Perfluoro-tagged, phosphine-free palladium nanoparticles supported on silica gel: application to alkynylation of aryl halides, Suzuki–Miyaura cross-coupling, and Heck reactions under aerobic conditions†
Roberta Berninia, Sandro Cacchi*b, Giancarlo Fabrizib, Giovanni Fortec, Francesco Petruccic, Alessandro Prastaroa, Sandra Niembrod, Alexandr Shafird and Adelina Vallribera*d
aDipartimento A. B. A. C., Università della Tuscia e Consorzio Universitario “La Chimica per l'Ambiente”, Via S. Camillo De Lellis, 01100, Viterbo, Italy
bDipartimento di Chimica e Tecnologie del Farmaco, Sapienza, Università di Roma, P.le A. Moro 5, 00185, Rome, Italy. E-mail: sandro.cacchi@uniroma1.it; Fax: +39 (06) 4991 2780; Tel: +39 (06) 4991 2795
cIstituto Superiore di Sanità, Viale Regina Elena 299, 00161, Rome, Italy
dDepartment of Chemistry, Universitat Autònoma de Barcelona, Cerdanyola, 08193, Spain. E-mail: adelina.vallribera@uab.cat; Tel: +34 93 581 3045
First published on 29th October 2009
Abstract
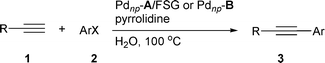
The utilization of perfluoro-tagged palladium nanoparticles immobilized on fluorous silica gel through fluorous–fluorous interactions (Pdnp–A/FSG) or through covalent bonding to silica gel (Pdnp–B) in the alkynylation of aryl halides, in the Suzuki–Miyaura cross-coupling, as well as in the Heck reaction between methyl acrylate and aryl iodides is described. The reactions are carried out under aerobic and phosphine-free conditions with excellent to quantitative product yields in each case. The catalysts are easily recovered and reused several times without significant loss of activity. The alkynylation of aryl halides (under copper-free conditions) and the Suzuki–Miyaura cross-coupling are carried out in water. The Heck reaction of methyl acrylate with aryl iodides is best performed in MeCN. The utilization of Pdnp–B in the synthesis of 2,3-disubstituted indoles from 2-(alkynyl)trifluoroacetanilides and aryl halides is also reported.
Introduction
Palladium catalysis has achieved an important place in the arsenal of the practising organic chemist.1 Palladium catalysts are expensive, however, and this may limit their utilization in some cases. Furthermore, their use might result in palladium contamination of the desired isolated product, a significant problem for the pharmaceutical industry, which has to meet strict specifications to limit the presence of heavy metal impurities in active substances.2 Immobilized palladium catalysts3 can provide a way to overcome these problems by allowing the palladium species to be separated, recovered, and reused, thus reducing palladium contamination of the isolated products. This approach would be exceedingly convenient in industrial applications as well as in cases when the reactions are carried out in multiple vessels for library generation.A number of studies have been devoted to immobilizing palladium on inert support materials, including activated carbon, silica gel, polymers containing covalently-bound ligands, metal oxides, porous aluminosilicates, clays and other inorganic materials, as well as microporous and mesoporous supports.4 Palladium has been microencapsulated in a polymeric coating,5 an efficient and cost-effective technique to ligate and retain palladium species. Aerogels, a new class of porous solids obtained via sol-gel processes coupled with supercritical drying of wet gels, have also been shown to exhibit a great potential for the preparation of heterogeneous catalysts.6
Recently, palladium nanoparticles have been shown to be stabilized by entrapment in perfluoro-tagged, phosphine-free compounds.7 This finding was somewhat unexpected, given that heavily fluorinated compounds are not expected to be the best constituents of protecting shields for nanoparticles (perfluorinated chains are indeed known to exhibit very small attractive interactions toward other materials and among themselves8). Thus, we were intrigued by the idea of immobilizing phosphine-free, perfluoro-tagged palladium nanoparticles on solid supports9 and evaluating the utilization of the immobilized catalyst in C–C bond forming reactions. A nice example of immobilizing perfluoro-tagged palladium has been reported by Bannwarth et al.10 who prepared several catalysts via adsorption of palladium(II) complexes containing perfluorinated phophine ligands on fluorous silica gel (FSG) and showed the advantages of their utilization (separation and recovery of perfluoro-tagged palladium) in the Suzuki–Miyaura cross-coupling reaction in comparison with fluorous biphasic catalysis approaches using expensive and environmentally-persistent perfluorinated solvents. Phosphines, however, are often air-sensitive. In that sense, interesting results have been achieved in some cases by employing more efficient phosphines.11 Such phosphines are not readily available, however, and some limits to their use in large-scale applications still remain. The development of efficient phosphine-free catalysts would provide obvious advantages in many synthetic applications. In this context, we observed and previously communicated that immobilized phosphine-free, perfluoro-tagged palladium nanoparticles can be extremely efficient in catalyzing the alknylation of aryl halides (with palladium nanoparticles immobilized through covalent bonding to silica gel)12 and the Heck reaction (with palladium nanoparticles immobilized on fluorous silica gel through fluorous–fluorous interactions13 as well as through covalent bonding to silica gel14).
We wish at this time to report full details of the utilization of this new immobilized phosphine-free palladium system in these two C–C bond-forming reactions. The alkynylation of aryl halides and the Heck reaction have been extended to a larger number of substrates. The utilization of immobilized phosphine-free, perfluoro-tagged palladium nanoparticles through covalent bonding to silica gel in the Heck reaction as well as in the synthesis of 2,3-disubstituted indoles from 2-(alkynyl)trifluoroacetanilides and aryl halides have also been investigated. Furthermore, perfluoro-tagged palladium nanoparticles immobilized on fluorous silica gel through fluorous–fluorous interactions and through covalent bonding to silica gel have been used in the Suzuki–Miyaura cross-coupling.
