Exploring the possibility of using a thermostable mutant of β-glucosidase for rapid hydrolysis of quercetin glucosides in hot water
Sofia Lindahla, Anna Ekmanc, Samiullah Khanb, Christina Wennerbergb, Pål Börjessonc, Per J. R. Sjöberga, Eva Nordberg Karlssonb and Charlotta Turner*a
aUppsala University, Department of Physical and Analytical Chemistry, Analytical Chemistry, P.O. Box 599, SE-751 24, Uppsala, Sweden. E-mail: Charlotta.Turner@kemi.uu.se
bLund University, Department of Biotechnology, P.O. Box 124, SE-221 00, Lund, Sweden
cLund University, Department of Technology and Society, P.O. Box 118, SE-221 00, Lund, Sweden
First published on 4th November 2009
Abstract
The antioxidant quercetin was extracted from yellow onion waste and converted to its aglycone form by a combination of subcritical water extraction and enzymatic hydrolysis. The hydrolytic step was catalysed by a double residue (N221S, P342L) mutant of the thermostable β-glucosidase (TnBgl1A), isolated from the thermophile Thermotoga neapolitana and cloned and produced in E. coli. The activity of wt TnBgl1A was shown to be dependent on the position of the glucosylation on the quercetin backbone, favouring hydrolysis of quercetin-4′-glucoside over quercetin-3-glucoside. The mutated variant of the enzyme harboured a mutation in the +2 sub-site (N221S) and showed increased catalytic efficiency in quercetin-3-glucoside hydrolysis and also to a certain extent hydrolysis of quercetin-4′-glucoside. The mutated enzyme was used directly in yellow onion extracts, prepared by subcritical water extraction, resulting in complete hydrolysis of the glucosylated flavonoids quercetin-3,4′-diglucoside, quercetin-4′-glucoside, quercetin-3-glucoside, isorhamnetin-4′-glucoside and isorhamnetin-3,4′-diglucoside. To complete hydrolysis within five minutes, 3 mg of TnBgl1A_N221S was used per gramme of onion (dry weight). A life cycle assessment was done to compare the environmental impact of the new method with a conventional solid–liquid extraction-and-hydrolysis method utilising aqueous methanol and hydrochloric acid. Comparison of the methods showed that the new method is preferable regarding primary energy consumption and global warming potential. Another advantage of this method is that handling of toxic chemicals (methanol and HCl) is avoided. This shows that combined subcritical water extraction/enzyme hydrolysis is both a fast and sustainable method to obtain quercetin from onion waste.
Introduction
The agricultural, forestry, and food industries produce tonnes of waste materials and by-products every year, such as onion skin, apple peel, carrot waste and birch bark. In the case of onion, the amount of onion waste annually produced within the European Union is almost half a million tons.1 The produced waste and by-products are today used in animal feed, or for composting, incineration or anaerobic digestion. However, these waste materials and by-products contain high-value compounds such as antioxidants that can be extracted before the waste reaches its final destination. Waste and by-products from fruits and vegetables are rich in antioxidants such as polyphenols, which have uses as additives in pharmaceuticals, food products and cosmetics. A large group of polyphenolic compounds are flavonoids. Quercetin (Q) is an example of a high-value flavonoid, and can for example be extracted from onion.2,3 Hence, it is of interest to be able to extract quercetin and other flavonoids from waste and by-products since they possess antioxidative properties – studies have shown that they have positive effects against, for example, cancer4 and neurodegenerative diseases.5 These compounds are mainly present in glycosylated forms in fruits and vegetables. In onion, they mainly occur as quercetin-4′-glucoside (Q-4′) and quercetin-3,4′-diglucoside (Q-3,4′) (Fig. 1), but other types of quercetin glucosides as well as the aglycone form (Fig. 1) are also present, but at much lower concentrations.6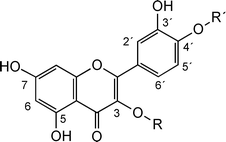 | ||
| Fig. 1 Chemical structures of quercetin (R, R′ = H), quercetin-3-glucoside (R = glucose and R′ = H), quercetin-4′-glucoside (R = H and R′ = glucose) and quercetin-3,4′-diglucoside (R, R′ = glucose). | ||
There are a number of different methods published on how to extract quercetin and quercetin glycosides from onion, of which the most commonly used technique is solid–liquid extraction with aqueous methanol as extraction solvent.6,7 The extraction of quercetin and quercetin glycosides is often combined with acid hydrolysis of the extracted glycosides. To catalyse the hydrolysis reaction, hydrochloric acid at high concentration is commonly used.7-10 Deglycosylation of the quercetin glycoside mixture obtained after extraction is advantageous for two reasons. Firstly, the glycosides are present in an extract at various concentrations, and by hydrolysing them to the quercetin aglycone, the quantitative determination is simplified. Secondly, the antioxidising power is in general higher for the deglycosylated form,11 which we have also measured for quercetin species using the DPPH (2,2-diphenyl-1-picrylhydrazyl) method (data not shown). Hence, by hydrolysing the quercetin glucoside mixture, the potential value of the material is increased. A problem with acid-catalysed hydrolysis is that the conditions are harsh, resulting in degradation of compounds such as phenolic acids in the extract, and even degradation of the target molecule quercetin.8 Another drawback is that the acidity of the sample can cause problems in the analysis step. This is commonly performed by high performance liquid chromatography (HPLC), and the HPLC-column may be damaged by the extremely low pH of the sample. To overcome these problems, enzymatic hydrolysis is an alternative.3,10,12β-Glucosidases catalyse hydrolysis of glucose linked to either a carbohydrate or non-carbohydrate moiety, and these enzymes have previously been shown to hydrolyse quercetin glucosides.3,10,13
Turner et al.3 have described a subcritical water extraction method, followed by hydrolysis by thermostable β-glucosidase from Thermotoga neapolitana that yielded quercetin in the aglycone form from a yellow onion extract. The pressurised liquid extraction with water as a solvent was run at 120 °C and 50 bar, and the following enzymatic hydrolysis in water at 90 °C and pH 5.0. This method gave the same yield as a conventional solid–liquid extraction method, although one limitation in the hydrolysis step was that the enzyme had a low conversion rate of quercetin-3-glucoside (Q-3) to the aglycone form (Q), which was speculated to be due to steric hindrance (Fig. 1). It is well known that water at elevated temperature and pressure is an efficient, less polar solvent, similar to methanol.14,15 Use of a thermostable enzyme may allow a combination of the extraction with the hydrolysis step, without extensive cooling in-between. Moreover, the hydrolysis reaction will go faster due to increased diffusion rates. Other advantages using subcritical water extraction combined with enzymatic hydrolysis compared to conventional methanol extraction and acid hydrolysis include: (i) lower environmental load, as water is a more sustainable solvent than methanol, and hence no production or destruction facilities are needed; (ii) use of the onion waste as animal feed or as feedstock in biogas production after extraction and hydrolysis is still possible, since only a small fraction of the biomass is removed, and (iii) a significantly shorter total processing time per sample.
