidazolium, pyridinium and pyrrolidinium ILs (R2 = 0.92). Garcia-Lorenzo et al.26 built a QSAR model, according to the Topological Sub-Structural Molecular Design (TOP-MODE) approach, which uses graph-based molecular descriptors to predict the cytotoxicity of 15 imidazolium-derived ILs in Caco-2 cells (R2 = 0.98). Recently, Torrecilla et al.27 used the empirical formulas (elemental composition) and molecular weights of 153 ILs (ammonium, imidazolium, morpholinium, phosphonium, piperidinium, pyridinium, pyrrolidinium and quinolinium salts) to estimate their toxicity (Log EC50) in a leukemia rat cell line (IPC-81) and acetylcholinesterase (AChE) by multiple linear regression (R2 = 0.87 and 0.81, respectively) and neural network (NN) models (R2 = 0.98 and 0.97, respectively).
Molecular simulation based on physically-based models, such as quantum mechanics, molecular dynamics or COSMO-RS, have been much applied as valuable tools to predict physical and thermodynamic properties of ILs, with the main advantage of providing fundamental understanding at the molecular level.28 However, to our knowledge, these reliable methods have hardly been used for the analysis of toxicological effects of ILs. Only one early attempt by Couling et al.29 developed a QSAR model for Vibrio fischeri toxicity of 25 ILs using molecular descriptors, calculated at low semi-empirical computational level, and genetic function approximations (R2 = 0.78). In this work, we will use a recently proposed COSMO-RS molecular descriptor (Sσ-profile)30 in a non-linear neural network analysis for the prediction of IL cytotoxicities. The Sσ-profile descriptor is an a priori quantum-chemical parameter, which quantifies the distribution of the polar electronic charge of a molecular structure on the polarity σ scale, obtained from the histogram function σ-Profile given by COSMO-RS methodology.31 We have recently shown its capability to describe qualitatively and quantitatively the electronic structure and molecular size of a wide variety of ionic liquids (IL).32 Specifically, the Sσ-profile molecular descriptor has been successfully related by NN to three solvent properties of neat ILs and their mixtures (density, IL solubility in hydrocarbons and partition coefficient of aromatic compounds between aliphatic and IL solvents),33 properties which depend on the molecular size and the intermolecular interactions of the mixture. In addition, the Sσ-profile descriptor has been used in quantitative structure–property relationships (QSPR) based on NN for the prediction of polarity/polarizability scales of pure solvents and their binary and ternary mixtures.34 This study represented the first a priori computational approach for a reliable prediction of the non-ideal behavior of solvent effects in mixtures. A remarkable advantage of the Sσ-profile parameter is its additive character, since Sσ-profile values result from the contribution of the different atomic groups on a σ-range.32,33 Thus, the Sσ-profile descriptor of a pure IL or a mixture of ILs can be successfully defined as the sum of those Sσ-profile values of their independent ions.
In the current study, we will develop a QSAR model to establish the toxicological effects of ILs based on the integration of the powerful Sσ-profile descriptor of the cations and anions on NN. In a preliminary section, Sσ-profile descriptor was validated by multilinear regression (MLR) relationships to the cytotoxicity of ILs in Leukemia Rat Cell Line (Log EC50 IPC-81).35 For this analysis, the available experimental Log EC50 IPC-81 data for 86 ILs and 9 closely related salts were used,7 including 35 different cations (ammonium, imidazolium, phosphonium, pyridinium, quinolinium, and sodium) and 18 different anions. Subsequently, a neural network was designed and optimized to estimate the LogEC50 IPC-81 values of ILs as a function of a restricted number of ten Sσ-profile input values (4 for cations and 6 for anions), revealed as statistically significant descriptors in previous MLR analysis. For the development of NN model the LogEC50 IPC-81 sample data were extended to 105 compounds (96 ILs and 9 salts), including additional cations and anions with very high and non-additive cytotoxicological effects. Then, the developed QSAR approach was used for the reliable LogEC50 IPC-81 predictions of ∼450 ILs, obtained using 53 cations – with 6 different head groups, different side chains (from 1 to 16 carbon atoms), and functional groups (ether, hydroxyl, benzyl and amino, among others) – and 20 different anions. As a result of this study, the new concept of a Quantitative Structure–Activity Map (QSAM) was established with the following main objectives: (i) the Quantitative Electronic Structure–Activity Map (QSAM-E) approach relates the estimated LogEC50 IPC-81 values to the molecular electronic information of IL compounds given by the COSMO-RS method. As a consequence, QSAM-E allows a deeper insight into the chemical features of toxicophore substructures and the toxicological impacts of ILs; and (ii) the Quantitative Chemical Structure–Activity Map (QSAM-C) approach relates the estimated LogEC50 IPC-81 values to the chemical structure of both cation and anion constituting the IL. Considering the non-linear toxicophore mixture effects,22 which prevent the use of additive predictive models for IL toxicity, QSAM-C is proposed as a useful guide for the prospective design of safer ILs.
Recently, we reported an a priori computational design tool to obtain ILs with suitable processing features for specific applications.33 QSAM approaches were presented as a complementary tool to also take into account the toxicological hazards in the product design of ILs. In this sense, the developed QSAR model was finally applied in this work to screen commercially available ILs in order to select those with comparatively low cytotoxicities.