Results and discussion
Preparation of phosphine-free, perfluoro-tagged palladium nanoparticles immobilzed on FSG (Pdnp–A/FSG) and covalently bound to silica gel (Pdnp–B)
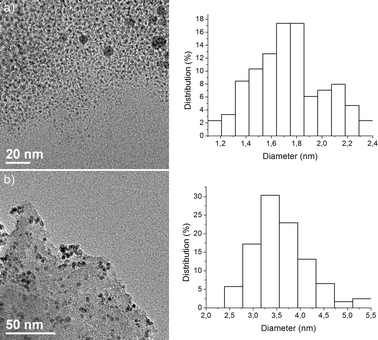
Phosphine-free, perfluoro-tagged palladium nanoparticles Pdnp–A (diameter 2.3 ± 0.7 nm; 13.4% palladium) were prepared as described previously for similar systems7e by reduction of Na2Pd2Cl6 (prepared in situ from PdCl2 and NaCl) with methanol at 60 °C in the presence of compound A, a stabilizing agent featuring long perfluorinated chains, followed by the addition of AcONa (Scheme 1 and Fig. 1a). To prepare the immobilized catalyst, nanoparticles, Pdnp–A, were dissolved in perfluorooctane, and to this solution, commercially available FSG (C8; 35–70 μm) was added and the solvent was evaporated, affording the immobilized catalyst (Pdnp–A/FSG) as an air stable powder. Transmission electron microscopy (TEM) of this material showed well-defined spherical particles dispersed in the silica matrix (Fig. 1b). The mean diameter of the nanoparticles was about 1.5 ± 0.7 nm.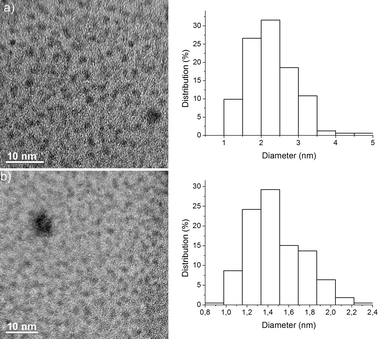 | ||
| Fig. 1 (a) TEM image and particle size distribution histogram of Pdnp–A (particle size 2.3 ± 0.7 nm). (b) TEM image and particle size distribution histogram of Pdnp–A/FSG (particle size 1.5 ± 0.7 nm). | ||
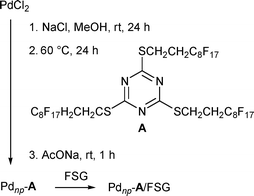 | ||
| Scheme 1 Synthesis of phosphine-free, perfluoro-tagged palladium nanoparticles Pdnp–A and their immobilization on fluorous silica gel (FSG). | ||
Palladium nanoparticles stabilized by a perfluorinated compound covalently bound to silica gel were also prepared. This catalyst system (Pdnp–B), containing 3.47% of palladium in the form of nanoparticles with an average particle size of 3.9 ± 0.9 nm, was prepared by the sol-gel process described previously14 (Scheme 2).
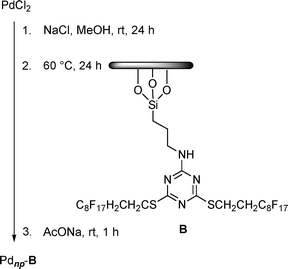 | ||
| Scheme 2 Synthesis of Pdnp–B. | ||
Alkynylation of aryl halides
We initiated our investigation by screening for reaction conditions under which the alkynylation reaction would proceed in the presence of oxygen, and in the absence of copper and phosphine. Copper salts can induce Glaser-type homocoupling15 of terminal alkynes due to the oxidative coupling of copper acetylide intermediates when exposed to oxidative agents or air. In addition, the presence of two metals hinders the recovery and reuse of the expensive palladium catalysts (its recovery would be the best way to overcome cost-related problems).Reactions were conducted in water. The use of water as the reaction medium is very attractive in organic synthesis for safety, economical, and environmental reasons.16 In addition, reactions involving water-insoluble substrates often benefit from the hydrophobic effect when carried out in water due to its high dielectric constant and density.17 There are only a few reports of alkynylation reactions of aryl halides in the presence of immobilized palladium catalysts under copper- and phosphine-free conditions in water18 or using water as cosolvent.19 Our recent report12 showed that palladium nanoparticles can be successfully employed in the alkynylation of aryl halides under these conditions.
Using the reaction of 3-(trifluoromethyl)iodobenzene with phenylacetylene as a probe for evaluating the reaction conditions (Table 1), we observed that the desired coupling product could be isolated in moderate yields after 5 h at 100 °C in water using Pdnp–A/FSG (catalyst loading 0.1 mol%), K2CO3 or KOAc as bases under aerobic conditions (entries 1 and 2). No homocoupling derivative was observed. We then tested nitrogen bases and found that an almost quantitative yield could be obtained with pyrrolidine (entry 6).
| ||
|---|---|---|
| Entry | Base | % Yield of 3ab |
| a Reactions were carried out under aerobic conditions in 2 mL of water using 1 mmol of 3-(trifluoromethyl)iodobenzene, 1 mmol of phenylacetylene and 2 mmol of base in the presence of 0.1 mol% of Pdnp–A/FSG.b Yields are given for isolated products. | ||
| 1 | K2CO3 | 50 |
| 2 | KOAc | 50 |
| 3 | Et2NH | 89 |
| 4 | Et3N | 91 |
| 5 | Piperidine | 95 |
| 6 | Pyrrolidine | 99 |
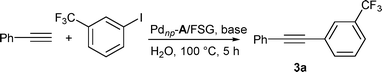
The coupled product was isolated in excellent yield under the optimized conditions [Pdnp–A/FSG (catalyst loading 0.1 mol%), H2O, 100 °C, 5 h]. However, recycling studies revealed that Pdnp–A/FSG has a limited capacity to be reused. Indeed, a significant loss of activity was observed in the third run (Table 2, entry 1). Increasing the catalyst loading resulted in only a marginal increase in the number of runs that could be performed without a significant loss of activity (entries 2–4).
| Entry | Catalyst | Loading | T/°C | Time/min | % Yield of 3ab | TON |
|---|---|---|---|---|---|---|
| a Reactions were carried out under aerobic conditions in 2 mL of water using 1 mmol of 3-(trifluoromethyl)iodobenzene, 1 mmol of phenylacetylene and 2 mmol of pyrrolidine in the presence of Pdnp–A/FSG or Pdnp–B.b Yields are given for isolated products. | ||||||
| 1 | Pdnp–A/FSG | 0.1 mol% | 100 | 120 | 95, 92, 50 | 2370 |
| 2 | 0.5 mol% | 100 | 45 | 95, 96, 93, 83, 15 | 764 | |
| 3 | 1.0 mol% | 80 | 45 | 97, 92, 90, 80, 60 | 427 | |
| 4 | 1.5 mol% | 80 | 40 | 85, 92, 91, 95, 55 | 279 | |
| 5 | Pdnp–B. | 0.5 mol% | 100 | 60 | 95, 90, 91, 86, 95, 92, 95, 90, 88, 86, 70 | 1956 |
Although only trace amounts of palladium were detected in the water fraction obtained after the reaction (0.05–0.08 ppm by SF-ICP-MS), high levels of palladium (39–240 ppm) were found in the crude isolated product. This is most likely due to the relative weakness of fluorous–fluorous interactions responsible for the binding of the Pdnp–A species to FSG, with the effect becoming most pronounced in the alkynylation reaction (not observed in the Suzuki–Miyaura cross-coupling and Heck reaction, vide infra). Confirming this assumption, the 19F NMR analysis of the crude mixture obtained from the reaction of phenylacetylene with 3-(trifluoromethyl)iodobenzene after filtration revealed the presence of significant amounts of A, corresponding to an approximately 50% loss of the fluorous stabilizer A per run.