However, it is important to not just analyse qualitative parameters, like the exchange of a solvent with another, but to also quantify the environmental load of the method. For this purpose a standardised life cycle assessment (LCA) is an excellent tool allowing quantification of primary energy consumption and emissions.16 In LCA, fluxes of materials and energy are followed from the extraction of raw materials, production and handling of the product when it has become waste. The flows are quantified and characterised to different environmental impact categories, of which Global Warming Potential (GWP) is one example. LCA can be used for several different purposes such as product and process development and for comparison of the environmental impact of different products and manufacturing processes. LCA is standardised according to ISO 14044.17
A long-term aim of our research is to develop a sustainable on-line extraction-biocatalysis system with high efficiency that can be used directly at the site of an industry that produces the waste, or alternatively at a plant taking care of waste products. As a first step, the aim of this study was to optimise the hydrolysis reaction with quercetin glucosides at an analytical level, and to improve the conversion of Q-3 to quercetin. A mutated β-glucosidase (TnBgl1A_N221S) was utilised in the hydrolysis and compared to the wild type (wt) enzyme, TnBgl1A. The optimised subcritical water extraction and enzymatic hydrolysis method was subsequently applied to a yellow onion extract, and the environmental performance of the developed processes, including greenhouse gas performance and energy efficiency, was assessed from a life cycle perspective.
Results and discussion
This paper focuses on use of the thermostable β-glucosidase, wt TnBgl1A, and a mutated variant of the same enzyme (termed TnBgl1A_N221S), as catalysts for deglucosylation of quercetin glucosides in a high-temperature step, coupled off-line to an environmentally sustainable, pressurised hot water method to extract the quercetin species. Previous work showed that the wild type, TnBgl1A, and a differently folded glucosidase (TnBgl3B), both efficiently hydrolysed the major quercetin glucoside in onion extracts, glucosylated at the 4′-position of the quercetin molecule (Q-4′).3 However, the Q-3 in the extract was not efficiently deglucosylated under the conditions used.3 To further investigate the possibility of the enzyme to hydrolyse quercetin species glucosylated at the 3-position (Fig. 1), this work involves deglucosylation studies of standards of quercetin glucosides (Q-4′, Q-3,4′, and Q-3, see Fig. 1) over time with a special emphasis on Q-3, using both the wt and a variant of TnBgl1A mutated at two positions (residues 221 (N→S) and 342 (P→L). Only one (residue 221) of the two mutated residues was judged to be important for specificity, as it is located at the substrate binding site (+2 sub-site), while the other (residue 342) is found on the surface on the reverse side (opposite the active site) of the enzyme.18 This variant was chosen from a set of mutants, as it both showed the highest turnover of the substrate para-nitrophenyl-β-D-glucopyranoside (pNPG), and based on a simple screening experiment, also higher conversion of Q-3 to quercetin.18 Finally, the mutated variant is utilized coupled with subcritical water extraction of onion waste, and the environmental performance of the method is evaluated.HPLC-UV analysis
In order to follow the hydrolysis process, high selectivity in the detection method is needed. Direct spectrophotometric detection could be employed if (for example) in the hydrolysis of pNPG to p-nitrophenol (pNP), the substrate and products have clearly different absorption maxima. For quercetin and quercetin glucosides there is no such difference in absorption maxima, and a separation step prior to detection with UV is necessary (although this is time-consuming).The number of samples to be analysed with HPLC-UV is high, and to minimise the analysis time of each sample an isocratic elution profile was selected, with methanol–water (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) containing 0.13 M (0.5 vol%) formic acid as mobile phase. The isocratic elution profile used resulted in base-line separation of the analytes: Q-3,4′; Q-3; Q-4′; and quercetin (see Fig. 5b).
:50) containing 0.13 M (0.5 vol%) formic acid as mobile phase. The isocratic elution profile used resulted in base-line separation of the analytes: Q-3,4′; Q-3; Q-4′; and quercetin (see Fig. 5b).
Hydrolysis of quercetin glucosides by wt TnBgl1A
In Fig. 2a,b, the relatively fast conversion of Q-4′ by wt TnBgl1A is clearly shown. Using a 1:100 enzyme:substrate molar ratio, Q-4′ was completely converted to Q within 5 min, while the same conditions for Q-3 resulted in only 70% conversion within 60 min. Enzymatic hydrolysis of the double glucosylated form (Q-3,4′), also showed that the 4′-glucosylation was completely hydrolysed within the 5 min interval, after which the remaining Q-3 was slowly deglucosylated to yield approximately 70% in the aglycone form (Fig. 2c). This shows that hydrolysis of the 4′-glucosylation is clearly favoured by wt TnBgl1A. Moreover, presence of Q-3 is not disturbing the deglucosylation at the 4′-position, and finally hydrolysis of Q-3 is possible but slow, which may be due to low affinity or steric hindrance for efficient binding of Q-3 in the active site. The homologous human β-glucosidase (hCBG), which has high specificity for Q-4′ and quercetin-7-glucoside, does not hydrolyse 3-linked flavonoid glucosides,19 hence hydrolysis of this substrate by TnBgl1A suggests an interesting difference in the catalytic site.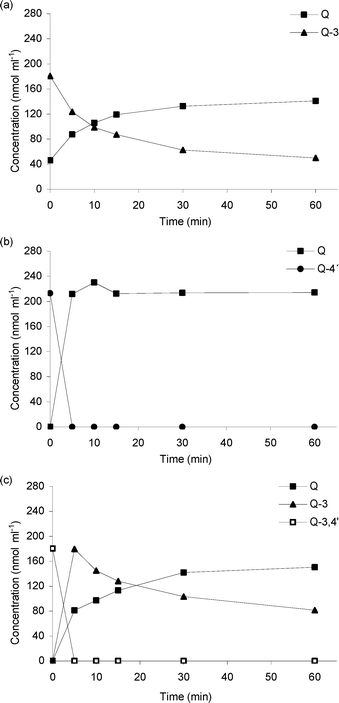 | ||
| Fig. 2 Catalysis by TnBgl1A with (a) Q-3, (b) Q-4′ and (c) Q-3,4′. 20 pmol of TnBgl1A and 200 nmol of substrate were used. Relative standard deviation (RSD) of the concentration values at the different sampling time points ranges between 0.3 and 20.6% (n = 3). | ||
Thermal stability and solubility of quercetin species
Self-hydrolysis of quercetin glucosides at high temperature was studied, and could be excluded since incubations of the quercetin glucosides at 95 °C for 1 h (the highest expected temperature for the enzyme catalysis), showed no thermal degradation of the glucosides (data not shown), and the biocatalyst was hence necessary.A drawback with production of the more potent antioxidant Q (the deglucosylated form), is a decreased water solubility, and initial hydrolysis data showed that this decrease led to losses of the produced quercetin (data not shown). To avoid this, solubility tests were made, and the results revealed that concentrations of maximum 25 nmol ml−1 of quercetin were soluble in the buffer at 95 °C. The highest concentration of quercetin species used in the remaining work was thus limited to 17 nmol ml−1, to ensure full solubility of the produced quercetin.