2. Computational details
2.1 Data set
All toxicity data used for this study was reported in successive works by Prof. Ranke's group and reviewed in ref. 7. The training sample includes the cytotoxicity values of 96 ILs and 9 closely related salts in a leukemia rat cell line (LogEC50 IPC-81). LogEC50 values are base-10 logarithms of EC50 values in μM, the sample data values ranging from −0.19 to 4.30. The external validation sample consists of LogEC50 IPC-81 values of 15 new ILs whose types of cation or anion were not previously used for developing the QSAR model.2.2 Molecular structures
The molecular geometries of independent cationic and anionic species of ILs were optimized at the B3LYP/6-31++G** computational level in the ideal gas phase using the quantum-chemical Gaussian03 package.36 Vibrational frequency calculations were performed for each case to confirm the presence of a minimum energy.2.3 Sσ-profile descriptor
Gaussian03 was used to compute the COSMO files.35 The ideal screening charges on the molecular surface for each species were calculated by the continuum solvation COSMO model using the BVP86/TZVP/DGA1 level of theory. In these calculations, the continuum solvation model COSMO is applied in order to simulate a virtual conductor environment for the molecule, inducing a polarization charge density σ on the interface of the molecule to the conductor, i.e. on the molecular surface, generating a more polarized electron density than in vacuum. The 3D distribution of the polarization charges σ of each molecule is converted into a surface composition function (σ-Profile) by the COSMOtherm program.37 Such a σ-Profile gives the relative amount of surface with polarity σ, px(σ), on the surface of the molecule. The Sσ-profile molecular descriptors of cations and anions were evaluated by the estimation of the probabilistic surface charge distribution on their σ-Profile at different polarity regions. The σ-Profile values of the IL counterions used in this work are provided as supplementary material, together with the nomenclature used to refer to the cations and anions included in the study.2.4 Neural network design
Neural networks are composed of neurons (information-processing units), and in each of them non linear algorithms are implemented (vide infra). The Multi-Layer Perceptron (MLP) used here has been designed by MATLAB version 7.01.24704 (R14).38 The MLP model is a feed-forward supervised network. Each neuron receives information of all the neurons from the previous layer. Every connection is controlled by parameters (called weights) that modulate the output of the neuron before inputting its numerical content to a neuron in the following layer. The process where the weights are optimized is called the learning or training process.38,39 Here, the training algorithm used is based on back-propagation algorithm (BP). As the NN estimates the LogEC50 IPC-81 of ILs using ten values of Sσ-Profiles, the learning, verification and validation samples consist of eleven rows (one for each Sσ-Profile and LogEC50 IPC-81) and as many columns as ILs tested (105 columns). The only difference between the verification and the learning samples is that the latter is composed of 86 columns (81.9% of data) and the former of 19 columns (18.1% of data). Taking into account that every datum of the verification sample should be interpolated within the learning range, the data were randomly distributed between both samples. The external validation sample is made up of the LogEC50 IPC-81 values of 15 new ionic liquids, specified in the ESI†.In order to guarantee the reliability of the estimations calculated by these models, the applicability domain has been evaluated selecting the compounds with cross-validated standardized residuals greater than three standard deviations.40–42 As the highest cross-validated standardized residual is 2.62 standard deviations (1-hexadecyl-3-methylimidazolium chloride), no set was considered as an outlier.40,41
The MLP model used consists of three layers (input, hidden and output), a topology widely used to deal with several problems.27,32–34 The input layer has as many nodes as Sσ-Profile used (10 input nodes) and one output neuron to estimate the LogEC50 IPC-81. The hidden neuron number (HNN) should be fixed by optimization techniques (vide infra). Given that the range of most Sσ-Profiles and the sigmoid function are between 0 and 1, the sigmoid function has been used as the MLP transfer function.38,39
The BP algorithm is based on the Bayesian Regularization (trainBR) training function. It was selected because its generalization power is higher than other training functions and avoids overfitting and overtraining when a small learning sample is used.39 Their specific parameters are learning coefficient (Lc), learning coefficient decrease, (Lcd), learning coefficient increase (Lci). The Lc parameter is similar to “h” in Newton's method (often called the Newton–Raphson method). Lcd and Lci control the value of Lc depending on the MLP model performance. To avoid overfitting of the NN model, learning and verification samples were used (vide supra) and the learning process was stopped when the error of prediction (mean square error, MSE) for the verification sample, defined by eqn (1), began to increase. A detailed description of the calculation process is described in the literature.43–46
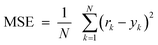 | (1) |
In eqn 1, N, yk and rk are the number of datasets of the data-base, the response of the output neuron and the corresponding real output response, respectively.
The HNN and NN parameters are optimized by an experimental design based on the Box-Wilson Central Composite Design 24 + Star Points. The experimental factors analyzed were Lc, Lcd (between 1 and 0.001) and Lci (between 2 and 100).47 Taking into account the learning sample size, the HNN range was selected between 3 and 10.48 The responses of the experimental design were the Mean Prediction Error (MPE), eqn 2, and correlation coefficient (predicted vs. real values, R2). Both indexes are easily computed and provide a good description of the predictive performance of the NN model.49 As the main objective is to have an NN which predicts results with the highest possible accuracy, the considerations taking into account to analyze the experimental design were the need to achieve the least MPE with the highest values of R2.
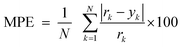 | (2) |
3. Results
3.1 Validation of COSMO-RS descriptor for IL toxicity
