New one-pot multistep process with multifunctional catalysts: decreasing the E factor in the synthesis of fine chemicals†
Maria J. Climent, Avelino Corma*, Sara Iborra, Maria Mifsud and Alexandra Velty
Instituto de Tecnología Quimica, Universidad Politécnica de Valencia, Avda. de los Naranjos s/n, 46022 Valencia, Spain. E-mail: acorma@itq.upv.es
First published on 26th October 2009
Abstract
The synthesis of a series of fine chemicals (4-(4-methoxyphenyl)-2-butanone, 4-(6,6-dimethylbicyclo[3,1,1]hept-2-en-2-yl)-2-butanone, and 2-cyclopentylcylopentanone) has been performed through a simple process that involves one-pot multi-step reactions (condensation–dehydration–reduction) using multifunctional base–acid-metal catalysts. The E factor for the specific process, as well as for the global process that includes the manufacturing of the reactants, has been calculated and compared with those obtained using conventional methods. In various cases, it is demonstrated that by means of process intensification with multifunctional solid catalysts, it is possible to decrease the E factor by one order of magnitude or more with respect to any of the manufacturing routes reported to date.
Introduction
In the late 1980s, Sheldon1 introduced the concept of the E (environmental) factor for assessing the environmental impact of manufacturing processes. The E factor is defined as the mass ratio of waste to the desired product (kg waste/kg product). To calculate the E factor, the chemical yield, reagents, solvent losses, all process aids, and even energy consumption (though this is frequently difficult to estimate) are taken into account. The water used in the process is not considered, but water formed is included in the calculations. Catalysis plays an important role in reducing or eliminating waste in chemical processes, particularly for the production of fine chemicals, where the E factors are especially high. Most of the waste generated in the manufacture of fine chemicals is inorganic salts formed by the use of stoichiometric inorganic reagents. Thus, the substitution of conventional stoichiometric methodologies by catalytic processes, combined with the possibility of intensifying processes by combining several catalytic steps into a one-pot catalytic system, provides a means to improve the economical and environmental aspects of chemical processes. In this sense, domino catalytic multi-step reactions with mono- or multifunctional catalysts are very efficient processes in terms of work up and catalyst utilization, with the additional advantage that isolation and purification of intermediates is not required.2–12 Moreover, the fact that the same catalyst can catalyze different consecutive steps in a single reaction vessel under the same reaction conditions decreases the operating time and the amount of waste produced. In this way, heterogeneous catalysts containing various active sites on their surface are promising candidates for promoting multiple reactions in a single pot.13-18Metal supported on alkaline earth layered double hydroxides (LDH) and its mixed oxides have been extensively applied as multifunctional base–acid-metal catalysts for cascade reactions such as the synthesis of methyl isobutyl ketone in one pot from acetone19-23 or from 2-propanol24-27 and for the preparation of fine chemicals such as 2-methyl-3-phenyl propanal,28,29α-alkylated nitriles,30 and the anti-inflamatory nabumetone.11,16
Here, we present that by means of multifunctional base–acid-metal catalysts (Pd-supported alkaline earth oxides and mixed oxides), it is possible to substitute existing processes for the production of a series of fine chemicals (4-(4-methoxyphenyl)-2-butanone, 4-(6,6-dimethylbicyclo[3,1,1]hept-2-en-2-yl)-2-butanone, and 2-cyclopentylcylopentanone) by a more simple process that involves one-pot multi-step reactions. We show that by means of process intensification with multifunctional solid catalysts, it has been possible, in various cases, to decrease the E factor by one order of magnitude or more, with respect to any of the manufacturing routes reported up to now. In this work, the E factor for the specific process as well as for the global process that includes the manufacturing of the reactants has been considered.