Enzymatic hydrolysis of the different quercetin glucosides
To obtain quantitative data, a number of enzyme:substrate ratios for the wt TnBgl1A and mutated variant, TnBgl1A_N221S, were tested, using the substrates, Q-3 and Q-4′, to find optimal ratios for determination of kinetic parameters. The detection method used requires sampling followed by quantification; hence the challenge was to find ratios where the initial reaction is neither too fast nor too slow. Indeed, the obtained results showed that different enzyme:substrate ratios were necessary for different enzyme/substrate combinations: for Q-3, 1:250-1250 and 1:2500-12500 for wt TnBgl1A and TnBgl1A_N221S, respectively, and for Q-4′, 1:2500-12500 and 1:5000-250000 for wt TnBgl1A and TnBgl1A_N221S, respectively. The higher concentration required of the wt was in accordance with the higher turnover and specific activity reported for TnBgl1A_N221S (1700 s−1 at 80 °C) as compared to the wt (485 s−1 at 80 °C) for hydrolysis of pNPG.18In Table 1 all kinetic data are summarised. As shown in Fig. 1, the glucose on Q-4′ is situated at a position resulting in an extended molecule, while for Q-3 the glucose is in the middle of the flavonoid backbone resulting in a different overall shape, which may be sterically more difficult to accommodate in the active site. This is reflected in the higher Km of Q-3 than for Q-4′. Furthermore, the Km (Michaelis–Menten constant) value of TnBgl1A_N221S is lower for both Q-3 and Q-4′ compared to the wt (Table 1), which indicates that the mutation has increased the affinity for both quercetin substrates. The effect is, however, especially pronounced for Q-3. The smaller size of the serine residue at position 221 may have reduced the steric hindrance of this molecule, leading to a better fit in the binding site resulting in higher catalytic efficiency. The maximal conversion rate (Vmax) and the turnover number (kcat) were in the same range for all enzyme and quercetin-substrate combinations, but as a consequence of the change in Km the catalytic efficiency (kcat/Km) was higher for the mutant (see Table 1).
| Enzyme | Substrate | Km/mM | Vmax/μmol min−1 mg−1 | kcat/s−1 | (kcat/Km)/s−1 M−1 |
|---|---|---|---|---|---|
| TnBgl1A | Q-3 | 0.129 ± 0.059 | 8.3 ± 2.1 | 7.2 | 55 |
| Q-4′ | 0.059 ± 0.026 | 8.3 ±1.4 | 7.2 | 122 | |
| TnBgl1A_N221S | Q-3 | 0.046 ± 0.020 | 8.1 ±1.3 | 7.1 | 154 |
| Q-4′ | 0.022 ± 0.015 | 7.1 ± 1.1 | 6.2 | 281 |
Despite the improved affinity for Q-3 using TnBgl1A_N221S, experiments with this variant at 95 °C showed a hydrolysis pattern with decreased deglucosylation rate at increasing incubation times (Fig. 3, 0–30 min). A second addition of the enzyme after 30 min restarted the reaction (Fig. 3, 30–65 min). To understand if this was a result of deactivation or product inhibition, thermal deactivation experiments of the enzyme in the absence of substrate, as well as incubations of the enzyme in the presence of products, were made (see below).
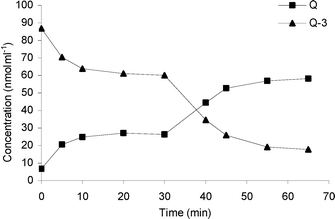 | ||
| Fig. 3 A second addition of 20 pmol TnBgl1A_N221S 35 min after the first addition of 20 pmol TnBgl1A_N221S. RSD of the concentration values at the different sampling time points ranges between 0.4 and 16.9% (n = 3). | ||
Thermal stability of the β-glucosidases TnBgl1A and TnBgl1A_N221S
The occurrence of irreversible thermal deactivation at 85, 90 and 95 °C was investigated to establish the tolerance of the enzymes to prolonged exposure to these temperatures (Table 2). Almost no enzymatic activity was lost for wt TnBgl1A during the first 30 min of incubation at 85 and 90 °C. Prolonged incubations up to 24 h fitted a first-order deactivation rate during the complete incubation period, resulting in a deactivation constant (k) of 0.027 h−1 (at 90 °C). The half-life of the enzyme (t1/2) could thus be calculated to exceed 10 h at 90 °C. A further 5 °C increase in temperature led to significant activity loss, giving a t1/2 of 28 min (0.46 h, Table 2). The mutant TnBgl1A_N221S showed a slightly higher deactivation rate, but both the wt and mutant were judged to be stable at temperatures up to 90 °C (t1/2 of the mutant was approximately 6 h).| T/°C | TnBgl1A | TnBgl1A_N221S | ||
|---|---|---|---|---|
| k/h−1 | t1/2/h | k (h−1) | t1/2/h | |
| 85 | 0.018 | 20 | 0.029 | 7.0 |
| 90 | 0.027 | 11 | 0.040 | 6.4 |
| 95 | 0.73 | 0.46 | 1.51 | 0.23 |
A larger difference was seen at 95 °C, where the deactivation constant (k) for the mutant was doubled, and its half-life reduced to 13 min (0.23 h, Table 2). Thermal transitions by differential scanning calorimetry have shown the apparent transition temperature (Tm) from the folded to unfolded state at 102.5 °C (wt) and 100.5 °C (mutant), respectively.18 However, inspection of the chromatograms showed that the starting point of the transition occurred around 95 °C (data not shown), explaining the reduced stability at this temperature. This sets a limitation in the time frame applicable for the hydrolysis reaction if a temperature of 95 °C is used, and should only be applied if the overall reaction time is short (≤10 min). This relatively high temperature was used in the kinetic study and the total reaction time was less than 5 min. A temperature of 95 °C was also used in the hydrolysis of the onion extract (with a total reaction time of 5 min) to minimise the temperature difference between extraction and hydrolysis temperatures in the process.