Klamt and co-workers30 have developed a quantum chemical approach (COSMO-RS) for the prediction of the thermodynamic properties of fluids using only the surface polarity distributions of individual molecules, which result from quantum chemical calculations. An important advantage of COSMO-RS methodology is that it also provides the 3D charge distribution (σ) on the molecular surface, easily visualized in the histogram function σ-Profile. Based on COSMO-RS methodology, the σ-Profile of one compound includes the main chemical information necessary to predict its possible interactions in a fluid.30 As an example, let us consider the surface polarization charge densities and the resulting σ-Profiles of some cations and anions of common ILs (Fig. 1). The COSMO-RS histogram can be qualitatively divided in three main regions upon next cut-off values: hydrogen bond donor (σHB < −0.0082 e Å−2) and acceptor (σHB > 0.0082 e Å−2) regions and non-polar region (−0.0082 < σ < 0.0082 e Å−2).30 Thus, the σ-Profile of the negatively charged Cl− anion corresponds to a single peak located at the strongly negative polar region (+0.019 e Å−2). This characteristic is represented in its polar surface by a deep red colour. Therefore, the chloride anion can be considered a hydrogen bond acceptor segment. Similarly, PF6− presents a unique peak in its σ-Profile, but at a less negative polar region (0.009 e Å−2), which is visualized in the polar surface by a light orange colour. It indicates a lower polar character in PF6− with respect to Cl−. NTf2− also shows a weak hydrogen bond acceptor fragment (peak located at 0.011 e Å−2 corresponding to –SO2 groups). In addition, NTf2− presents an electronic charge located in the non-polar σ regions, i.e., ±0.0082 e Å−2, mainly due to the –CF3 groups of the anion. The surface of these non-polar fragments is represented in green in Fig. 1.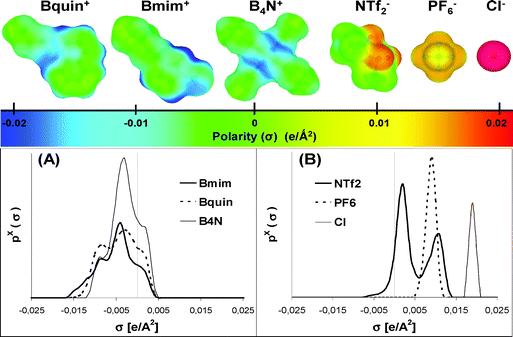 | ||
| Fig. 1 Surface polarization charge density and σ-Profile of some representative cations (A) and anions (B) of ionic liquids. | ||
The polarisable surface and the σ-Profiles of the imidazolium, ammonium and quinolinium cations with a common butyl side chain are shown in Fig. 1A. As can be seen, the surface of the cations is mainly non-polar, i.e., green, with a tendency to blue-green on the more polarized fragments. The σ-Profiles were dominated by a main peak with the charge distribution around zero (−0.0082 < σ < 0.0082 e Å−2), corresponding to the aliphatic groups of the cationic alkyl chains. In addition, some unresolved peaks along with low peaks were observed at lower values than the cutoff −0.0082 e Å−2. These peaks are related to the hydrogen atoms of the aromatic ring for the case of imidazolium and quinolinium cations and those of the ammonium cation closest to its nitrogen atom. According to COSMO-RS theory,31 these cationic fragments may contribute to hydrogen bonds as donors. These examples manifest how the σ-Profile qualitatively describes the different electronic nature of cations and anions. In fact, we have demonstrated that the charge distribution area (Sσ-profile) below σ-Profile can be used as a suitable molecular descriptor of solvent properties, with the remarkable advantages of being a quantum-chemical-derived parameter defined in a restricted scale of polarity (±0.025 e Å−2 for most compounds) and presenting an additive character (the σ-Profile of a IL can be reasonably defined as sum of cation and anion contributions).33
As the first step to develop a QSAR model for establishing IL toxicity based on COSMO-RS information, multilinear regression (MLR) models were constructed for prediction of LogEC50 IPC-81 values of ILs. We initially considered the 61 levels of charge distribution [px(σ)] defining the σ-Profile of IL ionic species in the range ±0.03 e Å−2, i.e., 61 Sσ-profile values. In order to design the best MLR model, the regression model selection (RMS) analysis was used (SPSS software version 15.0.1). The RMS analysis ranked the best subsets of the introduced explanatory variables, using the criteria of best adjusted correlation coefficient and MPE for all possible linear regression models (estimated vs. real LogEC50 IPC-81 values). Since Ranke et al.17,18 found a good correlation between the alkyl chain lengths in imidazolium-based ILs and their LogEC50 IPC-81 cytotoxicities, we initiated the RMS analysis introducing Sσ-profile descriptors of imidazolium cations for a sample data of 60 ILs (MLR Model 1 in Table 1). The RMS analysis revealed that only three Sσ-profile of cation were needed to obtain a good MLR model [eqn 3 in Table 1], located at −0.002, −0.008 and −0.011 e Å−2. The good quality of these simple relationships was indicated by a correlation coefficient R2 = 0.90 and a standard deviation of 0.31 (these being parameters based on a logarithmic scale). The results of LogEC50 IPC-81 predictions by eqn 3 are shown in Fig. 2A. As can be seen in Fig. 2C, the Sσ-profile descriptors used in eqn 3 were located at the peak maximum for alkyl fragment (−0.002 e Å−2) and hydrogen groups linked to head groups (−0.008 and −0.011 e Å−2). Interestingly, eqn 3 presents a negative coefficient for the two cationic Sσ-profile descriptors in the non polar range (−0.0082 < σ < 0.0082 e Å−2), i.e., the corresponding molecular fragments increase IL toxicity (lower LogEC50 IPC-81 values). In contrast, the coefficient of the Sσ-profile descriptor corresponding to the most polarized hydrogen group (−0.011 e Å−2) presents a positive signal in eqn 3, i.e. the more acidic groups of cation contribute to the reduction of the IL toxicity. As second step on COSMO-RS descriptor validation, we performed a RMS analysis for the LogEC50 ICP-81 values of 15 ILs with the common cation 1-butyl-3-methylimidazolium, using in this case the 61 Sσ-profile values of anions. A good MLR model (eqn 4 in Table 1) to describe anion effects was constructed using 6 statistically relevant Sσ-profile descriptors of anions, located at −0.002, 0.009, 0.011, 0.015, 0.017 and 0.019 e Å−2 on their σ-Profile (Fig. 2C). The analysis of eqn 4 indicated that higher polarized anionic fragments result in less toxic ILs. However, the presence of non-polar groups in the anion (see NTf2− in Fig. 2C) confers a toxicological effect on ILs, based on the negative coefficient in eqn 4.
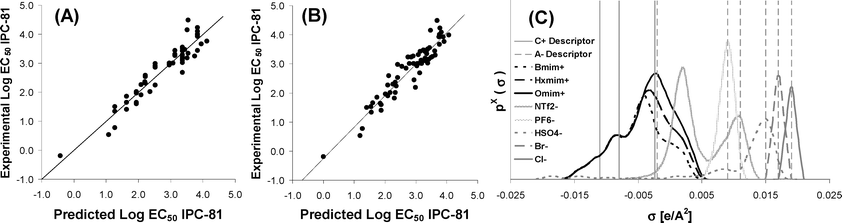
The next step was to achieve a general QSAR approach to analyze both cation and anion effects on IL cytotoxicity. For this purpose, we proposed a different MLR, Model 2 (Table 1), which estimates separately the contributions of cation and anion to LogEC50 IPC-81 value. The sample data included 65 imidazolium-based ILs, which incorporated anions with considerable toxic effects such as (C2F5)3PF3)−, N(CF3)2− and Co(CO)4−.19 Firstly, the main cationic effects were described for the 65 ILs by MLR Model 1 (eqn 5 in Table 1) using 3 Sσ-profile descriptors of cations (located at −0.002, −0.008 and −0.011 e Å−2). Then, the LogEC50 IPC-81 contribution from cation (eqn 5) was included as input in the MLR Model 2 (eqn 6 in Table 1), together with the 6 Sσ-profile descriptors of the above-mentioned anion. The predicted LogEC50 IPC-81 values by MLR Model 2 (eqn 6) were compared with the experimental values in Fig. 2B. Finally, the scope of the proposed MLR Model 2 was extended to 95 ILs with different head groups (imidazolium, pyridinium, phosphonium, ammonium, pyrrolidinium and sodium, as reference). The RMS analysis, introducing the 61 Sσ-profile descriptor of the cation, provided an optimized MLR Model 1 (eqn 7 in Table 1) with four Sσ-profile inputs, corresponding to those used in eqn 5, plus an additional input at −0.006 e Å−2, which presented a significant statistical influence in some head groups such as quinolinium. Then, MLR Model 2 (eqn 8 in Table 1) was constructed to predict both cation and anion effects by including the LogEC50 IPC-81 contribution from cation (eqn 7) and the 6 Sσ-profile descriptors of anions used in eqn 6.