Results and discussion
1. Preparation of 4-(4-methoxyphenyl)-2-butanone (2)
4-(4-Methoxyphenyl)-butan-2-one (2) (Scheme 1) is an odorous molecule with a raspberry scent which is extracted from aloe wood, and has been approved by the FDA for food uses at a level of 25 ppm. It has also been utilized as an insect attractant for the scarabaeid family and in the preparation of melanin formation inhibitors.31 One route for the preparation of 2 in one step (Scheme 1) is the Friedel–Crafts alkylation of anisole with 4-hydroxybutan-2-one or methyl vinyl ketone using acid catalysts such as H2SO4, H3PO4, HCl.32 However, many side reactions (transalkylations, isomerization, polyalkylation and polymerization) occur during Friedel–Crafts alkylation in the presence of Lewis or Brønsted homogeneous acid catalyst, resulting in a maximum selectivity for 2 of approximately 70%.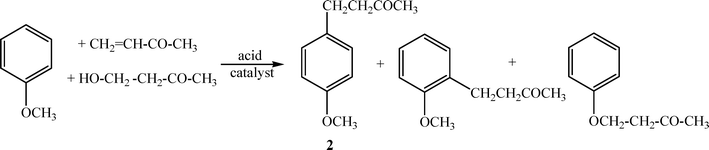 | ||
| Scheme 1 | ||
Heterogeneous catalysts, such as exchanged resins (Dowex-50),33 have also been used for this transformation, however low selectivity for 2 (60–70%) was obtained. Cation exchanged montmorillonite also containing Brønsted acid sites has been utilized, yielding only 25% of 2 at 100 °C after 48 h reaction time.34
There is another route to synthesize 2 (Scheme 2), that involves an aldol condensation between 4-methoxybenzaldehyde and acetone, yielding the α,β-unsaturated compound 1, which is followed by hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in a two-step reaction. When using homogenous catalysts, the condensation step has been carried out with NaOH as catalyst yielding 98% of the α,β-unsaturated ketone (1).35 While a one-pot process has been carried out under hydrogen atmosphere, in the presence of NaOH and 5% Pd/C, the two step process yields 80% of 4-(4-methoxyphenyl)-2-butanone (2).36
C bond in a two-step reaction. When using homogenous catalysts, the condensation step has been carried out with NaOH as catalyst yielding 98% of the α,β-unsaturated ketone (1).35 While a one-pot process has been carried out under hydrogen atmosphere, in the presence of NaOH and 5% Pd/C, the two step process yields 80% of 4-(4-methoxyphenyl)-2-butanone (2).36
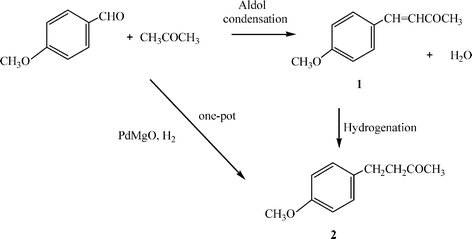 | ||
| Scheme 2 | ||
We propose here an alternative one-pot catalytic process to synthesize 2 that consists of reacting 4-methoxybenzaldehyde with acetone under hydrogen atmosphere in the presence of PdMgO catalyst to give an intermediate, 1, which is hydrogenated to give 2 (Scheme 2). The reaction between 4-methoxybenzaldehyde and acetone was performed using 1 wt% of Pd loaded on the nanocrystalline MgO (see Experimental) at 75 °C. The results in Table 1 show that PdMgO is able to convert 100% of 4-methoxybenzaldehyde, giving 99% yield and 99% selectivity for 2.
| Catalysts | r°× 104/mmol min−1 g−1 | Conversion (%) | Yield 1 (%) | Yield 2 (%) | Selectivity 2 (%) |
|---|---|---|---|---|---|
| r°: rate of disappearance of 4-methoxybenzaldehyde. Reaction conditions: 4-methoxybenzaldehyde (3.8 mmol), acetone (37 mmol); 75 °C, catalyst 38 wt% with respect to anisaldehyde. Conversion and yield at 75 min reaction time. The value in brackets is the percentage of Michael adduct. | |||||
| PdMgO | 12.0 | 100 | 1 | 99 | 99 |
| PdHTc | 5.0 | 89 | 25 | 62 | 68(2) |
| PdHTr | 9.7 | 95 | — | 95 | 100 |
It is known that the selection of the most appropriate type of centres for a given base-catalyzed process has a strong impact on the catalyst activity and selectivity.37 Thus, we have also prepared two Pd catalysts supported on Al/Mg calcined hydrotalcite (PdHTc) and on calcined–rehydrated Al/Mg hydrotalcite (PdHTr) which were tested under the same reaction conditions as PdMgO.
As can be seen in Table 1, PdHTc exhibits lower condensation and hydrogenation rates than PdMgO and, consequently, the yield and selectivity of 2 are lower. Moreover, a secondary product resulting from the Michael addition of acetone to 2 was also detected in minor amounts. When the calcined hydrotalcite containing palladium metal was rehydrated (PdHTr), a twofold increase in initial reaction rate was observed. This result is not surprising since by rehydration, a restoration of the layered structure occurs wherein the CO32− counter anions in the interlayer space are replaced by OH−, resulting in strong Brønsted base catalysts that efficiently catalyze a larger number of reactions.38–45 Results in Table 1 indicate that, together with PdMgO, PdHTr is a promising candidate for the preparation of 2, since 95% yield with 100% selectivity is obtained after 75 min reaction time.
Nevertheless, for a potential industrial implementation of the above catalysts (PdMgO and PdHTr) for the preparation of 2, the possibility of catalyst regeneration has to be considered. To study the reusability of the catalysts, the samples were regenerated after the reaction as described in the Experimental section and the catalyst was used again. As it can be seen in Table 2, an important decrease in activity upon regeneration was found with the PdHTr sample, which is probably due to an incomplete reconstruction and/or a decrease in the number of basic and hydrogenating active sites. In contrast, the PdMgO sample was subjected to four cycles of reaction–regeneration (calcination)–reaction, in all cases achieving 100% conversion and similar selectivity of 2 (Table 2).
| Catalysts | Cycles | Conversion (%) | Yield 1 (%) | Yield 2 (%) | Selectivity 2 (%) |
|---|---|---|---|---|---|
| PdHTr | 1 | 95 | — | 95 | 100 |
| 2 | 25 | 18 | 7 | 28 | |
| PdMgO | 1 | 100 | 1 | 99 | 99 |
| 2 | 100 | 2 | 98 | 98 | |
| 3 | 100 | 1 | 99 | 99 | |
| 4 | 100 | 2 | 98 | 98 |
In conclusion, we have shown that by using a multifunctional catalyst based on Pd supported on nanocrystalline MgO, it has been possible to develop a new green process for production of 2 in a one-pot cascade reaction. This new process allows yields of 99% with 99% selectivity for 2. The E factor involved is presented below.