A possible explanation is that the leaching of palladium and the loss of activity in this reaction depends on the nature of reagents and products. Relatively strong interactions20 might be established between palladium and alkyne (substrate/product). These interactions could favor the leaching of palladium that is removed from the solid support combined with the stabilizer A.
To circumvent the problems caused by the stabilizer leaching, we went on to investigate the use of material Pdnp–B, with the perfluoro-tagged palladium nanoparticles covalently linked to silica gel. The catalytic activity and stability of this catalyst was tested using our model system in water with a variety of bases (K2CO3, KOAc, pyrrolidine, piperidine, Et2NH, and Et3N). After some experimentation, we found that 3a could be isolated in 95% yield using 0.5 mol% of Pdnp–B and 2 equiv. of pyrrolidine at 100 °C for 1 h. Further studies revealed that the recyclability of this supported catalyst system was significantly superior to that of Pdnp–A/FSG (Table 2, entry 5). The Ostwald ripening process, where atoms detach from smaller clusters and reattach to bigger clusters,21 was not observed upon recycling. The material recovered after 11 runs was examined by TEM and showed nanoparticles of about 3.2 nm in diameter (Fig. 3).
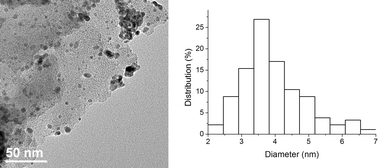 | ||
| Fig. 2 TEM image and particle size distribution histogram of Pdnp–B (particle size 3.9 ± 0.9 nm). | ||
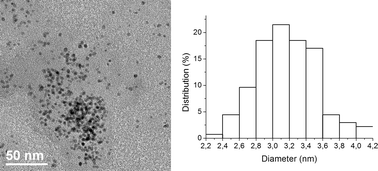 | ||
| Fig. 3 TEM image and particle size distribution histogram of Pdnp–B after 11 runs (particle size 3.2 ± 0.4 nm). | ||
A variety of terminal alkynes and aryl halides were then subjected to the optimized conditions using both Pdnp-A/FSG (procedure A) and Pdnp-B (procedure B). As shown in Table 3, acetylenes bearing electron-rich and electron-poor aryl groups as well as alkyl substituents give coupling products in excellent yields employing aryl iodides, which in turn can accommodate electron-withdrawing and electron-donating substituents. Ortho substituents in the aryl halide are also tolerated (entries 13, 16, 17). With aryl bromides, longer reaction times are required to obtain similar or slightly lower yields (compare entry 3 with entry 41, entry 5 with entry 40, entry 6 with entry 45, entry 7 with entry 42, entry 8 with entry 43, and entry 9 with entry 44).
| ||||||
|---|---|---|---|---|---|---|
| Entry | Terminal Alkyne 1 R | ArX 2 | Proc. | Time/h | % Yield of 3b | |
| a Reactions were carried out under aerobic conditions in 2 mL of water using 1 mmol of 2, 1 mmol of 1, 2 mmol of pyrrolidine with Pdnp-A/FSG (catalyst loading 0.1 mol%; procedure A) or with Pdnp–B (catalyst loading 0.5 mol%; procedure B).b Yields are given for isolated products.c In the presence of Pdnp–A/FSG (catalyst loading 0.5 mol%). | ||||||
| 1 | Ph | 3-CF3C6H4I | A | 3 | 3a | 95 |
| 2 | 3-CF3C6H4I | A | 3 | 3a | 95c | |
| 3 | 3-CF3C6H4I | B | 1 | 3a | 95 | |
| 4 | 4-EtO2CC6H4I | A | 5 | 3b | 95 | |
| 5 | 4-MeCOC6H4I | B | 6 | 3c | 80 | |
| 6 | 4-NO2C6H4I | A | 5 | 3d | 93 | |
| 7 | 4-MeOC6H4–I | B | 12 | 3e | 90 | |
| 8 | 4-CNC6H4I | A | 6 | 3f | 86 | |
| 9 | 4-CNC6H4I | B | 5 | 3f | 88 | |
| 10 | PhI | A | 24 | 3g | 94 | |
| 11 | PhI | B | 12 | 3g | 70 | |
| 12 | 3-Me-4-NO2C6H4I | A | 8 | 3h | 86 | |
| 13 | 2-NH2C6H4I | B | 24 | 3i | 75 | |
| 14 | 4-MeOC6H4 | 4-CNC6H4I | A | 5 | 3l | 85 |
| 15 | 4-CNC6H4I | B | 7 | 3l | 95 | |
| 16 | 2-MeC6H4I | A | 29 | 3m | 80 | |
| 17 | 2-MeC6H4I | B | 9 | 3m | 90 | |
| 18 | 4-CNC6H4 | 3-CF3C6H4I | A | 2 | 3n | 89 |
| 19 | 3-CF3C6H4I | B | 2 | 3n | 99 | |
| 20 | 4-MeCOC6H4 | 4-CNC6H4I | A | 4 | 3o | 90 |
| 21 | 4-CNC6H4I | B | 2 | 3o | 99 | |
| 22 | 4-CNC6H4 | 4-MeOC6H4–I | A | 3 | 3l | 85 |
| 23 | 4-MeOC6H4–I | B | 7 | 3l | 25 | |
| 24 | 3-CF3C6H4I | A | 2 | 3n | 89 | |
| 25 | 3-CF3C6H4I | B | 2 | 3n | 99 | |
| 26 | 2-MeC6H4 | 4-CNC6H4I | A | 44 | 3p | 84 |
| 27 | 4-MeOC6H4–I | A | 5 | 3m | 85 | |
| 28 | HOCH2 | 4-CNC6H4I | B | 24 | 3q | 89 |
| 29 | 4-MeOC6H4–I | B | 24 | 3r | 87 | |
| 30 | HO(Me)2C | 4-MeOC6H4I | A | 48 | 3s | 83 |
| 31 | 4-CNC6H4I | A | 24 | 3t | 75 | |
| 32 | HO(Me)(Et)C | 4-MeCOC6H4I | B | 22 | 3u | 90 |
| 33 | HO(Me)(Ph)C | 4-MeOC6H4I | B | 24 | 3v | 90 |
| 34 | 4-CNC6H4I | B | 12 | 3z | 95 | |
| 35 | 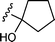 | 4-MeCOC6H4I | B | 14 | 3za | 85 |
| 36 | 4-MeOC6H4I | B | 24 | 3zb | 87 | |
| 37 | 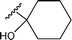 | 4-MeCO-C6H4I | B | 14 | 3zc | 92 |
| 38 | 4-MeOC6H4I | B | 18 | 3zd | 89 | |
| 39 | Ph | 4-MeCOC6H4Br | A | 44 | 3c | 50 |
| 40 | 4-MeCOC6H4Br | B | 10 | 3c | 80 | |
| 41 | 3-CF3C6H4Br | B | 9 | 3a | 91 | |
| 42 | 4-MeOC6H4Br | B | 48 | 3e | 65 | |
| 43 | 4-CNC6H4Br | A | 24 | 3f | 99 | |
| 44 | 4-CNC6H4Br | B | 8 | 3f | 75 | |
| 45 | 4-NO2C6H4Br | A | 24 | 3d | 92 | |
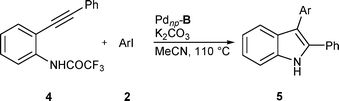
The formation of 3i (entry 13) is particularly attractive in that coupling products of this type are useful intermediates for the synthesis of indoles.22 We briefly investigated using Pdnp–B in the synthesis of 2,3-disubstituted indoles from 2-(phenylethynyl)trifluoroacetanilide and aryl iodides via the aminopalladation–reductive elimination protocol23 (Table 4). Excellent yields are obtained with electron-poor aryl iodides. Lower reaction rates and slightly lower yields are observed with electron-rich aryl iodides.