EC50 IPC-81 prediction using the Sσ-profile molecular descriptors of cation or/and anion obtained from the σ-Profilea
| MLR - Model 1
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aim/Sample | Eq. | C0 | Ccation−0.011 | Ccation−0.008 | Ccation−0.006 | Ccation−0.002 | Ccation−0.002 | Ccation0.009 | Ccation0.011 | Ccation−0.015 | Ccation−0.015 | Ccation−0.015 | R2 | σ | N |
| Cation effect /Imidazolium ILs | 3 | 4.8±0.8 | 0.2±0.1 | −0.15±0.03 | — | −0.085±0.004 | — | — | — | — | — | — | 0.90 | 0.31 | 60 |
| Anion effect /Imidazolium ILs | 4 | 2.3±0.1 | — | — | — | — | −0.012±0.006 | 0.015±0.006 | 0.025±0.008 | 0.047±0.009 | 0.038±0.007 | 0.047±0.009 | 0.91 | 0.19 | 15 |
| Cation effect /Imidazolium ILs + toxic anions | 5 | 3.8±0.9 | 0.3±0.2 | −0.13±0.04 | — | −0.081±0.005 | — | — | — | — | — | — | 0.81 | 0.42 | 65 |
| Anion effect /5 head group types of ILs | 7 | 4.9±0.4 | −0.03±0.01 | −0.12±0.03 | 0.06±0.02 | −0.077±0.004 | — | — | — | — | — | — | 0.81 | 0.43 | 95 |
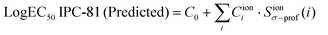
| MLR - Model 2
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a R2: Square correlation coefficient; σ = Standard deviation; N = number of IL in sample data. | |||||||||||||||
| Aim/Sample | Eq. | Ccation | IPC-81cationMLR![[hair space]](https://www.rsc.org/images/entities/b_char_200a.gif) Model1 Model1 | Canion−0.002 | Canion0.009 | Canion0.011 | Canion−0.015 | Canion−0.015 | Canion−0.015 | R2 | σ | N | |||
| Cation + Anioneffects /Imidazolium ILs | 6 | 0.96±0.04 | Eq. 5 | −0.025±0.006 | 0.005±0.003 | 0.002±0.005 | 0.03±0.01 | 0.007±0.006 | 0.013±0.005 | 0.91 | 0.31 | 65 | |||
| Cation + Anion effects / 5 head group types of ILs | 8 | 0.94±0.03 | Eq. 7 | −0.022±0.005 | 0.006±0.003 | 0.004±0.004 | 0.034±0.009 | 0.008±0.004 | 0.012±0.004 | 0.90 | 0.34 | 95 | |||
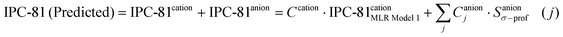
 | ||
| Fig. 2 Comparison of experimental LogEC50 IPC-81 values to those estimated by the MLR model (A) for 60 imidazolium -based ILs, using Eq. 1 with three Sσ-profile values of cation, located at −0.002, −0.008 and −0.011 e Å−2; and (B) for 65 imidazolium-based ILs, eqn 6 with nine Sσ-profile input values (cation: −0.002, −0.008, −0.011 e Å−2; and anion: −0.002, 0.009, 0.011, 0.015, 0.017, 0.019 e Å−2); and (C) Location of Sσ-profile descriptors of cations (used in eqn 3) and anions (used in eqn 4) at σ-Profile polarity range. | ||
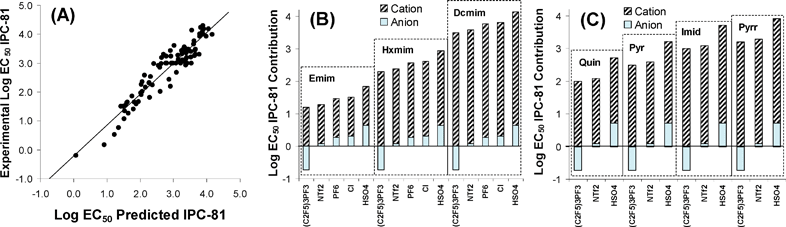
As can be seen in Fig. 3A, the proposed QSAR approach provided a reasonable prediction of LogEC50 IPC-81 values for a wide variety of ILs including 6 different head groups and 19 types of anions. In addition, MLR Model 2 (eqn 8) can be used for a simplistic estimation of cation and anion contribution to IL toxicity. Thus, Fig. 3B showed that the dominant contribution of cation to IL toxicity increases with the side chain length, in agreement with experimental evidence.17,18 On the other hand, current results indicate that most of anions (as Cl− or HSO4−) contribute to the reduction of the IL toxicity, whereas other anions (such as (C2F5)3PF3−, N(CF3)2− or Co(CO)4−) were described as strong toxicophores by eqn 8. Similarly, Fig. 3C presented the effect of head group (with common butyl side) on toxicity for some representative IL examples. Quinolinium and pyrrolidinium head groups exhibited, respectively, the highest and lowest hazardous effects, whereas imidazolium and pyridinium cations presented intermediate toxicities, in line with experimental trends with small anions [Experimental LogEC50 IPC-81 values: BquinBr (2.32) < BmimBr (3.43) < BpyrrBr (3.77)].7 However, the QSAR approach proposed by MLR Model 2 presents significant limitations to predict accurately IL cytotoxicity. For example, it failed to describe the non-linear behaviour of LogEC50 IPC-81 values with the alkyl chain length for highly hydrophobic ILs.21 In addition, the observed non-additive toxicophore effects for different cation and anion mixtures19,23 cannot be described by the QSAR model based on MLR approximation. Therefore, a main aim of this work was the generation of a non-linear model based on NN for the prediction of LogEC50 IPC-81 values of ILs using 4 Sσ-profile descriptors of cation and 6 Sσ-profile descriptors of anions. The sample data contained 105 different compounds, including ILs with very large cations (TetraDmim+ or P66614+) or anions (BBDB−). Following the aforementioned optimization process, the NN model was designed using the learning sample. The optimum values of HNN, Lc, Lcd and Lci were 9, 0.001, 1 and 2 respectively (see Table 2). Then, using these parameter values, the model was verified. In this process, the model was tested against a verification sample that had not been included in the neural network learning sample. Fig. 4A compared the LogEC50 IPC-81 estimations for learning and verification samples to the experimental values. The correlation coefficient R2 was obtained higher than 0.996 and MPE and σ were less than 0.47% and 0.12, respectively (these being statistical parameters based on a logarithmic scale of LogEC50 IPC-81). Finally, in order to carry out an external validation process of the optimized model,40,50,51 a new validation sample based on experimental data available in literature was employed. Taking into account that the external validation sample range must be within the learning sample range and belong to the same application domain (vide supra), the datasets used in the external validation of the MLP model were selected. The mathematical procedure followed was similar to the verification process described above. As can be seen in Fig. 4B, the MLP model presents an acceptable goodness of fit, with R2 > 0.96 and MPE < 5.7%. In terms of external explained variance (Q2ext), given that this parameter is close to 1 (Q2ext >0.94), the NN model is stable and predictive.41,50Fig. 4C reported the residuals from the NN model. Given that the R2 between residuals and LogEC50 IPC-81 values is less than 0.07, no mathematical dependence between them can be found. Therefore, in the light of the statistical results, the non-linear NN model (MLP) was adequate to estimate LogEC50 IPC-81 values of ILs, by only using 10 Sσ-profile descriptor values of counterion components, which are derived solely from quantum-chemical COSMO-RS calculations. The diversity of cations (>40) and anions (>20) used to design the NN model, together with the high variability of LogEC50 IPC-81 values (between −0.19 and 4.30), guaranteed the generality of the developed non-linear QSAR model based on NN, validating the a priori Sσ-profile parameter for the description of IL cytotoxicities.