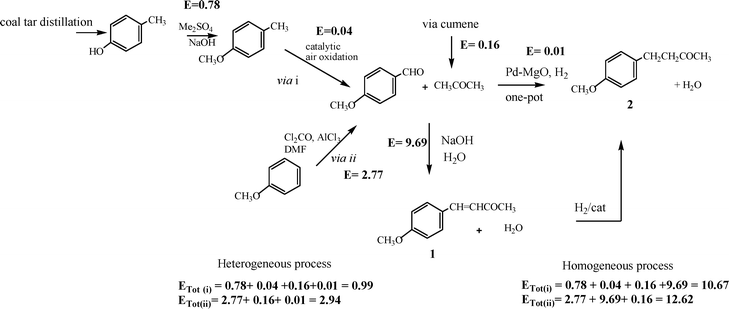 | ||
| Scheme 3 | ||
Considering that acetone is an easily recoverable solvent and the hydrogenation process does not produce any waste, the E factor for the one-pot reaction starting from p-methoxybenzaldehyde is 0.01, water being the main waste. The E factors for the syntheses of the reagents, acetone and 4-methoxybenzaldehyde, have also been calculated. For the synthesis of acetone we have selected the cumene route starting from benzene and propylene since this is the main industrial method used to produce acetone (see the ESI†). The total E factor for the production of acetone is 0.16.
On the other hand, the value for 4-methoxybenzaldehyde has been obtained according to two different routes (Scheme 3):
(i) by methylation of p-cresol giving p-methoxytoluene followed by catalytic oxidation with air, which has an E factor of 0.82 (see the ESI†).
(ii) by formylation of anisole through a Vilsmeier reaction,46 which has an E factor of 2.77 (see the ESI†). If we consider the global process, via i and via ii, the global E factors are 0.99 and 2.94, respectively (see Table 3).


 2
2If we follow the same methodology for the commercially accepted process where the aldol condensation is carried out using a 10% aqueous solution of NaOH35 (see Scheme 3), the specific E factor is 9.69, and when considering the global process, via i and via ii, the global E factors are 10.67 and 12.62, respectively (see Table 3).
These results indicate that considering the global process starting from p-cresol, the E factor for the procedure presented here with PdMgO is one order of magnitude lower than that of the conventional process (see Table 3). In Table 3, the specific E factors of all compounds involved in the production of compound 2 are summarized, as well as the global E factors following routes i and ii, performed using heterogeneous and homogeneous catalysts in the specific path for the synthesis of 2.
We have also considered here another potential procedure for the synthesis of 2 through two consecutive steps, involving a Heck coupling reaction between p-iodoanisole and methyl vinyl ketone (MVK) followed by hydrogenation (see Scheme 4). It has been reported that the synthesis of 2viaScheme 4 could be performed in a one pot, two-step process using palladium supported on titanium dioxide as a catalyst. Under optimized reaction conditions, this catalytic route gave 2 with 97% yield.12 Thus, for comparison purposes we have calculated the E factor for the synthesis of compound 2 through the Heck coupling reaction under heterogeneous and homogeneous catalysis, and the result, considering that the solvent can be fully recycled, is 1.96 (Scheme 4).
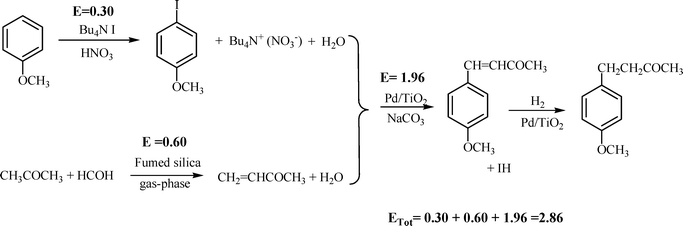 | ||
| Scheme 4 | ||
The waste produced during the synthesis of the reagents in Scheme 4 is also considered and, since the synthesis of 4-iodoanisole can be carried out by different methodologies, for calculation of the E factor we have selected a green process recently reported by Earle et al.47 which has an E factor of 0.30 (see the ESI†) This value is lower than E = 0.80 calculated when the iodation of anisole was performed with N-iodosuccinimide48 as the iodating agent in acetonitrile as solvent.
The second reactant, i.e. methyl vinyl ketone, is produced through the aldol condensation between acetone and formaldehyde. The process can be carried out in either the gas phase or liquid phase. In the liquid phase,49 the E factor is 0.99, while in the gas phase50 the E factor is 0.60 (see the ESI†). If we accept the greener, gas phase process for calculating the global E factor for the formation of the 4-(4-methoxyphenyl)-2-butanone via Heck coupling, the calculated value is E = 2.86 (see the ESI†). When this value is compared to that obtained during the aldol condensation with the solid catalyst (Scheme 3) in a one-pot reaction, it can be concluded that the heterogeneous Heck process produces about three times more waste than the heterogeneous aldol condensation.