| ||||
|---|---|---|---|---|
| Entry | ArI 2 | Time/h | % Yield of 5b,c | |
| a Reactions were carried out under aerobic conditions in 2 mL of MeCN using 1 mmol of 2, 1 mmol of 4, and 2 mmol of K2CO3 in the presence of Pdnp–B (catalyst loading 0.1 mol%).b Yields are given for isolated products.c Yields in parentheses refer to successive runs carried out with the recovered catalyst. | ||||
| 1 | PhI | 9 | 82 | 5a |
| 2 | 3-CF3C6H4I | 2 | 91 (90, 86, 87) | 5b |
| 3 | 4-CNC6H4I | 4 | 84 (92, 90,86) | 5c |
| 4 | 4-MeCOC6H4I | 2 | 89 (91, 87) | 5d |
| 5 | 3-MeC6H4I | 40 | 77 | 5e |
| 6 | 4-MeOC6H4I | 48 | 70 | 5f |
| 7 | 4-ClC6H4I | 5 | 96 (92, 83) | 5g |
| 8 | 4-NO2C6H4I | 4 | 90 | 5h |
Suzuki–Miyaura cross-coupling
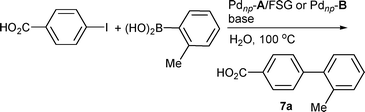
We started this part of the study by examining the reaction of 4-iodobenzoic acid with 2-tolylboronic acid as the model system, using a material containing 20 mg of Pdnp–A per g of FSG (0.26% palladium) and catalyst loading down to 0.1 mol% in the presence of K3PO4 in water at 100 °C for 5 h.24 Under these conditions, the corresponding cross-coupling product 7a was isolated in 80% yield. The influence of bases on the reaction outcome was next investigated (Table 5). K2CO3 gave a higher yield (entry 2) while 7a was isolated in moderate yield using KF (entry 3). Complete conversion could be obtained when the reaction was carried out in the presence of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 K2CO3/KF combination26 (entry 4). An even higher reaction rate was observed under the same conditions using 0.1 mol% of Pdnp–B, giving 7a in 99% yield after 1 h (entry 5).
:1 K2CO3/KF combination26 (entry 4). An even higher reaction rate was observed under the same conditions using 0.1 mol% of Pdnp–B, giving 7a in 99% yield after 1 h (entry 5).
| ||||
|---|---|---|---|---|
| Entry | Catalyst | Base | Time/h | % Yield of 7ab |
| a Reactions were carried out under aerobic conditions in 2 mL of water using 1 mmol of 4-iodobenzoic acid, 1 mmol of 2-tolylboronic acid, 2 mmol of base in the presence of Pdnp–A/FSG or Pdnp–B (catalyst loading 0.1 mol%).b Yields are given for isolated products. | ||||
| 1 | Pdnp–A/FSG | K3PO4 | 5 | 80 |
| 2 | K2CO3 | 5 | 87 | |
| 3 | KF | 5 | 64 | |
| 4 | K2CO3/KF (1:1) | 5 | 99 | |
| 5 | Pdnp–B | K2CO3/KF (1:1) | 1 | 99 |
Recycling studies were then performed and revealed that both Pdnp–A/FSG and Pdnp–B could be reused several times without significant loss of activity (Table 6, entries 1 and 4). Further recycling studies were performed using lower palladium loading. Using materials containing 2 mg of Pdnp–A per g of FSG (0.026% palladium) with a palladium loading down to 0.01 mol% and 0.2 mg of Pdnp–A per g of FSG (0.0026% palladium) with a palladium loading down to 0.001 mol%, the cumulative turn-over number (TON) over six and four runs is 55300 and 333000, respectively (entries 2 and 3). With Pdnp–B and a palladium loading down to 0.01% the cumulative turnover number over is 37000 (entry 5).
| Entry | Catalyst | Loading | % Yield of 7a | TON |
|---|---|---|---|---|
| a Reactions were carried out under aerobic conditions in 2 mL of water using 1 mmol of 4-iodobenzoic acid, 1 mmol of 2-tolylboronic acid, 2 mmol of K2CO3/KF (1:1) at 100 °C in the presence of Pdnp–A/FSG (5 h) or Pdnp–B (1 h).b Yields are given for isolated products. | ||||
| 1 | Pdnp–A/FSG | 0.1 mol% | 99, 86, 84, 88, 91, 90, 86, 92, 87, 90, 90, 85, 84, 91, 93 | 13360 |
| 2 | 0.01 mol% | 98, 95, 99, 92, 99, 70 | 55300 | |
| 3 | 0.001 mol% | 89, 85, 87, 72 | 333000 | |
| 4 | Pdnp–B | 0.1 mol% | 99, 99, 91, 99, 97, 99, 97, 100, 95, 90, 92, 95, 90, 85, 85 | 14130 |
| 5 | 0.01 mol% | 97, 95, 90, 87 | 37000 |
The resistance of Pdnp–A/FSG and Pdnp–B to leaching was assessed for the same reaction (4-iodobenzoic acid and 2-tolylboronic acid). SF-ICP-MS analysis indicated the level of palladium species in water and in the raw product to be in the range of 1–2.4 and 3–23 ppb, respectively (Table 7). In addition, TEM analysis showed that, after 15 runs, the palladium nanoparticles are very similar in shape and size to those of the original catalyst system (compare Fig. 4a with Fig. 1 and Fig. 4b with Fig. 2).
 | ||
| Fig. 4 (a) TEM image and particle size distribution histogram of Pdnp–A/FSG after the 15th run (particle size 1.7 ± 0.3). (b) TEM image and particle size distribution histogram of Pdnp–B after the 15th run (particle size 3.6 ± 0.6). | ||
The crucial role of fluorous–fluorous interactions with Pdnp–A/FSG was assessed by immobilizing Pdnp–A on standard reversed phase silica gel and using the resultant catalyst in our model reaction (4-iodobenzoic acid with 2-tolylboronic acid). Compound 7a was obtained in 87% yield in the first run. However, a pronounced loss of activity was observed in the second run, which gave the cross-coupling product in 62% isolated yield.