| Parameters | |
|---|---|
| Transfer function | Sigmoid |
| Input nodes number | 10 |
| Hidden neurons number | 9 |
| Output neuron number | 1 |
| Learning coefficient, Lc | 0.001 |
| Learning coefficient decrease, Lcd | 1 |
| Learning coefficient increase, Lci | 2 |
 | ||
| Fig. 3 (A) Comparison of experimental LogEC50 IPC-81 values to those estimated by the MLR model for 95 ILs using eqn 8 with ten Sσ-profile input values (cation: −0.002, −0.006, −0.008, −0.011 e Å−2; and anion: −0.002, 0.009, 0.011, 0.015, 0.017, 0.019 e Å−2); Cation and anion contribution to estimated LogEC50 IPC-81 values given by eqn 8 for (B) the 1-alkyl-3-methylimidazolium series and (C) the head group series (with a common butyl side chain). | ||
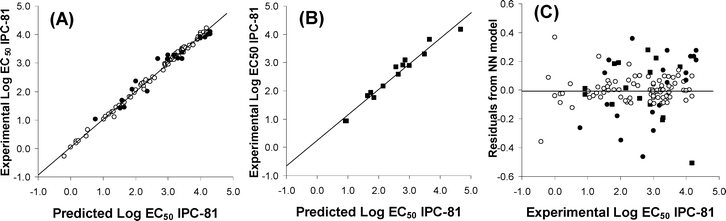 | ||
| Fig. 4 Comparison of experimental LogEC50 IPC-81 values to those estimated by the NN model for 105 ILs using ten Sσ-profile input values (cation: −0.002, −0.006, −0.008, −0.011 e Å−2; and anion: −0.002, 0.009, 0.011, 0.015, 0.017, 0.019 e Å−2). (A) Learning and verification samples and (B) Validation sample. (C) Graphical analysis of residuals from NN model. (○ Learning sample; ● Verification simple; ■ External validation sample). | ||
3.2 Quantitative structure–activity maps (QSAM) for IL toxicity
In this section, the new concept of Quantitative Structure–Activity Map is presented as a systematic way of examining IL compounds from their structural components (alkyl side chain, head group, anion type and linked functional groups) determining their effects on a biological system (leukemia rat cell line, IPC-81). For this purpose, firstly, an extensive screening of IL cytotoxicity for ∼450 possible cation–anion combinations was performed using the reliable QSAR model based on optimized NN model with 10 Sσ-profile descriptors (LogEC50 IPC-81 estimations of ILs were given as ESI†). To establish our hypothesis, Fig. 5A shows the polar charge distribution given by the σ-Profiles of a 1-alkyl-3-methylimidazolium series, which can be represented by a 3-D graph or by a 2-D contour plot. It seems to be clear that the alkyl side effects on the IL toxicities may be rationalized on a molecular basis by relating them to quantum-chemical COSMO-RS information of the cation. With this aim, Fig. 5B proposes a QSAM-E for describing the alkyl chain effects on a series of 1-alkyl-3-methylimidazolium tetrafluoroborate compounds. QSAM-E approach visually relates the estimated LogEC50 IPC-81 values to the electronic polar structure of cation, distributed along the σ-polarity scale. Thus, the progressive increase of IL toxicity with the length of cation side chain can be ascribed to the increasing amount of charge distribution on the non-polar range (−0.0082 < σ < 0.0082 e Å−2) of σ-Profile. Continuing the analysis, Fig. 5C presents a QSAM-E for the study of the effect of cationic head group on LogEC50 IPC-81 values for a subset of different core structures, all carrying the common anion (BF4−) and butyl side chain. The conclusion of the analysis was again that the increasing addition of non-polar fragments on cation contributes to the hazardous effect of ILs. Interestingly, the more polarized the head group, the less toxic it is. Thus, the reported different behaviour of the quinolinium head group, as a strong toxicophore substructure,21 can be assigned to its largely delocalized non-polar electronic charge. Finally, the QSAM-E approach was applied to the systematic study the anion effect on LogEC50 IPC-81 estimations for a series of 1-butyl-3-methylimidazolium compounds (Fig. 6). It was observed that the toxicological effects increased progressively with the loss of anion polarity. In fact, we identified three class of anions in QSAM-E analysis: (i) those with high basicity, i.e., with polar nucleophilic fragments present at σ > 0.015 e Å−2, and low toxicophore features, as Cl− or CH3SO3−, with LogEC50 IPC-81 > 3.4; (ii) those with medium basicity, i.e., polar density located in the range 0.005 < σ < 0.015 e Å−2, and low or intermediate toxicological effects (3.0 < LogEC50 IPC-81 < 3.4); and (iii) those with strong non-polar characteristics, described by charge density located at σ < 0.005 e Å−2, and a marked contribution to IL toxicity, as (C2F5)3PF3−, N(CF3)2−, Co(CO)4− or BBDB−, with LogEC50 IPC-81 < 2.5. To sum up, NN estimations confirmed strong intrinsic cytotoxicity effects of both cation and anion moieties in IL compounds. In fact, the QSAM-E tool seemed to offer a coherent description of IL toxicity as a function of the polar charge distribution of ionic substructures of the compound.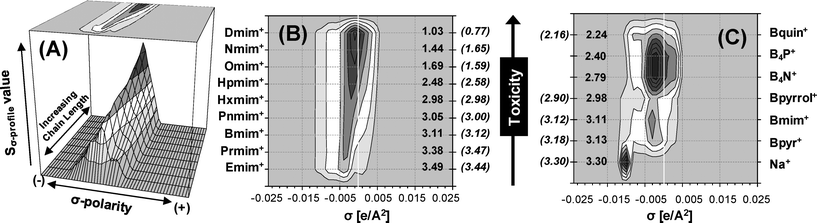 | ||
| Fig. 5 (A) 3D Graph of σ-Profile for the 1-alkyl-3-methylimidazolium cation series; (B) Quantitative Electronic Structure–Activity Map (QSAM-E) of the alkyl chain effect for the 1-alkyl-3-methylimidazolium tetrafluoroborate series; and (C) QSAM-E of the head-group effect for IL series with a common BF4− anion and butyl side chain. Experimental and estimated LogEC50 IPC-81 values are given in parentheses and without parentheses, respectively. | ||
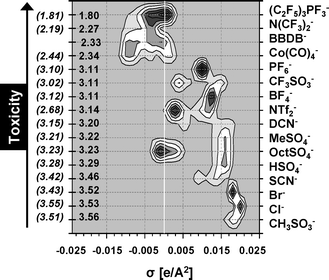 | ||
| Fig. 6 QSAM-E of the anion effect for the 1-butyl-3-methylimidazolium series. Experimental and estimated LogEC50 IPC-81 values are given in parentheses and without parentheses, respectively. | ||
These results were basically consistent with a previous QSAR model to establish IL toxicity mainly driven by the lipophilicity of the compound.19,22 However, the authors reported some cases with significant deviations from the general linear trend between cytotoxicity and lipophilicity: (i) ILs containing anions with clear intrinsic toxicities (as NTf2− or BBDB−); (ii) ILs based on quinolinium or 4-(dimethylamino)pyridinium cations; and (iii) ILs containing very lipophilic cations with very long alkyl chains. In this work, the lipophilicity parameter logk0, derived from reverse-phase gradient HPLC retention times,21 was found to be directly related to the Sσ-profile descriptor of the cation located at the non-polar region σ = −0.002 e Å−2, as it is shown in Fig. 7A for 32 ILs including 7 different head groups. The amount of charge estimated by Sσ-profile (−0.002 e Å−2) was mainly provided by aliphatic fragments of the structure. This result was relevant because it indicated that the lipophilicity estimations by the logk0 parameter of ILs would be assigned to those aliphatic fragments of the cation. As a consequence, the deviations from the lipophilicity–cytotoxicity relationship19–23 must be attributed to the interactions of other toxicophore fragments with biological systems, which seem to be underestimated by lipophilicity parameter logk0 in HPLC. For example, QSAM-E in Fig. 7B demonstrated that the different behaviour evidenced for the quinolinium head group21 must be assigned to the toxicological effects of the aromatic fragment, which presents a strongly delocalized charge in the negative region of low polarity (−0.0082 < σ < −0.002 e Å−2). In addition, the greater toxicity of 4-(dimethylamino)pyridinium with respect to the imidazolium head group22,23 can be ascribed not only to the higher amount of non-polar groups at −0.002 e Å−2 region but also to the presence of polarized charge density at the σ region between −0.0082 and −0.002 e Å−2. For its part, the intrinsic cytotoxic effects of NTf2−, compared to the reference chloride anion,19,22 must be ascribed to the presence of non-polar groups in its structure (Fig. 7C and 7D). On the other hand, the observed significant reduction of imidazolium IL toxicity by the introduction of an oxygenated side chain23 was explained by QSAM-E on the basis of the charge shift of cation structure towards more polarized regions (Fig. 7E), resulting in less toxicophore features.