Finally, we have considered that the synthesis 4-(4-methoxyphenyl)-2-butanone (2) can also be achieved through the Heck reaction by coupling an aryl halide with a α-methylene-β-hydroxy compound in the presence of a Pd complex catalyst (Scheme 5). Specifically, the process involves the reaction between p-bromoanisole and methyl-3-hydroxy-2-methylene butyrate using a tetraphosphine palladium complex as a catalyst, in DMF as a solvent and in the presence of K2CO3 or NaHCO3. The yield reported for 4-(4-methoxyphenyl)-2-butanone is 87%.51 The calculated E factor for the specific process, considering the solvent recoverable, is 3.11 (Scheme 5). The E factor involved in the preparation of the tetraphosphine palladium catalyst has not been considered here.
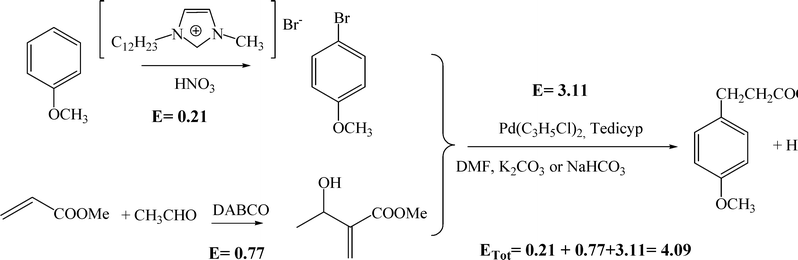 | ||
| Scheme 5 | ||
On the other hand, the calculated E factors for the preparation of p-bromoanisole and methyl-3-hydroxy-2-methylene butyrate are 0.21 and 0.77, respectively (see the ESI†). Considering these two values, plus the value for the reaction in Scheme 5, the global E factor for the preparation of raspberry scent following Scheme 5 is 4.09. This is about 1.5× higher than the heterogeneous process described above, and approximately 5× higher than the one-pot process using the PdMgO catalyst.
In Table 4 the specific E factors of each compound involved in the synthesis of 2 are summarized as well as the global E factor for the heterogeneous and homogeneous Heck coupling rection.




In conclusion, a new synthetic process for 4-(4-methoxyphenyl)-2-butanone with raspberry scent has been outlined that involves a one-pot process with a multifunctional (metal acid–base) solid catalyst and which has a much lower E factor than any reported before.
2. Preparation of 4-(6,6-dimethylbicyclo[3,1,1]hept-2-en-2-yl)-2-butanone (3)
Terpenes are important compounds for the fragrance and flavour industry. The terpenic compound 4-(6,6-dimethylbicyclo[3,1,1]hept-2-en-2-yl)-2-butanone (3) (Scheme 6), is used as perfume and possesses activity as an insect juvenile hormone.52 The conventional preparation of 3 is carried out through the acetoacetic reaction between myrthenyl bromide and ethyl acetoacetate in the presence of stoichiometric amounts of sodium ethoxide at the reflux temperature of ethanol yielding, after 4 h, 78% of ethyl 3-oxo-2-(2-pinen-10-yl) butyrate. Thus, in a second synthetic step, the ketoester is decarboxylated by refluxing for 18 h with barium hydroxide in aqueous ethanol giving 3 in 85% yield.53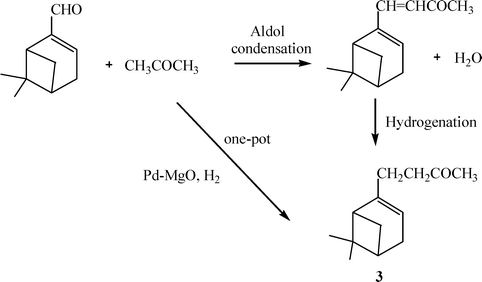 | ||
| Scheme 6 | ||
We have calculated the E factor of each step of the process given in Scheme 7, starting from β-pinene, whose bromination to myrthenyl bromide can be accomplished with N-bromosuccinimide in refluxing CCl4, yielding 51% of myrthenyl bromide.54 Taking into account that solvent and unreacted β-pinene are recoverable, we have calculated an E factor of 1.18 for the production of myrthenyl bromide. On the other hand, the second reactant involved, i.e., ethyl acetoacetate, can be produced by a solvent-free Claisen condensation of ethyl acetate in the presence of tBuOK that yields 75% of ethyl acetoacetate. The calculated E factor for this reaction is 2.78. Thus, the E factor of the global process presented in Scheme 7 to prepare the desired product, 3, is 5.45.