Using the optimized conditions (K2CO3/KF 1:1, 100 °C, H2O), we next explored the efficiency of Pdnp–A/FSG and Pdnp–B with other aryl iodides and bromides as well as arylboronic acids. As shown in Table 8, both precatalyts gave good to excellent results with aryl halides and arylboronic acids containing both electron-donating and electron-withdrawing substituents. The success with the electron-deficient arylboronic acids (entries 5, 30, 32, 33, and 34) is particularly noteworthy given that these compounds are often reluctant to give Suzuki–Miyaura products,27 a tendency usually attributed to their lower nucleophilicity (in comparison to their electron-neutral or electron-rich analogues) and, consequently, slower transmetalation. Ortho methyl substituents are tolerated both in the aryl halide and the arylboronic acid component (entries 1, 2, 4, 13, 23, 29, 32). In addition, ortho functional groups such as –Br, –COOH, and –NO2 are also well tolerated (entries 25, 33, 34).
| ||||||
|---|---|---|---|---|---|---|
| Entry | ArX 2 | Arylboronic acid 6 | Proc. | Time/h | % Yield of 7b,c | |
| a Unless otherwise stated, reactions were carried out under aerobic conditions in 2 mL of water using 1 mmol of aryl halide, 1 mmol of arylboronic acid, 2 mmol of K2CO3/KF (1:1) at 100 °C in the presence of Pdnp–A/FSG (procedure A) or Pdnp–B (procedure B) (catalyst loading 0.1 mol%).b Yields are given for isolated products.c Yields in parentheses refer to successive runs carried out with the recovered catalyst. | ||||||
| 1 | 4-HO2CC6H4I | 2-MeC6H4B(OH)2 | A | 5 | 7a | 99 |
| 2 | 2-MeC6H4B(OH)2 | B | 1 | 7a | 99 | |
| 3 | 4-F,3-MeC6H3B(OH)2 | A | 5 | 7b | 72 | |
| 4 | 4-MeOC6H4I | 2-MeC6H4B(OH)2 | A | 24 | 7c | 92 |
| 5 | 4-MeCOC6H4B(OH)2 | B | 5 | 7d | 91 | |
| 6 | 4-HO2CC6H4Br | 4-F,3-MeC6H3B(OH)2 | A | 44 | 7b | 80 |
| 7 | 4-CF3C6H4B(OH)2 | A | 7 | 7e | 71 | |
| 8 | 4-CF3C6H4B(OH)2 | B | 8 | 7e | 90(92) | |
| 9 | PhB(OH)2 | A | 8 | 7f | 72 | |
| 10 | PhB(OH)2 | B | 8 | 7f | 91 | |
| 11 | 4-MeOC6H4B(OH)2 | A | 6 | 7g | 91(90) | |
| 12 | 4-MeOC6H4B(OH)2 | B | 8 | 7g | 87 | |
| 13 | 2-MeC6H4B(OH)2 | B | 5 | 7a | 90 | |
| 14 | 4-MeC6H4B(OH)2 | B | 5 | 7h | 87 | |
| 15 | 4-HO2CC6H4 I | 4-MeOC6H4B(OH)2 | A | 5 | 7g | 95(99) |
| 16 | 4-CF3C6H4B(OH)2 | A | 2 | 7e | 94(99, 84) | |
| 17 | 4-MeC6H4B(OH)2 | A | 5 | 7h | 99 | |
| 18 | PhB(OH)2 | A | 24 | 7f | 83 | |
| 19 | 4-MeOC6H4I | 4-CF3C6H4B(OH)2 | A | 24 | 7i | 80 |
| 20 | 4-MeOC6H4Br | 4-CF3C6H4B(OH)2 | B | 24 | 7i | 88(85) |
| 21 | 4-MeCOC6H4B(OH)2 | B | 48 | 7d | 80 | |
| 22 | PhI | 4-CF3C6H4–B(OH)2 | A | 24 | 7l | 50 |
| 23 | 2-MeC6H4B(OH)2 | B | 9 | 7m | 99 | |
| 24 | 4-MeCOC6H4I | 4-CF3C6H4B(OH)2 | A | 8 | 7n | 91 |
| 25 | 2-BrC6H4B(OH)2 | B | 48 | 7o | 62 | |
| 26 | 4-MeCOC6H4Br | 4-CF3C6H4B(OH)2 | B | 8 | 7n | 92(93) |
| 27 | 4-MeOC6H4B(OH)2 | B | 24 | 7d | 84 | |
| 28 | 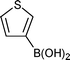 | B | 5 | 7p | 90 | |
| 29 | 4-FC6H4I | 2-MeC6H4B(OH)2 | A | 6 | 7q | 89 |
| 30 | 4-CNC6H4I | 4-MeCOC6H4B(OH)2 | B | 24 | 7r | 87 |
| 31 | 4-MeOC6H4Br | 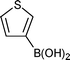 | B | 16 | 7s | 89 |
| 32 | 2-MeC6H4I | 4-MeCOC6H4B(OH)2 | B | 6 | 7t | 93 |
| 33 | 2-HO2CC6H4I | 4-MeCOC6H4B(OH)2 | B | 2 | 7a | 80 |
| 34 | 2-NO2,4-MeC6H4I | 4-MeCOC6H4B(OH)2 | B | 2 | 7u | 87 |
Heck reaction
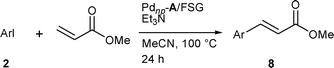
To explore the utilization of Pdnp–A/FSG and Pdnp–B in the Heck reaction, we decided to study the synthesis of cinnamate esters from methyl acrylate and aryl iodides. Iodobenzene was selected as the model system. A silica gel containing 100 mg of Pdnp–A per g of FSG (1.22% palladium) and a 0.6 mol% palladium loading was initially evaluated. The reaction, carried out in water in the presence of Et3N, in air at 80 °C for 24 h, gave no evidence of 8a. A slightly better result was obtained with t-butyl acrylate, the corresponding vinylic substitution product being isolated in 35% yield. Similarly, employing a 40% EtOH aqueous solution gave only a 3% conversion after 24 h. Gratifyingly, using DMF led to the formation of methyl cinnamate in almost quantitative yield. A similar result was obtained in the second run. However, a loss of activity was observed in the third run (85% yield) which became even more substantial in the fourth run (75% yield). Assuming that these results might be due to the leaching of palladium in DMF, we switched to the utilization of MeCN. After some experimentation we found that complete conversion could be reached in MeCN at 100 °C after 24 h using 20 mg of Pdnp–A per g of FSG and a catalyst loading down to 0.1 mol%. Methyl cinnamate was isolated in 90% yield. A similar result was obtained using 0.1 mol% of Pdnp–B under the same conditions. Methyl cinnamate was isolated in 93% yield.Recycling studies showed that both Pdnp–A/FSG and Pdnp–B can be reused several times without significant loss of activity (Table 9). Using a material containing 3.3 mg of Pdnp–A per g of FSG (0.04% palladium) and a palladium loading down to 0.001 mol%, the cumulative turn-over number over three runs is 265000 (entry 3). With Pdnp–B and a palladium loading down to 0.01 mol%, the cumulated turnover number over four runs is 36100 (entry 4).
| Entry | Catalyst loading | Time/h | T/°C | Yield% of 8a | TON |
|---|---|---|---|---|---|
| a Reactions were carried out under aerobic conditions in 2 mL of MeCN using 1 mmol of iodobenzene, 2 mmol of methyl acrylate, 3 mmol of Et3N at 100 °C for 24 h in the presence of Pdnp–A/FSG or Pdnp–B.b Yields are given for isolated products.c Yields in parentheses refer to a second set of experiments | |||||
| 1 | Pdnp–A/FSG 0.1 mol% | 24 | 100 | 90(91), 96(90, 96(90), 93(86), 87(88), 89(95), 97(100), 96(99), 95(94), 98(95), 97, 90, 90, 84, 80 | 13780 |
| 2 | Pdnp–A/FSG 0.01 mol% | 24 | 120 | 100, 96, 81, 97, 85 | 45900 |
| 3 | Pdnp–A/FSG | 24 | 140 | 100, 82, 83 | 265000 |
| 0.001 mol% | |||||
| 4 | Pdnp–B | 24 | 120 | 97, 94, 86, 84 | 36100 |
| 0.01 mol% |
The resistance of Pdnp–A/FSG to leaching was assessed for the model reaction. SF-ICP-MS analysis indicated the level of palladium in the crude mixtures to be in the 2–7 ppm range. Agglomeration of nanoparticles was not observed upon recycling. The recovered material after the 15th run was examined by TEM showing nanoparticles of about 1.9 ± 0.3 nm (Fig. 5). Control experiments were also carried out to assess whether leaching of the stabilizer A might take place under reaction conditions. 19F NMR analysis of the crude mixture produced in the reaction of methyl acrylate with 3-(trifluoromethyl)iodobenzene after filtration revealed the presence of small amounts of A, corresponding to a loss of about 5%. No evidence of fluorine was observed in the isolated vinylic substitution product.
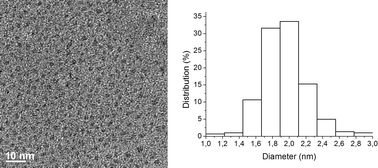 | ||
| Fig. 5 TEM image and particle size distribution histogram of Pdnp–A/FSG after the 15th run (paticle size 1.9 ± 0.3 nm). | ||
As was the case with the Suzuki–Miyaura cross-coupling, the reusability of the catalyst system is strongly dependent on fluorous–fluorous interactions. Immobilizing Pdnp–A on standard reversed phase silica gel and using the resultant catalyst in the reaction of methyl acrylate with iodobenzene gave methyl cinnamate in 93% yield in the first run. However, a remarkable loss of activity was observed in the second run where the Heck product was isolated only in 59% yield.
We next evaluated the efficiency of Pdnp–A/FSG and Pdnp–B with other aryl iodides. The preparative results are summarized in Table 10.
| |||||
|---|---|---|---|---|---|
| Entry | Aryl iodide 2 | Procedure | Time/h | % Yield of 8b,c | |
| a Reactions were carried out under aerobic conditions in 2 mL of MeCN using 1 mmol of aryl iodide, 2 mmol of methyl acrylate, 3 mmol of Et3N at 100 °C in the presence of Pdnp–A/FSG (procedure A) or Pdnp–B (procedure B) (catalyst loading 0.1 mol%.)b Yields are given for isolated products.c Yields in parentheses are for the second run carried out with the recovered catalyst. | |||||
| 1 | PhI | A | 24 | 90 | 8a |
| 2 | PhI | B | 24 | 93 | 8a |
| 3 | 4-MeCOC6H4I | A | 5 | 89 (90) | 8b |
| 4 | 4-MeCOC6H4I | B | 5 | 92 | 8b |
| 5 | 4-MeOC6H4I | A | 23 | 90 (70) | 8c |
| 6 | 4-MeOC6H4I | B | 18 | 89 | 8c |
| 7 | 4-MeC6H4I | A | 23 | 90 | 8d |
| 8 | 4-NO2C6H4I | A | 48 | 74 (65) | 8e |
| 9 | 3-CF3C6H4I | A | 6 | 93 (90) | 8f |
| 10 | 4-EtO2CC6H4I | A | 5 | 98 | 8g |
| 11 | 3-Me-4-NO2C6H3I | A | 23 | 96 | 8h |
| 12 | 2-NH2C6H4I | A | 48 | 84 | 8i |
| 13 | 2-MeO2CC6H4I | A | 7 | 89 | 8j |
We have not investigated in detail whether the nanoparticles on the solid surface are the actual catalyst or they are just a source that leaches active catalyst species28 in all the reactions described in the present manuscript. Nevertheless, we observed that when methyl acrylate, p-iodoanisole and Et3N were added to the crude mixture derived from the reaction of methyl acrylate with iodobenzene (after separation of the solid material), the corresponding vinylic substitution derivative could be isolated in 40% yield after 23 h at 100 °C. This result supports the notion that palladium species leached from the solid surface are, at least in part, responsible for the catalytic activity.
Conclusions
In conclusion, we have demonstrated that perfluoro-tagged palladium nanoparticles immobilized on fluorous silica gel by fluorous–fluorous interactions (Pdnp–A/FSG) or linked to silica gel by covalent bonds (Pdnp–B) can be successfully used under aerobic and phosphine-free conditions in the alkynylation of aryl halides (under copper-free conditions), in the Suzuki–Miyaura cross-coupling, and in the Heck reaction of methyl acrylate with aryl iodides. The alkynylation of aryl halides and the Suzuki–Miyaura cross-coupling are carried out in water. The Heck reaction of methyl acrylate with aryl iodides is best performed in MeCN. Pdnp–B can be successfully used in the synthesis of 2,3-disubstituted indoles from 2-(alkynyl)trifluoroacetanilides and aryl halides.Both catalysts allow for the isolation of the desired products, usually in high to excellent yields. However, in the alkynylation of aryl halides the use of Pdnp–B gives the best results in term of recovery and reuse.