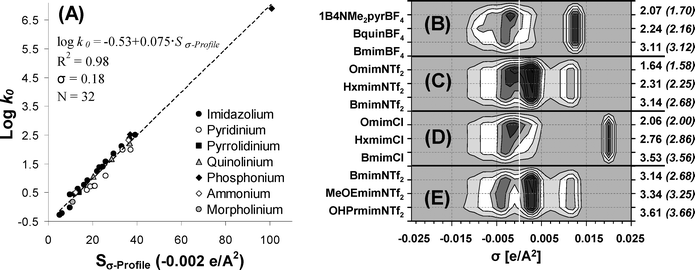 | ||
| Fig. 7 (A) Comparison of the values for Sσ-profile descriptor of cations at −0.002 e Å−2 against lipophilicity parameter logk0 derived from reverse-phase gradient HPLC retention times;19,22 (B) QSAM-E for ILs with different toxicity-lipophilicity relationships. Experimental LogEC50 IPC-81 values are in parentheses. | ||
As noted in the previous section, the current QSAR study confirmed non-additive mixture effects of toxicophores constituting the IL structures. This is shown in Fig. 8, where the general agreement between the experimental and NN estimated values of LogEC50 IPC-81 of ILs contrasted with the results of MLR Model 2. Thus, the increasing side chain lengths generally imply a decrease of LogEC50 IPC-81 values of ILs (higher toxicity), being the non-linear effects due to the well-known biological cut-off effect correctly described by NN model. It is noteworthy, however, that the scope of the alkyl chain effect is strongly modified by the anion nature (Fig. 8A). For example, (C2F5)3PF3− clearly behaves as stronger toxicophore than Cl− (the latter being used as a non-toxic and bio-compatible reference for anions52) for IL with short cationic alkyl chains;however, it presents the opposite behaviour with very long side chains. Mixture effects were also found to be drastically different for different head groups (Fig. 8B). Thus, the contribution to toxicity of different anions is significantly modified by the cationic head group (see the case of CF3SO3− in Fig. 8B). In conclusion, current results show the difficulty of assigning an intrinsic toxicity to an anion, since its toxicophore behaviour is heavily dependent on cationic structure. In other words, a reliable additive treatment based on the intrinsic quantification of cation and anion toxicities is no longer suitable. As a suitable alternative, we proposed the QSAM-C approach to consider the IL cytotoxicity in terms of mixture toxicity. QSAM-C relates the chemical structures of both cation and anion of ILs to the LogEC50 IPC-81 estimations in a handy contour plot, as shown Fig. 9 for the families of ILs based on imidazolium (A), pyridinium (B), ammonium (C) and phosphonium (D). The QSAM-C graphics allow a visual quantitative evaluation of IL cytotoxicity in function of the non-linear cation and anion interactions. Firstly, it was clear that the three main structural elements of an IL (cation side chain, cation head group and the kind of anion) have a significant impact on its toxicity, even when the influence of cationic moiety was found to dominate. Thus, the range of LogEC50 IPC-81 for a commonly considered low toxic type of IL, such as 1-alkyl-3-methylimidazolium methylsulfate,22 spans from 3.9 to 1.2 by increasing the alkyl side in 7 carbon atoms (Fig. 9A). The non-linear mixture effects were also evident: ILs with intermediate polar anions, such as CF3SO3− or DCN−, presented the highest (0.6) and lowest (3.9) toxicity for, respectively, the longest and shortest alkyl imidazolium chains (Fig. 9A). On the other hand, a small anion such as Cl− seemed to increase its toxicological effects with the length of alkyl chains, whereas a large anion such as BBDB− presented stronger toxicophore effects in low toxic imidazolium or pyridinium cations, i.e., cations with short side chains (Fig. 9A and 9B). As a general contribution, current QSAM-C revealed that the ammonium-based ILs present lower cytotoxicity than those based on imidazolium, pyridinium or phosphonium head groups, particularly those synthesized with short alkyl chains and alkylsulfate anions (estimated LogEC50 IPC-81 values ∼4.2).
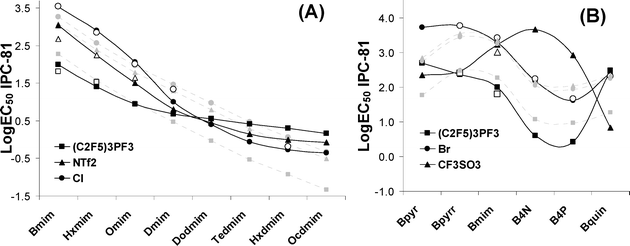 | ||
| Fig. 8 Non-additive toxicophore effects described by NN estimated (black symbols), MLR estimated (grey symbols) and experimentally available (white symbols) LogEC50 IPC-81 values in (A) anion+cation side chain mixture effect and (B) anion+cation head group mixture effects. | ||
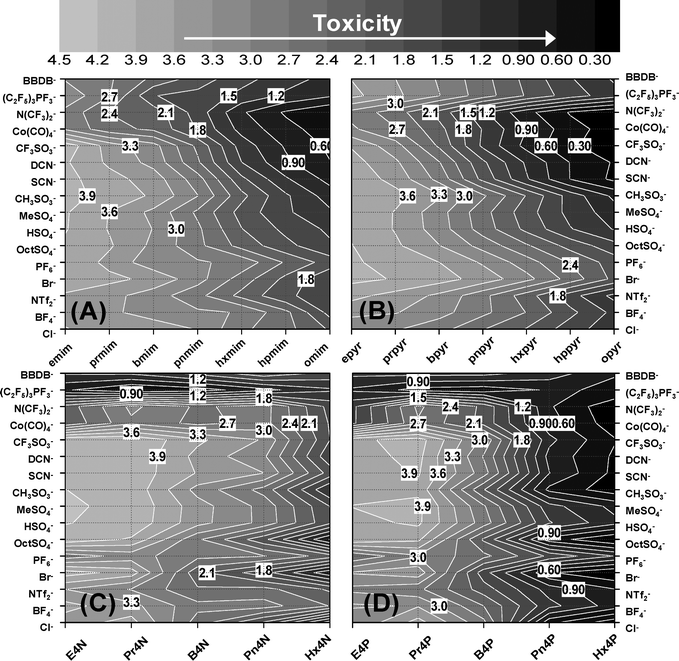 | ||
| Fig. 9 Quantitative Chemical Structure–Activity Map (QSAM-C) of IL cytotoxicity for (A) 1-alkyl-3-methylimidazolium; (B) 1-alkylpyridinium; (C) tetraalkylammonium and (D) tetraalkylphosphonium series. Estimated LogEC50 IPC-81 values for iso-toxicity contour lines are shown with a white background. | ||
Finally, in order to carry out a useful application, the developed QSAR model was easily used in this work to filter environmentally benign ILs by screening the commercially available compounds, using the criteria of low IL cytotoxicity given in Fig. 6 (LogEC50 IPC-81 > 3.4). Table 3 reports 30 ILs with comparatively low hazardous effects, together with their CAS reference number. In fact, 10 commercial ILs in Table 3 were estimated to have lower toxicity than 1-ethyl-3-methylimidazolium ethylsulfate, a compound used in the literature as a reference for low IL toxicity. It should be noted, however, that the examined commercial ILs still have cytotoxicities higher than those of common solvents [LogEC50 IPC-81 of methyl tert-butyl ether (5.6), methanol (6.2) or acetone (6.8)].17 As a result, further QSAM analysis, based on quantum-chemical data, is currently being performed to select the toxicologically favourable structural elements for designing more sustainable ILs.