However, in longer studies of the deglucosylation, deactivation will be significant at temperatures above 90 °C. This means that the slow deglucosylation rate of Q-3 seen within the 30 min interval for the wt cannot be a consequence of deactivation, as this was run at 90 °C (Fig. 2), while in the reaction performed with the mutated variant at 95 °C, deactivation should be significant (Fig. 3).
Activation/inhibition of the enzyme
An experiment was set up to evaluate if the mutated enzyme was product-inhibited. In this experiment, glucose and quercetin, respectively, were added to TnBgl1A_N221S 18 min prior to adding a hydrolysable substrate (in this case Q-4′). Product inhibition acts on the enzyme, and the presence of substrate can hence be used to detect if inhibition has occurred after the pre-incubation. To avoid thermal deactivation, the temperature was set to 90 °C. The results in Fig. 4 show that most of the Q-4′ was hydrolysed after 10 min with either quercetin or the control (just the buffer). Interestingly, incubations in the presence of glucose led to complete Q-4′ hydrolysis after just 5 min. The conclusion is that there is no inhibition effect by either quercetin or glucose on the enzyme, but rather that glucose is an activator of the enzyme. An activating effect of glucose on the β-glucosidases from the glycoside hydrolase family 1 (GH1) is in accordance with results reported by other researchers,20-22 and has previously been reported for wt TnBgl1A.18,21 This work shows that the activating effect of glucose also works for the mutated variant of the enzyme.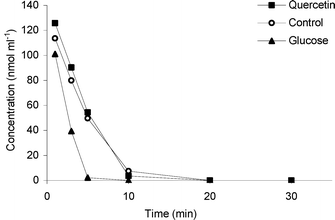 | ||
| Fig. 4 The effect of incubation of TnBgl1A_N221S with glucose, quercetin and buffer (control) on the hydrolysis of Q-4′. RSD of the concentration values at the different sampling time points ranges between 0.3 and 6.1% (n = 3). | ||
The activating effect of glucose may also explain the improved conversion of Q-3 to Q seen after the second addition of TnBgl1A_N221S (Fig. 3), where glucose released after the first enzyme addition is present in the reaction mixture. Currently, one can only speculate that glucose may prevent non-productive binding of the Q-3 or Q-4′ substrate, for example by blocking a high affinity sub-site. To obtain proof of this suggestion, detailed studies on the binding affinity of the subsites of TnBgl1A will be necessary.
Application to an onion sample
Whole yellow onion was chopped and both the skin and the pulp was mixed and extracted using subcritical water at 120 °C and 50 bar as solvent. The result of the HPLC-UV analysis of the extract was that seven flavonoids were identified in the yellow onion extract – Q-4′, Q-3, Q-3,4′, quercetin, isorhamnetin-4′-glucoside (I-4′), isorhamnetin-3,4′-diglucoside (I-3,4′) and isorhamnetin. Quercetin and the three quercetin glucosides were identified by comparing the retention time and elution order with standards, and isorhamnetin, I-4′ and I-3,4′ were identified based on the results by Turner et al.3 One of the peaks labelled “unknown” is most likely kaempferol, and this assumption is based on a previous study.3 Q-4′ and Q-3,4′ are the most abundant flavonoids identified in the yellow onion extract and this corresponds well to other publications of flavonoid content in yellow onion.3,6 Three different amounts of TnBgl1A_N221S were tested (20 pmol, 100 pmol and 3 nmol) (data not shown) resulting in the following method optimised for 1.2 ml of onion extract corresponding to 0.4 g of onion (50 mg dw); 3 nmol of TnBgl1A_N221S, reaction temperature of 95 °C, pH 5.0 and 5 min reaction time. Chromatograms of yellow onion extracts before and after enzymatic treatment at 95 °C can be seen in Fig. 5b and 5c. In Fig. 5a the amounts of quercetin and quercetin glucosides have been plotted as a function of sampling time points. Almost all the Q-4′ and Q-3,4′ have been hydrolysed after 1 min, while Q-3 concentration has increased, and after 5 min only trace amounts of glucosides are detected (Fig. 5c). Within the few minutes it takes to conduct the reaction of the yellow onion extract, TnBgl1A_N221S maintains its activity, even though the reaction is done at 95 °C. The quercetin aglycone concentration increased at the same rate as the concentration of glucosides decreased. The increase in Q-3 concentration (after 1 min) shows that the presence of the glucosylation at this position does not limit the activity of the enzyme, but that accommodation of the substrate favours hydrolysis of glucose bound at the 4′-position. By comparing the peaks in Fig. 5b and 5c it is seen that after 5 min of reaction no I-4′ or I-3,4′ peaks are detected, but instead an isorhamnetin peak can be found. This indicates that TnBgl1A_N221S also catalyses the hydrolysis of isorhamnetin glucosides to isorhamnetin. However, to confirm if it the enzyme catalyses the reaction, a study using isorhamnetin species standards should be done.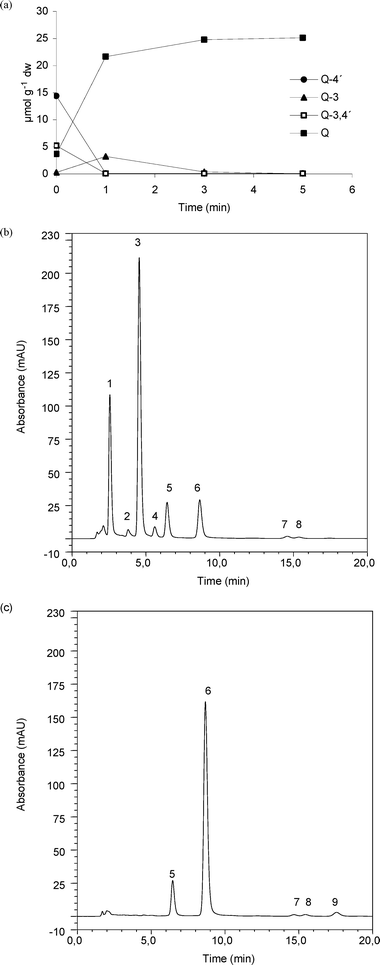 | ||