The supported palladium can be easily recovered and reused. The recovery involves centrifugation and decanting the solution in the presence of air, without any particular precaution.
Acknowledgements
This work was carried out in the framework of the National Projects “Stereoselezione in Sintesi Organica. Metodologie ed Applicazioni” supported by the Ministero dell'Università e della Ricerca (MUR) and by Sapienza, Università di Roma. Financial support from Ministerio de Ciencia e Innovación of Spain (Projects CTQ2008-05409-C02-01 and CTQ2005-04968-C02-01 and Consolider Ingenio 2010 (CSD2007-00006)) and DURSI-Generalitat de Catalunya (SGR 2005-00305 and SGR 2009-1441) are gratefully acknowledged. A.S. has been supported through a Ramon y Cajal contract from the Ministerio de Educación y Ciencia of Spain. S.N. acknowledges MICINN for a predoctoral fellowship.Notes and references
- (a) J. Tsuji, Palladium Reagents and Catalysts - Innovation in Organic Synthesis, John Wiley & Sons, New York, 1995 Search PubMed; (b) Handbook of Organopalladium Chemistry for Organic Synthesis, E. Negishi, Ed.; John Wiley & Sons, New York, 2002, Vols. 1 and 2 Search PubMed; (c) J. Tsuji, Palladium Reagents and Catalysts - New Perspectives for the 21st Century, John Wiley & Sons, New York, 2004 Search PubMed.
- C. E. Garret and K. Prasad, Adv. Synth. Catal., 2004, 346, 889 CrossRef CAS.
- For a review, see: S. V. Ley, I. R. Baxendale, R. N. Brem, P. S. Jackson, A. G. Leach, A. Longbottom, M. Nesi, J. S. Scott, R. I. Storer and S. J. Taylor, J. Chem. Soc., Perkin Trans. 1, 2000, 3815 Search PubMed.
- For some excellent recent reviews on the use of heterogeneous palladium catalysts in C–C bond forming reactions, see: (a) L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133 CrossRef CAS; (b) M. Moreno-Mañas and R. Pleixats, Acc. Chem. Res., 2003, 36, 638 CrossRef CAS.
- For selected references, see: (a) R. Akiyama and S. Kobayashi, Angew. Chem., Int. Ed., 2001, 40, 3469 CrossRef CAS; (b) C. Ramarao, S. V. Ley, S. C. Smith, I. M. Shirley and N. DeAlmeida, Chem. Commun., 2002, 1132 RSC; (c) S. V. Ley, C. Ramarao, R. S. Gordon, A. B. Holmes, A. J. Morrison, I. F. McConvey, I. M. Shirley, S. C. Smith and M. D. Smith, Chem. Commun., 2002, 1134 RSC; (d) N. Bremeyer, S. V. Ley, C. Ramarao, I. M. Shirley and S. C. Smith, Synlett, 2002, 1843 CAS; (e) S. V. Ley, C. Mitchell, D. Pears, C. Ramarao, J.-Q. Yu and W. Zhou, Org. Lett., 2003, 5, 4665 CrossRef CAS.
- (a) T. F. Baumman, G. A. Fox and J. H. Satcher, Jr., Langmuir, 2002, 18, 7073 CrossRef; (b) T. F. Baumman and J. H. Satcher, Jr., Chem. Mater., 2003, 15, 3745 CrossRef CAS; (c) F. J. Maldonado-Hódar, C. Moreno-Castilla and A. F. Pérez-Cadenas, Microporous Mesoporous Mater., 2004, 69, 119 CrossRef CAS; (d) C. Moreno-Castilla and F. J. Maldonado-Hódar, Carbon, 2005, 43, 455 CrossRef CAS; (e) S. Martinez, A. Vallribera, C. L. Cotet, M. Popovici, L. Martin, A. Roig, M. Moreno-Mañas and E. Molins, New J. Chem., 2005, 29, 1342 RSC; (f) L. C. Cotet, M. Gich, A. Roig, I. C. Popescu, V. Cosoveanu, E. Molins and V. Danciu, J. Non-Cryst. Solids, 2006, 352, 2772 CrossRef CAS; (g) S. Cacchi, C. L. Cotet, G. Fabrizi, G. Forte, A. Goggiamani, L. Martin, S. Martinez, E. Molins, M. Moreno-Mañas, F. Petrucci, A. Roig and A. Vallribera, Tetrahedron, 2007, 63, 2519 CrossRef CAS; (h) R. Soler, S. Cacchi, G. Fabrizi, G. Forte, L. Martín, S. Martínez, E. Molins, M. Moreno-Mañas, F. Petrucci, A. Roig, R. M. Sebastián and A. Vallribera, Synthesis, 2007, 3068 CAS; (i) A. Vallribera, E. Molins, Aerogel supported nanoparticles in catalysis, in Nanoparticles and Catalysis, Wiley-VCH, 2008 Search PubMed.
- (a) M. Moreno-Mañas, R. Pleixats and S. Villarroya, Organometallics, 2001, 20, 4524 CrossRef CAS; (b) M. Moreno-Mañas, R. Pleixats and S. Villarroya, Chem. Commun., 2002, 60 RSC; (c) M. Tristany, J. Courmarcel, P. Dieudonne, M. Moreno-Mañas, R. Pleixats, A. Rimola, M. Sodupe and S. Villarroya, Chem. Mater., 2006, 18, 716 CrossRef CAS; (d) A. Serra-Muns, R. Soler, E. Badetti, P. de Mendoza, M. Moreno-Mañas, R. Pleixats, R. M. Sebastián and A. Vallribera, New J. Chem., 2006, 30, 1584 RSC; (e) S. Niembro, A. Vallribera and M. Moreno-Mañas, New J. Chem., 2008, 32, 94 RSC.
- (a) Organofluorine Chemistry. Principles and Commercial Applications, R. E. Banks, B. E. Smart, J. C. Tatlow, Eds: Plenum, New York, 1994 Search PubMed; (b) Modern Fluoroorganic Chemistry, P. Kirsch, Ed.: Wiley-VCH, Weinheim, 2004 Search PubMed.
- For selected recent reviews on fluorous catalysts, see: (a) J. A. Gladysz, R. Correa de Costa, In The handbook of fluorous chemistryJ. Gladysz, A. D. P. Curran, I. T. Horvath, Eds.; Wiley-VCH, Weinheim, 2004, pp. 24-40, see especially sections 4.6 - 4.9 Search PubMed; (b) D. P. Curran, In The handbook of fluorous chemistry, J. A. Gladysz, D. P. Curran, I. T. Horvath, Eds.; Wiley-VCH, Weinheim, 2004, pp. 101-127, see section 7.6 Search PubMed; (c) S. Schneider, C. C. Tzschucke, W. Bannwarth, In The handbook of fluorous chemistry, J. A. Gladysz, D. P. Curran, I. T. Horvath, Eds.; Wiley-VCH, Weinheim, 2004, pp. 257-272 Search PubMed; (d) D. P. Curran, K. Fischer and G. Moura-Letts, Synlett, 2004, 1379 CrossRef CAS.