EC50 IPC-81 of commercially available ionic liquids with comparatively low cytotoxicity (LogEC50 IPC-81 > 3.4)
| CAS | Cation | Anion | LogEC50 IPC-81 | |
|---|---|---|---|---|
| Estimated | Experimental | |||
| a Ionic liquid in the Io-Li-Tec catalogue, presented as new without CAS number | ||||
| 479500-35-1 | Bpyrr+ | Cl− | 4.30 | — |
| 35895-69-3 | E4N+ | CF3SO3− | 4.19 | — |
| 145022-44-2 | Emim+ | CF3SO3− | 4.08 | 4.09 |
| 145022-45-3 | Emim+ | CH3SO3− | 4.03 | — |
| 331717-63-6 | Emim+ | SCN− | 4.00 | — |
| 1906-79-2 | Epyr+ | Br− | 3.98 | — |
| 370865-89-7 | Emim+ | DCN− | 3.94 | — |
| 103173-73-5 | Epyr+ | PF6− | 3.94 | — |
| 342573-75-5 | Emim+ | MeSO4− | 3.91 | 3.93 |
| 412009-61-1 | Emim+ | HSO4− | 3.92 | 3.99 |
| 65039-08-9 | Emim+ | Br− | 3.89 | — |
| 155371-19-0 | Emim+ | PF6− | 3.86 | 3.92 |
| 93457-69-3 | Bpyrr+ | Br− | 3.63 | 3.77 |
| 874-80-6 | Bpyr+ | Br− | 3.73 | — |
| 85100-76-1 | Prmim+ | Br− | 3.68 | — |
| 712354-97-7 | Epyr+ | NTf2− | 3.68 | — |
| 186088-50-6 | Bpyr+ | PF6− | 3.60 | — |
| 3878-80-6 | Epyr+ | CF3SO3− | 3.57 | — |
| 342789-81-5 | Bmim+ | CH3SO3− | 3.51 | 3.56 |
| 65039-09-0 | Emim+ | Cl− | 3.56 | — |
| 174899-82-2 | Emim+ | NTf2− | 3.54 | — |
| 79917-90-1 | Bmim+ | Cl− | 3.53 | 3.55 |
| 258273-75-5 | B1M3N+ | NTf2− | 3.52 | 3.61 |
| 85100-78-3 | Bmim+ | Br− | 3.52 | 3.43 |
| 216300-12-8 | Prmim+ | PF6− | 3.51 | — |
| —a | Prpyr+ | NTf2− | 3.50 | — |
| 143314-16-3 | Emim+ | BF4− | 3.49 | 3.44 |
| 344790-87-0 | Bmim+ | SCN− | 3.46 | 3.42 |
| 350-48-1 | Epyr+ | BF4− | 3.42 | — |
| 216299-72-8 | Prmim+ | NTf2− | 3.40 | — |
4. Conclusions
Predictive relationships between quantum-chemical Sσ-profile descriptors of IL compounds and their cytotoxicities (LogEC50 IPC-81) were effectively established by non-linear NN analysis for a wide range of cations (>40) and anions (>20), by using a training set of 105 compounds (96 ILs and 9 closely related salts). The reliable non-linear QSAR model was used for the estimation of cytotoxicities of ∼450 IL compounds, with the aim of systematically analyzing the toxicological effects of their substructural elements (alkyl side chain, head group and type of anion). As result of this study, the non-additive behaviour of IL toxicophores was generally observed. This implied that the cytotoxicity of ILs cannot be systematically estimated as the sum of intrinsic toxicities of independent cations and anions. As an alternative predictive model, a new concept of the Quantitative Structure–Activity Map (QSAM) was proposed as a useful tool to design ILs that are not only task-specific, but also inherently safer. Firstly, the QSAM-E approach allowed the analysis of the toxicological behaviour of the components of ILs on a molecular basis, since LogEC50 IPC-81 values were understandably related to the polar charge distribution of cations and anions given by the quantum-chemical COSMO-RS method. Secondly, the QSAM-C approach was presented as a simple and reliable graphical guide for the selection of cation and anion, by taking into account the non-linear range of toxicophore mixture effects when the aim is a sustainable computational design of safer ILs. Finally, based on these favourable results, the QSAR model was applied to amplify the current knowledge of IL cytotoxicities by screening commercially available compounds with comparatively low toxicity (LogEC50 IPC-81 > 3.4).
Acknowledgements
The authors are grateful to the “Ministerio Ciencia e Innovación” for financial support (projects CTQ2008-01591 and CTQ2008-05641). José S. Torrecilla was supported by a Ramón y Cajal research contract from the “Ministerio Ciencia e Innovación” in Spain. We are very grateful to the “Centro de Computación Científica de la Universidad Autónoma de Madrid” for computational facilities.References
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS.
- P. Wasserscheid, T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2003 Search PubMed.
- R. D. Rogers and K. R. Seddon, Ionic Liquids as Green Solvents, ACS Symposium Series 856, American Chemical Society, 2003 Search PubMed.
- R. D. Rogers and K. R. Seddon, Ionic Liquids IIIB: Fundamentals, Properties, Challenges and Opportunities, ACS Symposium Series 902, American Chemical Society, 2005 Search PubMed.
- X. Han and D. W. Armstrong, Acc. Chem. Res., 2007, 40, 1079 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- J. Ranke, S. Stolte, R. Störmann, J. Arning and B. Jastorff, Chem. Rev., 2007, 107, 2183 CrossRef CAS.
- A. Garcia-Lorenzo, E. Tojo, J. Tojo, M. Teijeira, F. J. Rodriguez-Berrocal, M. P. Gonzalez and V. S. Martinez-Zorzano, Green Chem., 2008, 10, 508–516 RSC.
- K. J. Kulacki and G. A. Lamberti, Green Chem., 2008, 10, 104–110 RSC.
- A. Romero, A. Santos, J. Tojo and A. Rodriguez, J. Hazard. Mater., 2008, 151, 268–273 CrossRef CAS.
- L. Carson, P. K. W. Chau, M. J. Earle, M. A. Gilea, B. F. Gilmore, S. P. Gorman, M. T. McCann and K. R. Seddon, Green Chem., 2009, 11, 492–497 RSC.
- A. Latala, M. Nedzi and P. Stepnowski, Green Chem., 2009, 11, 580–588 RSC.
- M. Rebros, H. Q. N. Gunaratne, J. Ferguson, K. R. Seddon and G. Stephens, Green Chem., 2009, 11, 402–408 RSC.
- B. Jastorff, R. Stormann and J. Ranke, Clean: Soil, Air, Water, 2007, 35, 399–405 Search PubMed.
- B. Jastorff, K. Molter, P. Behrend, U. Bottin-Weber, J. Filser, A. Heimers, B. Ondruschka, J. Ranke, M. Schaefer, H. Schroder, A. Stark, P. Stepnowski, F. Stock, R. Stormann, S. Stolte, U. Welz-Biermann, S. Ziegert and J. Thoming, Green Chem., 2005, 7, 362–372 RSC.
- B. Jastorff, R. Stormann, J. Ranke, K. Molter, F. Stock, B. Oberheitmann, W. Hoffmann, J. Hoffmann, M. Nuchter, B. Ondruschka and J. Filser, Green Chem., 2003, 5, 136–142 RSC.