| Fig. 5 HPLC-UV analysis of a yellow onion extract before and after enzyme treatment. Concentration change of Q-4′, Q-3, Q-3,4′ and Q during enzymatic reaction (a), RSD of concentration at the different sampling time points ranges between 0.8 and 10.6%, within the calibration curve range (n = 3). HPLC-UV chromatogram of yellow onion extract before addition of enzyme (b) and 5 min after addition of TnBgl1A_N221S (c). Peak labels; 1 (mix of Q-3,4′ and I-3,4′), 2 (Q-3), 3 (Q-4′), 4 (I-4′), 5 (morin (IS)), 6 (Q), 7-8 (unknowns) and 9 (isorhamnetin). | ||
Environmental performance
The results from the environmental impact assessment are shown in Table 3 and 4. The traditional method with organic solvents leads to a significantly higher contribution to the GWP than the extraction with subcritical water. This is mainly due to the use of fossil methanol as a solvent, but also due to the larger amount of solvents needed per gramme of onion for the traditional method. Although clear differences in energy consumption can be seen for the two methods, energy for heating gives relatively small contributions to the overall environmental impact. The major input of primary energy is in the manufacture of HCl and methanol.| Extraction method | Heating/MJ | Extraction/kJ | Total energy/MJ | GWP (kg CO2-eq) |
|---|---|---|---|---|
| Traditional method | 13 (9.3–25) | 70 (60–80) | 13 (9.4–25) | 1.3 (0.94–2.5) |
| New method | 8.4 (4.2–15) | 11 (8–13) | 8.4 (4.2–15) | 0.85 (0.42–1.5) |
The environmental impact is summarised in Fig. 6. There are several uncertainities involved in the environmental assessment performed, such as the linear scaling-up of the laboratory methods. Furthermore, depending on the primary fuel used for producing heat (in this case natural gas), the contribution to the GWP will differ. However, despite these uncertainties, there are some “hot-spots” identified, supporting the conclusion that subcritical water extraction is more favourable. These “hot-spots” are the effect of the use of fossil-fuel-derived methanol on GWP, and in addition the effect of the use of HCl on the primary energy consumption.
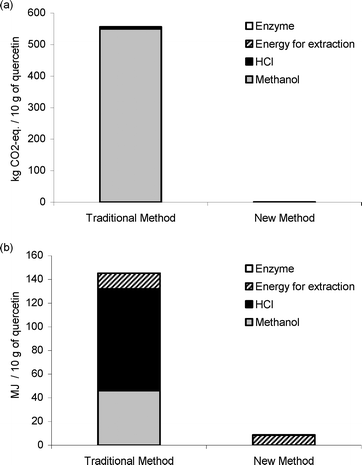 | ||
| Fig. 6 Total contribution to the GWP expressed as kg CO2-equivalents per 10 g of quercetin, for the two extraction methods (a), and as total primary energy consumption, expressed as MJ per 10 g of quercetin, for the two extraction methods (b). | ||
Experimental
Materials
Methanol and formic acid were purchased from Merck (Darmstadt, Germany). Quercetin dihydrate, quercetin 3-β-D-glucoside (Q-3), citric acid monohydrate, disodium hydrogen phosphate and pNPG were purchased from Sigma-Aldrich (Steinheim, Germany). Quercetin 4′-O-β-glucopyranoside (Q-4′) and quercetin 3,4′-di-O-β-glucopyranoside (Q-3,4′) were purchased from Polyphenols Laboratories AB (Sandnes, Norway). Ultrapure water (MilliQ) was used at all times.Enzyme production and purification
The gene encoding wt TnBgl1A was cloned in pET22b(+), under control of the T7lac-promoter, as described by Turner et al.3 The TnBgl1A_N221S was obtained by site-directed mutagenesis using megaprimer PCR.18 The fragment encoding the mutated TnBgl1A was cloned in pET22b(+) (Novagen, Madison, WI, USA) under control of the T7lac-promoter, and transformed to E. coli Bl21(DE3) for production.Both enzymes were produced in 2.5 l batch cultivations, in strain Bl21(DE3) after induction with isopropyl-1-thio-β-D-galactopyranoside (IPTG) (2 h), as described by Abou-Hachem et al.26 The cells were harvested by centrifugation (10![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000g, 10 min, 4 °C). Cell pellets were resuspended in buffer (20 mM citrate–phosphate pH 5.6 for a single step heat treatment (80 °C, 30 min, used only for the irreversible deactivation studies)/or binding buffer (20 mM Tris-HCl, 20 mM imidazole, 0.75 M NaCl, pH 7.5) for a two-step procedure involving heat treatment (70 °C, 30 min) and immobilized metal ion affinity chromatography). The cell pellets were disintegrated by sonication (60 W cm−2, 0.5 cycle, for 5 × 3 min) with a UP400S sonicator (Dr. Hielscher, Stahnsdorf, Germany) while cooling in an ice–water mixture. The sonicated extracts were centrifuged at 25000g (20 min, 4 °C), and the resulting supernatants stored at 4 °C until purified.