- (a) C. C. Tzschucke, C. Markert, H. Glatz and W. Bannwarth, Angew. Chem., Int. Ed., 2002, 41, 4500 CrossRef CAS; (b) C. C. Tzschucke and W. Bannwarth, Helv. Chim. Acta, 2004, 87, 2882 CrossRef CAS.
- See, for example: (a) V. P. W. Böhm and W. A. Herrmann, Eur. J. Org. Chem., 2000, 3679 CrossRef CAS; (b) T. Hundertmark, A. F. Littke, S. L. Buchwald and G. C. Fu, Org. Lett., 2000, 2, 1729 CrossRef CAS; (c) M. Eckhardt and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 13642 CrossRef CAS; (d) K. W. Anderson and S. L. Buchwald, Angew. Chem., Int. Ed., 2005, 44, 6173 CrossRef CAS.
- R. Bernini, S. Cacchi, G. Fabrizi, G. Forte, F. Petrucci, A. Prastaro, S. Niembro, A. Shafir and A. Vallribera, Org. Biomol. Chem., 2009, 7, 2270 RSC.
- R. Bernini;, S. Cacchi;, G. Fabrizi;, G. Forte, S. Niembro, F. Petrucci, R. Pleixats, A. Prastaro, R. M. Sebastian, R. Soler and M. Tristany, Org. Lett., 2008, 10, 561 CrossRef CAS.
- S. Niembro, A. Shafir, A. Vallribera and R. Alibes, Org. Lett., 2008, 10, 3215 CrossRef CAS.
- For selected references, see: (a) C. Glaser, Ber. Dtsch. Chem. Ges., 1869, 2, 422 CrossRef; (b) A. S. Hay, J. Org. Chem., 1962, 27, 3320 CrossRef CAS; (c) R. Rossi, A. Carpita and C. Bigelli, Tetrahedron Lett., 1985, 26, 523 CrossRef CAS; (d) N. G. Kundu, M. Pal and C. Chowdhury, J. Chem. Res., 1993, 432 Search PubMed; (e) A. Lei, M. Srivastava and X. Zhang, J. Org. Chem., 2002, 67, 1969 CrossRef CAS and references therein; (f) I. J. S. Fairlamb, P. S. Bauerlein, L. R. Marrison and J. M. Dickinson, Chem. Commun., 2003, 632 RSC; (g) A. S. Batsanov, J. C. Collings, I. J. S. Fairlamb, J. P. Holland, J. A. K. Howard, Z. Lin, T. B. Marder, A. C. Parsons and R. M. Ward, J. Org. Chem., 2005, 70, 703 CrossRef CAS; (h) for a review of alkyne coupling, see: P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632 Search PubMed.
- For a recent review on C–C bond forming reactions in aqueous media, see: C.-J. Li, Chem. Rev., 2005, 105, 3095 Search PubMed.
- M. Cai, Q. Xu, J. Sha and J. Molec, J. Mol. Catal. A: Chem., 2007, 272, 293 CrossRef CAS.
- (a) J. Gil-Moltó, S. Karlström and C. Nájera, Tetrahedron, 2005, 61, 12168 CrossRef CAS; (b) Z.-W. Ye and W.-B. Yi, J. Fluorine Chem., 2008, 129, 1124 CrossRef CAS.
- L. Djakovitch and P. Rollet, Tetrahedron Lett., 2004, 45, 1367 CrossRef CAS.
- M. Ahlquist, G. Fabrizi, S. Cacchi and P.-O. Norrby, Chem. Commun., 2005, 4196 RSC.
- (a) A. Imre, D. L. Beke, E. Gontier-Moya, I. A. Szabo and E. Gillet, Appl. Phys., A, 2000, 71, 19 CAS; (b) A. Howard, C. E. J. Mitchell and R. G. Egdell, Surf. Sci., 2002, 515, L504 CrossRef CAS; (c) R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2003, 125, 8340 CrossRef CAS.
- For some recent reviews, see: (a) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285 CrossRef CAS; (b) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079 CrossRef CAS; (c) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS; (d) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef CAS; (e) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS; (f) L. Ackermann, Synlett, 2007, 5607; (g) K. Krüger, A. Tillack and M. Beller, Adv. Synth. Catal., 2008, 350, 2153 CrossRef.
- (a) G. Battistuzzi, S. Cacchi and G. Fabrizi, Eur. J. Org. Chem., 2002, 2671 CrossRef CAS; (b) See also: S. Cacchi, G. Fabrizi and A. Goggiamani, Adv. Synth. Catal., 2006, 348, 1301 Search PubMed and the references cited therein.
- While this manuscript was in preparation, a study of cross-coupling using the same catalyst system, Pdnp–A/FSG, was reported (L. Wang, C. Cai, J. Mol, Catal. A: Chem. 2009, 306, 97). Reactions, however, were performed in the presence of Bu4NBr (which may limit the degree of recycling of the catalyst system25) and an increase of palladium nanoparticles to 5–10 nm (from the initial 2–3 nm) and a palladium leaching of 10 ppm was observed.
- For some leading references on tetraalkylammonium-stabilized palladium nanoparticles, see: (a) M. T. Reetz, R. Breinbauer and K. Wanninger, Tetrahedron Lett., 1996, 37, 4499 CrossRef; (b) M. Beller, H. Fischer, K. Kühlein, C.-P. Reisinger and W. A. Herrmann, J. Organomet. Chem., 1996, 520, 257 CrossRef CAS; (c) M. T. Reetz and M. Maase, Adv. Mater., 1999, 11, 773 CrossRef CAS; (d) M. T. Reetz and E. Westermann, Angew. Chem., Int. Ed., 2000, 39, 165 CrossRef CAS.
- Interestingly, a similar result was obtained subjecting 4-iodobenzoic acid and potassium 4-methoxyphenyltrifluoroborate to 0.1 mol% of Pdnp–A/FSG at 100 °C in water in the presence of K2CO3, omitting KF. The corresponding diaryl derivative 7i was isolated in 99% yield after 5 h (compare with the result reported in Table 8, entry 15).
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS.
- For a recent review on solid catalysts and on evidence for and against catalysis by solid surfaces vs. soluble species, see: (a) N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef CAS; (b) see also: ref. 9a and M. B. Thathagar, J. E. ten Elshof and G. Rothenberg, Angew. Chem., Int. Ed., 2006, 45, 2886 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and physical data of compounds. See DOI: 10.1039/b915465e |
| This journal is © The Royal Society of Chemistry 2010 |