- J. Ranke, K. Molter, F. Stock, U. Bottin-Weber, J. Poczobutt, J. Hoffmann, B. Ondruschka, J. Filser and B. Jastorff, Ecotoxicol. Environ. Saf., 2004, 58, 396–404 CrossRef CAS.
- J. Ranke, K. Molter, F. Stock, U. Bottin-Weber, J. Poczobutt, J. Hoffmann, B. Ondruschka, J. Filser and B. Jastorff, Ecotoxicol. Environ. Saf., 2005, 60, 350–350 CrossRef CAS.
- S. Stolte, J. Arning, U. Bottin-Weber, M. Matzke, F. Stock, K. Thiele, M. Uerdingen, U. Welz-Biermann, B. Jastorff and J. Ranke, Green Chem., 2006, 8, 621–629 RSC.
- M. Matzke, S. Stolte, K. Thiele, T. Juffernholz, J. Arning, J. Ranke, U. Welz-Biermann and B. Jastorff, Green Chem., 2007, 9, 1198–1207 RSC.
- J. Ranke, A. Muller, U. Bottin-Weber, F. Stock, S. Stolte, J. Arning, R. Stormann and B. Jastorff, Ecotoxicol. Environ. Saf., 2007, 67, 430–438 CrossRef CAS.
- S. Stolte, J. Arning, U. Bottin-Weber, A. Muller, W. R. Pitner, U. Welz-Biermann, B. Jastorff and J. Ranke, Green Chem., 2007, 9, 760–767 RSC.
- J. Arning, S. Stolte, A. Boschen, F. Stock, W. R. Pitner, U. Welz-Biermann, B. Jastorff and J. Ranke, Green Chem., 2008, 10, 47–58 RSC.
- E. F. Borra, O. Seddiki, R. Angel, D. Eisenstein, P. Hickson, K. R. Seddon and S. P. Worden, Nature, 2007, 447, 979–981 CrossRef CAS.
- P. Luis, I. Ortiz, R. Aldaco and A. Irabien, Ecotoxicol. Environ. Saf., 2007, 67, 423–429 CrossRef CAS.
- A. Garcia-Lorenzo, E. Tojo, J. Tojo, M. Teijeira, F. J. Rodriguez-Berrocal, M. P. Gonzalez and V. S. Martinez-Zorzano, Green Chem., 2008, 10, 508–516 RSC.
- J. S. Torrecilla, J. Garcia, E. Rojo and F. Rodriguez, J. Hazard. Mater., 2009, 164, 182–194 CrossRef CAS.
- J. Palomar, V. R. Ferro, M. A. Gilarranz and J. J. Rodriguez, J. Phys. Chem. B, 2007, 111, 168–180 CrossRef CAS.
- D. J. Couling, R. J. Bernot, K. M. Docherty, J. K. Dixon and E. J. Maginn, Green Chem., 2006, 8, 82–90 RSC.
- J. Palomar, V. R. Ferro, J. S. Torrecilla and F. Rodriguez, Ind. Eng. Chem. Res., 2007, 46, 6041–6048 CrossRef CAS.
- A. Klamt, COSMO-RS: From Quantum Chemistry to Fluid Phase Thermodynamics and Drug Design, Elsevier, Amsterdam, 1st edn, 2005 Search PubMed.
- J. Palomar, J. S. Torrecilla, V. R. Ferro and F. Rodriguez, Ind. Eng. Chem. Res., 2008, 47, 4523–4532 CrossRef CAS.
- J. Palomar, J. S. Torrecilla, V. R. Ferro and F. Rodriguez, Ind. Eng. Chem. Res., 2009, 48, 2257–2265 CrossRef CAS.
- J. Palomar, J. S. Torrecilla, J. Lemus, V. R. Ferro and F. Rodriguez, Phys. Chem. Chem. Phys., 2008, 10, 5967–5975 RSC.
- N. Lacaze, G. Gombaud-Saintonge and M. Lanotte, Leuk. Res., 1983, 7, 145–154 CAS.
- Gaussian 03, Revision C.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- F. Eckert and A. Klamt, COSMOtherm, Version C2.1, Release 01.06, COSMOlogic, Leverkusen, Germany, 2006 Search PubMed.
- A. J. Maren, T. Harston and R. P. Pap, Handbook of neural computing applications, Academic Press Inc., San Diego, 1990, pp. 323–324 Search PubMed.
- H. Demuth, M. Beale and M. Hagan, Neural Network Toolbox For Use with MATLAB User's Guide, version 4.0.6, ninth printing, 2005; revised for version 4.0.6; release 14SP3 Search PubMed.
- P. Gramatica, E. Giani and E. Papa, J. Mol. Graphics Modell., 2007, 25, 755–766 CrossRef CAS.
- P. Gramatica, QSAR Comb. Sci., 2007, 26, 694–670 Search PubMed.
- T. I. Netzeva, A. P. Worth, T. Aldenberg, R. Benigni, M. T. D. Cronin, P. Gramatica, J. S. Jaworska, S. Kahn, G. Klopman, C. A. Marchant, G. Myatt, N. Nikolova-Jeliazkova, G. Y. Patlewicz, R. Perkins, D. W. Roberts, T. W. Schultz, D. T. Stanton, J. J. M. van de Sandt, W. D. Tong, G. Veith and C. H. Yang, Alternatives to Laboratory Animals, 2005, 33, 155–173 Search PubMed.
- J. S. Torrecilla, M. Deetlefs, K. R. Seddon and F. Rodríguez, Phys. Chem. Chem. Phys., 2008, 10, 5114–5120 RSC.
- J. S. Torrecilla, F. Rodríguez, J. L. Bravo, G. Rothenberg, K. R. Seddon and I. Lopez-Martin, Phys. Chem. Chem. Phys., 2008, 10, 5826–5831 RSC.
- J. S. Torrecilla, L. Otero and P.D. Sanz, J. Food Eng., 2005, 69, 299–306 CrossRef.
- J. S. Torrecilla, M. L. Mena, P. Yáñez-Sedeño and J. García, J. Agric. Food Chem., 2007, 55, 7418–7426 CrossRef CAS.
- V. Vacic, Summary of the training functions in Matlab's NN toolbox, 2005, http://www.cs.ucr.edu/~vladimir/cs171/nn_summary.pdf Search PubMed.
- Y. Sun, Y. Peng, Y. Chen and A. J. Shukla, Adv. Drug Delivery Rev., 2003, 55, 1201–1215 CrossRef CAS.
- L. B. Sheiner and S. Beal, J. Pharmacokinet. Biopharm., 1981, 9, 503–509 Search PubMed.
- Guidance Document on the Validation of (Quantitative) Structure Activity Relationship [(Q)SAR] Models, No. 69 (OECD Series on Testing and Assessment), Organisation of Economic Cooperation and Development, Paris, France, 2007, http://www.oecd.org Search PubMed.
- J. C. Dearden, M. T D. Cronin and K. L. E. Kaiser, SAR QSAR Environ. Res., 2009, 20, 241–266 Search PubMed.
- M. Matzke, S. Stolte, A. Boschen and J. Filser, Green Chem., 2008, 10, 784–792 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Nomenclature of ionic liquid units and Sσ-profile descriptors for cations and anions. See DOI: 10.1039/b919806g |
| This journal is © The Royal Society of Chemistry 2010 |