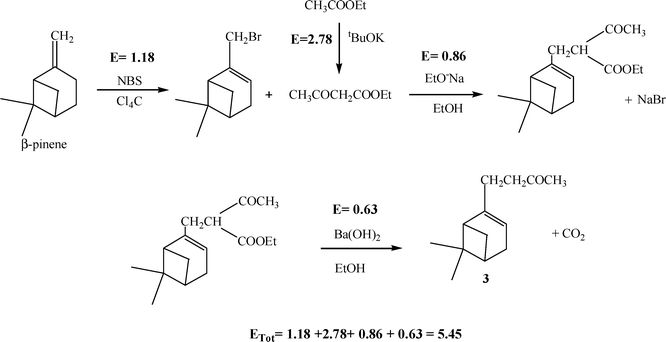 | ||
| Scheme 7 | ||
An alternative synthetic approach is presented here (Scheme 8), in which 3 can be prepared in a one-pot cascade reaction which involves an aldol condensation between myrthenal and acetone, followed by dehydration of the aldol formed, and selective double bond hydrogenation. For calculating the E factor of the process, it has been considered that myrthenal can be obtained by oxidation of α-pinene with SeO2.55 The oxidation of α-pinene is carried out with equimolar amounts of SeO2 in the presence of a solvent such as ethanol yielding 50% of myrthenal. The E factor for this step (see Scheme 8), considering that both ethanol and the remaining α-pinene can be recycled, is 1.3. The E factor for the global process given in Scheme 8 is therefore 1.6. This value is 3.4× lower than the homogeneous process described previously for the preparation 4-(6,6-dimethylbicyclo[3,1,1]hept-2-en-2-yl)-2-butanone (3).
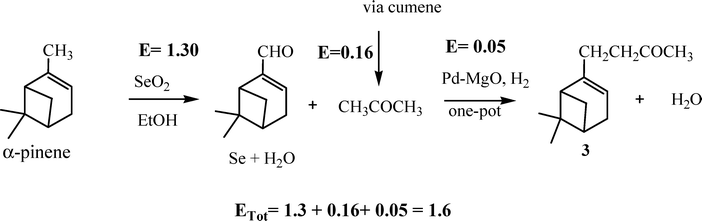 | ||
| Scheme 8 | ||
3. Preparation of 2-cyclopentylcylopentanone (4)
The product 2-cyclopentylcylopentanone (4) with a jasmine-like smell is used for perfume or flavouring56 and as a wood preservative.57 2-Cyclopentylcyclopentanone can be synthesized by different multi-steps methods, for instance by the acyloin condensation of dimethyl glutarate in the presence of sodium (Scheme 9).58 However, the preferred method in terms of yield and availability of starting materials, involves the condensation of cyclopentanone in the presence of a basic catalyst (NaOH, KOH or sodium) followed by the hydrogenation of the resulting 2-cyclopentilydene-cyclopentanone in the presence of a hydrogenating catalyst such as Pd/C59 (Scheme 10). Thus, by reacting cyclopentanone at 90 °C in the presence of a 10% aqueous solution of NaOH, a 54% yield of 2-cyclopentilydene-cyclopentanone (5) was obtained after 3 h reaction time. After purification by distillation at reduced pressure, the compound is hydrogenated using palladium (5 wt%) on activated carbon as catalyst, yielding 89% of 2-cyclopentylcyclopentanone (4). The main drawback associated with this methodology is, in general, the low yields of 2-cyclopentilydene-cyclopentanone (6) achieved, due to the fact that the self-condensation product can further condense with a second cyclopentanone molecule to form the highly conjugated trimer adduct (2,5-dicyclopentylidenecyclopentanone) (6). Due to the high thermodynamic stability of the conjugated trimer adduct, this compound is formed in important amounts that, depending on reaction conditions, can reach to between 30 and 50%.60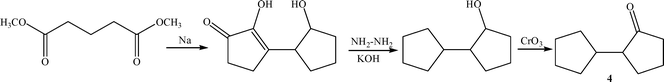 | ||
| Scheme 9 | ||
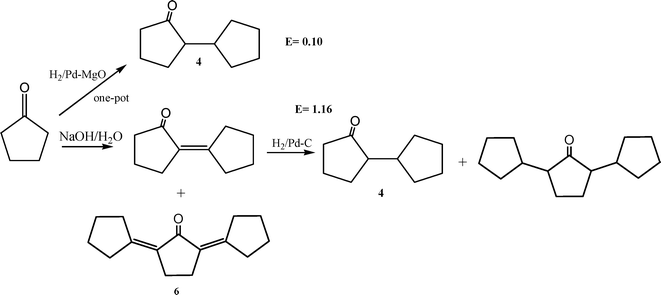 | ||
| Scheme 10 | ||
It appeared to us that if the reaction was performed in a one-pot system in which the rate of hydrogenation of the condensation product was very high, then the second condensation that leads to formation of the trimer (6) could be avoided or, at least, could be strongly diminished, giving a higher final selectivity to 2-cyclopentylcylopentanone (5) (Scheme 10). To do this, cyclopentanone was reacted in the presence of PdMgO under a constant hydrogen pressure at 140 °C. After 8 h reaction time, the conversion was 98% with 91% selectivity for 4. Under our reaction conditions, the hydrogenated trimer (2,5-dicyclopentylcyclopentanone) (6) and 2-cyclopentylcylopentanol were also detected, with yields of 10 and 2%, respectively. The higher selectivity towards 4 achieved in this case, with respect to the conventional procedure, demonstrates the advantage of the one-pot cascade process using the PdMgO catalyst. In this case, it appears that the fast hydrogenation of the intermediary, 2-cyclopentilydene-cyclopentanone, diminishes the rate of the subsequent aldol condensation that leads to the undesired highly-conjugated trimer.