000g, 10 min, 4 °C). Cell pellets were resuspended in buffer (20 mM citrate–phosphate pH 5.6 for a single step heat treatment (80 °C, 30 min, used only for the irreversible deactivation studies)/or binding buffer (20 mM Tris-HCl, 20 mM imidazole, 0.75 M NaCl, pH 7.5) for a two-step procedure involving heat treatment (70 °C, 30 min) and immobilized metal ion affinity chromatography). The cell pellets were disintegrated by sonication (60 W cm−2, 0.5 cycle, for 5 × 3 min) with a UP400S sonicator (Dr. Hielscher, Stahnsdorf, Germany) while cooling in an ice–water mixture. The sonicated extracts were centrifuged at 25000g (20 min, 4 °C), and the resulting supernatants stored at 4 °C until purified.
After the heat treatment, the samples were again centrifuged (30 min at 25000g and 4 °C) for removal of denatured aggregated proteins. The supernatants obtained after heat treatment at 80 °C were used directly to study irreversible deactivation, while those treated at 70 °C were filtered (0.4 μm pore size) and further purified by immobilised metal-ion affinity chromatography (IMAC).27
Enzyme assays
The total protein concentrations were estimated using the bicinchoninic acid method (Sigma-Aldrich, Steinheim, Germany) or the Bradford protein assay (BioRad, Hercules, CA, USA) according to the manufacturers’ instructions. Bovine serum albumin (BSA) was used as standard (in concentrations from 0.2 to 1.0 mg ml−1). The absorbance was measured at 562 nm (bicinchoninic acid method) and 595 nm (Bradford method) using an Ultrospec 1000 (Pharmacia, Uppsala, Sweden). To estimate the purity of the enzyme, SDS-PAGE according to Laemmli28 was used.For thermal inactivation tests, the β-glucosidase activity was measured as the release of pNPG after 5 min incubations at 85 °C. To 960 μl preheated substrate (2.92 mM pNPG dissolved in citrate–phosphate buffer pH 5.6), 40 μl of enzyme solution was added, and the assay was performed as described in Turner et al.29 Absorbance was measured at 405 nm using Ultrospec 1000 (Pharmacia). One Unit (U) for this substrate corresponds to the amount of enzyme that will release 1 μmol p-nitrophenol per minute under the described conditions.
Enzymatic hydrolysis of quercetin standards
In an initial activity screening, 200 nmol sample of Q-3, Q-4′ and Q-3,4′ dissolved in methanol (in triplicate) were evaporated, 1.0 ml of 100 mM citrate–phosphate buffer, pH 5.0, was added and the vials were heated at 90 °C until substrate dissolved. A 50 μl fraction was collected and added to 450 μl of mobile phase composed of methanol–water (50:50) and 0.13 M (0.5 vol%) formic acid (0 min fraction in Fig. 2). The reaction took place at 90 °C and the reaction was started by adding 20 pmol of wt TnBgl1A. 50 μl fractions were collected and added to 450 μl of mobile phase 1–60 min after addition of enzyme. Samples were analysed by HPLC-UV. Collection of fractions and enzyme addition was done with a 50 μl GC-syringe. The fraction-collection procedure and enzyme addition were similar for all experiments described below, unless otherwise noted.Thermal stability of the quercetin standards
Samples (150 nmol) of Q-3, Q-4′ and Q-3,4′ dissolved in methanol were evaporated, 1.5 ml of 100 mM citrate–phosphate buffer was added (triplicate samples for each glucoside) and the vials were heated at 95 °C until substrate was dissolved. Fractions were collected 10, 30 and 60 min after the first fraction collection. Fractions were collected and diluted as above. The samples were analysed by HPLC-UV.Enzyme kinetics of the hydrolysis of Q-3 and Q-4′
To determine the kinetics of the hydrolysis reactions, 50–250 nmol samples of Q-3 and Q-4′ dissolved in methanol were evaporated and re-dissolved as described above, and the volume of buffer used was 1.5 ml. Fractions were collected at 0, 1, 3, and 5 min after enzyme addition. The enzyme amounts used were 200 pmol of wt TnBgl1A and 20 pmol of TnBgl1A_N221S to Q-3 and 20 pmol of wt TnBgl1A and 10 pmol of TnBgl1A_N221S to Q-4′. Samples were analysed by HPLC-UV. The enzyme activity using Q-3 and Q-4′ is expressed in Units (U). One U corresponds to the amount of enzyme required to release 1 μmol quercetin per minute under the described conditions. Obtained data was analysed using Enzpack (Biosoft, UK), using a time-range allowing fit to the Michaelis–Menten curve. Kinetic constants were calculated using the Wilkinson non-linear regression method, supplied in the program.Repeated enzyme additions at the applied conditions
A sample of Q-3 (triplicate of 150 nmol) in methanol was evaporated and re-dissolved as above at 95 °C. Fractions were collected at 0, 5, 10, 20 and 30 min after addition of 20 pmol TnBgl1A_N221S. 35 min after the first enzyme addition, the same amount of TnBgl1A_N221S was added and fractions were collected 40, 45, 55 and 65 min after the first enzyme addition. Samples were analysed by HPLC-UV.Thermal stability of the enzymes in absence of substrate
Thermal stability of the enzymes was measured as irreversible deactivation. The heat-treated wt (TnBgl1A, 1.5 mg ml−1, 29 μM) and mutant (TnBgl1A_N221S, 2.0 mg ml−1, 38 μM) glucosidase were aliquoted into microcentrifuge tubes. These samples were incubated at 85, 90 and 95 °C, at time intervals ranging from 0 to 24 h. During the first 30 min, duplicate samples were withdrawn every 5 min, then samples were taken after 1, 2, 3, 4, 5, 18 and 24 h, and were chilled on ice (at least 30 min), centrifuged (13000g, 30 min, 4 °C) and stored at 4 °C until activity analysis was performed. The inactivation kinetics of the different constructs was analysed assuming a first-order reaction rate. For a constant temperature:| A/A0 = e−kt | (1) |
| lnA− lnA0= −kt | (2) |
Product inhibition studies