As shown in Scheme 10, the E factor for the heterogeneous-catalyzed, one-pot cascade process is one order of magnitude lower than the conventional method.
Table 5 summarizes the E factors for the synthesis of 4 using heterogeneous and homogeneous catalysts.
| Compound | E factor (heterogeneous) | E factor (homogeneous) |
|---|---|---|
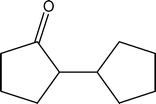 4 4 | 0.10 | 1.16 |
Conclusions
We have illustrated here that process intensification by means of multifunctional solid catalysts that allow one-pot cascade reactions, opens new, more environmentally benign routes than those currently used for the synthesis of fine chemicals.Since, in many cases, the new routes proposed require starting from different reactants than the conventional routes, we have also calculated the E factor for the global process starting from the primary products. It has been seen that, in most cases, the new one-pot cascade reactions have E factors which are one order of magnitude lower. The differences in the E factor can be even larger if one considers the energy involved and even the investments, since the one-pot processes do not require intermediate separations.
Finally, it has been shown that, in some cases, higher selectivities can be achieved in the one-pot cascade reactions with multifunctional catalysts, by adjusting the relative rates of the different consecutive steps.
Experimental
Preparation of catalysts
A nanocrystalline MgO sample was purchased from NanoScale Materials Inc.The magnesium–aluminium layered double hydroxide sample (Mg–Al LDH) was prepared from a gel produced by mixing two solutions, A and B. Solution A contained (3 −x) mol of Mg(NO3)2·6H2O and x mol of Al(NO3)3·9H2O in the (Al + Mg) concentration of 1.5 mol L−1 for an Al/(Al + Mg) molar ratio of 0.25. Solution B was formed by (6 + x) mol of NaOH and 2 mol of Na2CO3 dissolved in the same volume as solution A. Both solutions were co-added at a rate of 1 mL min−1 under vigorous mechanical stirring at room temperature. The gel was aged under autogeneous pressure conditions at 333 K for 12 h. Then, the hydrotalcite was filtered and washed until pH = 7 and the solid was dried. The Mg–Al mixed oxide (HTc) base catalyst was obtained by calcination of Mg–Al-LDH at 723 K for 6 h in a dry nitrogen flow.
Pd catalysts at 1 wt% loading were obtained by exposing the Mg–Al-LDH or MgO samples to an anhydrous toluene solution with the required amount of palladium acetylacetonate (Pd(OAc)2) for 12 h. After evaporation of toluene at reduced pressure, the solid was dried overnight at 80 °C in vacuum and then calcined in nitrogen flow at 550 °C for 3.5 h. These samples were denoted as PdHTc (calcined hydrotalcite) and PdMgO
The PdHTc sample was rehydrated at room temperature by direct addition of decarbonated water (MilliQ) (36 wt%) just before their use as catalyst, and was labelled as PdHTr.
Samples of PdMgO and PdHTc were activated before reaction by heating the solid at 450 °C under air for 5 h and then 5 h under nitrogen. Reduction of the Pd(II) was performed by heating the solid at 450 °C in a flow of H2/N2 (90/10) for 2 h.
The catalysts prepared were characterized by XRD (Fig. 1), TEM and nitrogen adsorption. N2 adsorption/desorption isotherms were performed at 77 K in an ASAP 2010 apparatus from Micromeritics, after pre-treating the samples under vacuum at 673 K overnight and the BET surface areas were obtained using the BET methodology. The main characteristics of the catalysts are given in Table 6.