Quercetin samples (triplicates of 100 nmol each) in methanol were evaporated and re-dissolved in 750 μl of 100 mM citrate–phosphate buffer pH 5.0, and the vials were heated at 90 °C until the quercetin had dissolved. Glucose (triplicates of 100 nmol) dissolved in 750 μl 100 mM citrate–phosphate buffer, pH 5.0, and controls containing 750 μl of 100 mM citrate–phosphate buffer pH 5.0, (triplicate) were also heated at 90 °C, and treated in the same way as the quercetin samples. TnBgl1A_N221S (10 pmol) was added to the vials and the samples were heated at 90 °C. Then, 18 min after addition of enzyme, pre-heated 100 nmol of Q-4′ dissolved in 750 μl of 100 mM citrate–phosphate buffer, pH 5.0, was added to the vials. Fractions were collected as above, 1, 3, 5, 10, 20 and 30 min after addition of Q-4′. The first point in Fig. 4 is the 1 min fraction. Samples were analysed by HPLC-UV.Subcritical water extraction
15–18 g of onion samples, in triplicate, were weighed into 33 ml stainless steel extraction cells containing a cellulose filter at the bottom. Extractions were performed on a Dionex ASE®-200 subcritical water extraction system (Dionex, Sunnyvale, CA, USA) using water as the only solvent as described by Turner et al.3 The pressure was set to 50 bar and the initial heating lasted for 5 min. Extraction temperature was 120 °C and extraction time 15 min (three extraction cycles of 5 min each). Purging between extractions was performed with nitrogen. The extract was collected in 100 ml clear glass vials, cooled down to room temperature and diluted with degassed water to a total volume of 50 ml.Enzymatic hydrolysis of onion extract
A 1.2 ml aliquot of onion extract, prepared by subcritical water extraction, was mixed with 300 μl of 100 mM citrate–phosphate buffer, pH 5.0, and heated at 95 °C until the sample components dissolved. Fractions were collected at 0, 1, 3, 5 and 10 min after addition of 3 nmol of TnBgl1A_N221S. Samples were analysed by HPLC-UV.HPLC analysis
HPLC-UV analysis was performed using the chromatographic system UltiMate 3000 from Dionex (Germering, Germany). An Agilent Zorbax SB-C18 column (100 × 2.1 mm, 3.5 μm) was used for isocratic separation with a methanol–water (50:50) and formic acid (0.13 M, 0.5 vol%) mobile phase at a flow rate of 0.15 ml min−1. The injection volume was 10 μl and detection was accomplished at 350 nm. Quantification of quercetin and its glucosides was performed using a five-point calibration curve of a quercetin dihydrate, Q-3, Q-4′ and Q-3,4′ standards at concentrations between 0.5 and 25 μg ml−1. Each vial taken to analysis had a total volume of 500 μl.Environmental impact assessment
The global warming potential (GWP) and primary energy consumption for the new and a traditional extraction method were assessed from a gate-to-gate perspective.16,30 The calculations are based on a functional unit of 10 g of quercetin. The new, subcritical water extraction method is described by Turner et al.3 (see Subcritical water extraction, above), while the enzymatic reaction method is described in this study (see Enzymatic hydrolysis of onion extract, above). The traditional method using methanol extraction and HCl-catalysed hydrolysis (80 °C for 2 h) is described by Hertog et al.7 and Nuutila et al.8 Extractions were scaled up linearly to produce 10 g of quercetin. The dry matter content in onion is around 13.5% and the quercetin content in onion is, on average, 13 mg g−1 dry matter, varying between 7 and 25 mg g−1 (see Table 5).3| Extraction method | Onion/kg | Amount of solvent/kg | Amount of catalyst/kg | Total volume (dm3) |
|---|---|---|---|---|
| Traditional method | 5.7 (3.0–10.6) | Methanol: 31 (16–56) | HCl: 3.2 (1.7–5.9) | 87 (41–150) |
| New method | 5.7 (3.0–10.6) | Water: 13 (6.5–23) | Enzyme: 0.003 (0.002–0.006) | 21 (9.4–33) |
Energy demand is calculated for heating and heat losses during extraction according to Welty et al.31 Extraction is set to be carried out in vessels of stainless steel insulated with mineral wool. Production of extraction equipment and generation of pressure are not considered in this study. A natural gas burner with 60% efficiency is set to be used for heating. Emissions due to production and combustion of natural gas are from Uppenberg et al.32 The environmental impact of water is assumed to be insignificant.
Conclusions
In this paper, we have developed analytical methods to study the kinetics of a thermostable β-glucosidase (wt TnBgl1A), and a mutant of the same enzyme (TnBgl1A_N221S) towards Q-3 and Q-4′. We have also developed an off-line biocatalysis method, in which the hydrolysis of all quercetin glucosides present in yellow onion extract is completed within 5 min at 95 °C using only 3 nmol of enzyme per 1.3 μmol of quercetin species. This study shows that TnBgl1A_N221S has higher activity towards both Q-3 and Q-4′ compared to the wt TnBgl1A, i.e. a mutation of residue 221 at the +2 binding site of the enzyme does significantly affect substrate affinity, but this effect is higher for the Q-3 substrate. Another conclusion that can be drawn is that deactivation is significant at 95 °C, and to avoid this, the temperatures should be kept below 95 °C, if reactions are more than a few minutes long. To increase the hydrolysis rate of quercetin glucosides, addition of glucose can be considered. Future aspects include immobilisation of TnBgl1A_N221S onto a support material allowing recycling of the enzyme, and the use of an on-line flow system of hot water extraction and enzymatic hydrolysis of quercetin and quercetin glucosides from yellow onion waste and other potential by-products and waste. The immobilisation of the enzymes may also improve the thermostability of the enzymes.From an environmental point of view, extraction with subcritical water followed by an enzymatic hydrolysis is to be preferred regarding primary energy consumption and global warming potential. Shorter extraction time and smaller volumes makes the process more energy-efficient, although a higher temperature and pressure have to be generated. Furthermore, avoiding the use of organic solvents from fossil origin, such as methanol, makes the subcritical water extraction more resource efficient and favourable from a greenhouse gas perspective. Handling of large quantities of toxic chemicals, such as HCl and methanol, is also avoided.