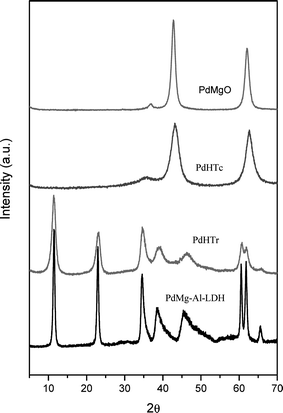 | ||
| Fig. 1 XRD patterns of the solid catalysts. | ||
Reaction procedure
For a typical experiment, the reactants were placed in a batch slurry reactor which was immersed in a thermostated silicone oil bath and magnetically stirred. When the temperature was reached, the catalyst was added and hydrogen was applied at a constant hydrogen pressure of 5 bar. The course of the reaction was followed by gas chromatography (GC) using a Hewlett Packard GC 5988 A with FID detector and a HP-5 capillary column of 5% phenylmethylsilicone. In all the experiments, dodecane was used as an internal standard. At the end of the reaction, after cooling, the reaction mixture was filtered to remove the catalyst. The solid was extracted thoroughly in a Soxhlet apparatus using methanol as a solvent and the extract considered in the mass balance. The reaction mixture was concentrated under reduced pressure and the reaction products were analyzed by GC-MS (Varian Saturn II, using the same column as GC) and 1H NMR spectroscopy (Varian Gemenis- 300 MHz, CDCl3 as a solvent).One-pot synthesis of 4-(4-methoxyphenyl)2-butanone (2)
4-methoxybenzaldehyde (3.8 mmol), was reacted with acetone (37 mmol) in the presence of PdMgO (1 wt% of Pd) (38 wt% with respect to 4-methoxybenzaldehyde) under constant hydrogen pressure (5 bar) at 75 °C. After 75 min reaction time, 100% conversion of 4-methoxybenzaldehyde with 99% selectivity for 2 was obtained.One-pot synthesis of 4-(6,6-dimethylbicyclo[3,1,1]hept-2-en-2-yl)-2-butanone (3)
Myrthenal (3.9 mmol) was reacted with acetone (48 mmol) in the presence of PdMgO (1 wt% of Pd) (34 wt% with respect to myrthenal) under constant hydrogen pressure (5 bar) at 75 °C for 3 h. The target molecule was obtained in 95% yield.One-pot synthesis of 2-cyclopentylcylopentanone (4)
Cyclopentanone (10.7 mmol) was reacted in the presence of PdMgO (21 wt%) under a constant hydrogen pressure (5 bar) at 140 °C during 8 h. A 98% conversion of cyclopentanone with 91% selectivity for 4 was achieved.Regeneration of the catalysts
PdHTr and PdMgO samples were regenerated after each use by calcination of the solids at 450 °C under air atmosphere for 5 h and then under nitrogen for 5 h. Reduction of the Pd was performed by heating the solid at 450 °C in a flow of H2/N2 (90/10) for 2 h.Acknowledgements
The authors thank CICYT for financial support (Project MAT 2006-14274-C02-01). The contribution of Mr Pablo Ramos to the experimental work reported herein is also gratefully acknowledged.References
- R. A. Sheldon, Chem. Ind. (London), 1992, 903 CAS.
- A. Bruggink, R. Schoevaart and T. Kieboom, Org. Process Res. Dev., 2003, 7, 622 Search PubMed.
- N. Hall, Science, 1994, 266, 32 CrossRef.
- K. M. Koeller and C. H. Wong, Chem. Rev., 2000, 100, 4465 CrossRef CAS.
- F. Gelman, J. Blum and D. Avnir, J. Am. Chem. Soc., 2000, 122, 11999 CrossRef CAS.
- J. C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001 CrossRef CAS.
- A. Corma and M. Renz, Angew. Chem., Int. Ed., 2007, 46, 298 CrossRef CAS.
- A. Corma, M. Renz and C. Schaverien, ChemSusChem, 2008, 1, 739 CrossRef CAS.
- A. Corma, V. Fornes, S. Iborra, M. Mifsud and M. Renz, J. Catal., 2004, 221, 67 CrossRef CAS.
- X. Zhang and A. Corma, Angew. Chem., Int. Ed., 2008, 47, 4358 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and M. Mifsud, J. Catal., 2007, 247, 223 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and M. Mifsud, Adv. Synth. Catal., 2007, 349, 1949 CrossRef CAS.
- B. M. Choudary, N. S. Chowdari, S. Madhi and M. L. Kantam, J. Org. Chem., 2003, 68, 1736 CrossRef CAS.
- F. Zhang, G. Liu, W. He, H. Yin, X. Yang, H. Li, J. Zhu, H. Li and Y. Lu, Adv. Funct. Mater., 2008, 18, 3590 CrossRef CAS.
- F. X. Felpin and E. Fouquet, ChemSusChem, 2008, 1, 718 CrossRef CAS.
- M. J. Climent, A. Corma and S. Iborra, ChemSusChem, 2009, 2, 500 CrossRef CAS.
- A. Corma, P. Serna and H. Garcia, J. Am. Chem. Soc., 2007, 129, 6358 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra, L. L. Santos, Chem. A Eur. J., in press Search PubMed.
- N. Das, D. Tichit, R. Durand, P. Graffin and B. Coq, Catal. Lett., 2001, 71, 181 CrossRef CAS.
- R. Unnikrishnan and S. Narayanan, J. Mol. Catal. A: Chem., 1999, 144, 173 CrossRef CAS.
- Y. Z. Chen, C. M. Hwang and C. W. Liaw, Appl. Catal., A, 1998, 169, 207 CrossRef CAS.
- W. Ferry, A. J. van Pillen and K. P. de Jong, J. Mol. Catal. A: Chem., 2004, 219, 273 CrossRef CAS.
- M. J. Martínez-Ortiz, D. Tichit, P. Gonzalez and B. Coq, J. Mol. Catal. A: Chem., 2003, 201, 199 CrossRef CAS.
- J. I. Di Cosimo, G. Torres and C. R. Apesteguía, J. Catal., 2002, 208, 114 CrossRef CAS.
- C. Carlini, C. Flego, M. Marchionna, M. Noviello, A. M. Raspolli Galletti, G. Sbrana, F. Basile and A. Vaccari, J. Mol. Catal. A: Chem., 2004, 220, 215 CrossRef CAS.
- C. Carlini, C. Flego, M. Marchionna, M. Noviello, A. M. Raspolli Galletti, G. Sbrana, F. Basile and A. Vaccari, J. Mol. Catal. A: Chem., 2005, 232, 13 CrossRef CAS.
- G. Torres, C. R. Apesteguia and J. I. Di Cosimo, Appl. Catal., A, 2007, 317, 161 CrossRef CAS.
- D. Tichit, M. J. Martinez-Ortiz, D. Francova, C. Gerardin, B. Coq, R. Durand, F. Prinetto and G. Ghiotti, Appl. Catal., A, 2007, 318, 170 CrossRef CAS and references cited therein.
- D. Tichit, B. Coq, S. Cerneaux and R. Durand, Catal. Today, 2002, 75, 197 CrossRef CAS.
- K. Motokura, N. Fujita, K. Mori, T. Mizugapi, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2005, 46, 5507 CrossRef CAS.
- K. Kasahara, H. Nakanishi, JP Pat., 2002370929, 2002.
- Badische Anilin and Soda Fabrick, A. G. Ger, Offen. Pat., 2145308, 1973.
- Chisso Corp Jpn. Kokai Tokkyo Koho JP Pat. 55151530 (1980).
- J. Tateiwa, H. Horiuchi, K. Hashimoto, T. Yamauchi and S. Uemura, J. Org. Chem., 1994, 59, 5901 CrossRef CAS.
- S. Paul and M. Gupta, Synth. Commun., 2005, 35, 213 CrossRef CAS.
- Y. Kido, T. Hamazaki, K. Yoneda and T. Ohnishi, JP Pat., 2000103754, 2000.
- A. Corma and S. Iborra, Adv. Catal., 2006, 49, 239 CAS.
- R. Teissier, D. Tichit, F. Figueras, and J. Kervennal, F Pat., 2729137, 1995.
- M. J. Climent, A. Corma, S. Iborra and A. Velty, J. Catal., 2004, 221, 474 CrossRef CAS.
- (a) A. Guida, M. H. Lhouty, D. Tichit, F. Figueras and P. Geneste, Appl. Catal., A, 1997, 164, 251 CrossRef CAS; (b) K. K. Rao, M. Gravelle, J. Valente and F. Figueras, J. Catal., 1998, 173, 115 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and A. Velty, Catal. Lett., 2002, 79, 157 CrossRef CAS.
- J. A. A. Roelofs, D. J. Lensveld, A. J. van Dillen and K. P. De Jong, J. Catal., 2001, 203, 184 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra and A. Velty, Green Chem., 2002, 4, 474 RSC.
- M. L. Kantam, B. M. Choudary, C. V. Reddy, K. K. Rao and F. Figueras, Chem. Commun., 1998, 1033 RSC.
- B. M. Choudary, M. L. Kantam, C. V. Reddy, K. K. Rao and F. Figueras, J. Mol. Catal. A: Chem., 1999, 146, 279 CrossRef CAS.
- G. Vergne, O. Dabard and F. Cornille, FR Pat., 2824555, 2001 (assigned to Societe Nationale des poudres et explosives).
- M. Earle and S. P. Katdare, WO Pat., 0230852, 2002.
- A. S. Castanet, F. Colobert and P. E. Broutin, Tetrahedron Lett., 2002, 43, 5047 CrossRef CAS.
- J. Pugach and J. S. Salek, US Pat., 5004839, 1990 (assigned to Aristech Chemical Corp).
- I. Gummin and P. Noesberger, WO Pat., 2007115832, 2007 (assigned to DSM IP ASSETS B.V.).
- F. Berthiol, H. Doucet and M. Santelli, Tetrahedron, 2006, 62, 4372 CrossRef CAS.
- L. Borowiecki, A. Kazubski, E. Reca and W. Wlodzimierz, Liebigs Ann. Chem., 1985, 1985, 929 CrossRef.
- L. Borowiecki, A. Kazubski and E. Reca Liebigs, Ann. Chem., 1982, 1766 CAS.
- G. Zweifel and C. C. Whitney, J. Org. Chem., 1966, 31, 4178 CrossRef CAS.
- (a) J. A. Retamar, Bull. Soc. Chim. Fr., 1996, 1227; (b) J. A. Retamar, Essenze Derivati Agrumari, 1989, 59, 159 Search PubMed.
- S. ShiozakiM. Senuma, S. Furumai and H. Kawashima, E Pat., 16650, 1980.
- Nippon Zeon Co., Jpn. Kokai Tokkyo Koho JP Pat., 57056401, 1982.
- M. Cordon, J. D. Knight and D. J. Cram, J. Am. Chem. Soc., 1954, 76, 1643 CrossRef CAS.
- S. Shiozaki, M. Senuma, S. Furumai and H. Kawashima, E Pat., 16650 19801001, 1980.
- (a) P. Rollin, Bull. Soc. Chim. Fr., 1973, 4, 1509; (b) S. V. Svetozarskii, G. A. Razuvaev, E. N. Zilberman and G. S. Volkov, Zh. Obshch. Khim., 1960, 30, 2042 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of E factor calculations. See DOI: 10.1039/b919660a |
| This journal is © The Royal Society of Chemistry 2010 |