Abbreviations
| Q | Quercetin |
| Q-3 | Quercetin-3-glucoside |
| Q-4′ | Quercetin-4′-glucoside |
| Q-3,4′ | Quercetin-3,4′-diglucoside |
| pNPG | p-Nitrophenyl-β-D-glucopyranoside |
| pNP | p-Nitrophenol |
| BSA | Bovine serum albumin |
| GWP | Global warming potential |
| LCA | Life cycle assessment |
| I-4′ | Isorhamnetin-4′-glucoside |
| I-3,4′ | Isorhamnetin-3,4′-diglucoside |
| IPTG | Isopropyl-1-thio-β-D-galactopyranoside |
| dw | Dry weight |
| DPPH | 2,2-Diphenyl-1-picrylhydrazyl |
| RSD | Relative standard deviation |
| IMAC | Immobilised metal-ion affinity chromatography |
| wt | Wild type |
| Tm | Transition temperature |
| GH1 | Glycoside hydrolase family 1 |
Acknowledgements
The Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS, 2006-1346); the Swedish Research Council (VR, 2006-4084 and 2006-6048); the Swedish International Development Cooperation Agency (SIDA, SWE-2005-473); the Swedish Foundation for Strategic Research (SSF, 2005:0073/13) and the Foundation for Strategic Environmental Research (MISTRA, Greenchem) are greatly acknowledged.References
- A. Schieber, F. C. Stintzing and R. Carle, Trends Food Sci. Technol., 2001, 12, 401–413 CrossRef CAS.
- M. G. L. Hertog, P. C. H. Hollman and M. B. Katan, J. Agric. Food Chem., 1992, 40, 2379–2383 CrossRef CAS.
- C. Turner, P. Turner, G. Jacobson, K. Almgren, M. Waldeback, P. Sjöberg, E. N. Karlsson and K. E. Markides, Green Chem., 2006, 8, 949–959 RSC.
- A. Murakami, H. Ashida and J. Terao, Cancer Lett., 2008, 269, 315–325 CrossRef CAS.
- K. Ono, T. Hamaguchi, H. Naiki and M. Yamada, Biochim. Biophys. Acta, Mol. Basis Dis., 2006, 1762, 575–586 CrossRef CAS.
- K. R. Price and M. J. C. Rhodes, J. Sci. Food Agric., 1997, 74, 331–339 CrossRef CAS.
- M. G. L. Hertog, P. C. H. Hollman and D. P. Venema, J. Agric. Food Chem., 1992, 40, 1591–1598 CrossRef CAS.
- A. M. Nuutila, K. Kammiovirta and K. M. Oksman-Caldentey, Food Chem., 2002, 76, 519–525 CrossRef CAS.
- S. H. Hakkinen, S. O. Karenlampi, I. M. Heinonen, H. M. Mykkanen and A. R. Torronen, J. Sci. Food Agric., 1998, 77, 543–551 CrossRef CAS.
- J. B. Harborne, Phytochemistry, 1965, 4, 107–120 CrossRef CAS.
- Y. K. Do, J. M. Kim, S. M. Chang, J. H. Hwang and W. S. Kim, in International Conference on Biorefinery, Elsevier Science Bv, Beijing, JAPAN, 2007, pp. 173-178 Search PubMed.
- F. Siewek and R. Galensa, J. Chromatogr., A, 1984, 294, 385–389 CrossRef CAS.
- N. Lambert, P. A. Kroon, C. B. Faulds, G. W. Plumb, W. R. McLauchlan, A. J. Day and G. Williamson, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1999, 1435, 110–116 Search PubMed.
- E. Ibanez, A. Kubatova, F. J. Senorans, S. Cavero, G. Reglero and S. B. Hawthorne, J. Agric. Food Chem., 2003, 51, 375–382 CrossRef CAS.
- K. Hartonen, K. Inkala, M. Kangas and M. L. Riekkola, J. Chromatogr., A, 1997, 785, 219–226 CrossRef CAS.
- A. Azapagic, Handbook of Green Chemistry and Technology, Blackwell Science, Oxford, 2002, 62-85 Search PubMed.
- I. S. Organization, Enviromental management - Life cycle assesment - Requirements and guidelines, ISO 14044, 2006 Search PubMed.
- S. Khan, T. Pozzo, C. Paul, S. Lindahl, C. Turner, and E. Nordberg Karlsson, 2009, unpublished work.
- J. G. Berrin, W. R. McLauchlan, P. Needs, G. Williamson, A. Puigserver, P. A. Kroon and N. Juge, Eur. J. Biochem., 2002, 269, 249–258 CrossRef CAS.
- R. M. Wright, M. D. Yablonsky, Z. P. Shalita, A. K. Goyal and D. E. Eveleigh, Appl. Environ. Microbiol., 1992, 58, 3455–3465 CAS.
- D. A. Yernool, J. K. McCarthy, D. E. Eveleigh and J. D. Bok, J. Bacteriol., 2000, 182, 5172–5179 CrossRef CAS.
- J. A. Perezpons, X. Rebordosa and E. Querol, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1995, 1251, 145–153 Search PubMed.
- Å. Furnander, Life Cycle Assessment of Dimethyl Ether as a Motor Fuel, Chalmers University of Technology, Gothenburg, 1996 Search PubMed.
- I. Boustead, Eco-Profiles of the European Plastic Industry - Hydrogen Chloride, PlasticEurope - Association of Plastics Manufacturers, 2005 Search PubMed.
- P. H. Nielsen, K. M. Oxenboll and H. Wenzel, Int. J. Life Cycle Assess., 2007, 12, 432–438 CrossRef CAS.
- M. Abou-Hachem, F. Olsson and E. N. Karlsson, Extremophiles, 2003, 7, 483–491 CrossRef CAS.
- P. Turner, D. Svensson, P. Adlercreutz and E. N. Karlsson, J. Biotechnol., 2007, 130, 67–74 CrossRef CAS.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CAS.
- P. Turner, A. Pramhed, E. Kanders, M. Hedstrom, E. N. Karlsson and D. T. Logan, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2007, 63, 802–806 Search PubMed.
- A. E. V. Petersson, L. M. Gustafsson, M. Nordblad, P. Borjesson, B. Mattiasson and P. Adlercreutz, Green Chem., 2005, 7, 837–843 RSC.
- J. R. Welty, C. H. Wicks, R. E. Wilson and G. Rorrer, Fundamentals of momentum, Heat and Mass Transfer, John Wiley & Sons, Inc., 2001, 201-218 Search PubMed.
- S. Uppenberg, M. Almemark, M. Brandel, L.-G. Lindfors, H.-O. Marcus, H. Stripple, A. Wachtmeister and L. Zetterberg, Miljöfaktabok för Bränslen Del 2, IVL Swedish Enviromental Research Institute Ltd., 2001 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |