High performance magnetic separation of gold nanoparticles for catalytic oxidation of alcohols†
Rafael L. Oliveiraa, Pedro K. Kiyoharab and Liane M. Rossi*a
aInstituto de Química, Universidade de São Paulo, 05508-000, São Paulo, SP, Brazil. E-mail: lrossi@iq.usp.br; Fax: +55 11 3815 5579; Tel: +55 11 30912181
bInstituto de Física, Universidade de São Paulo, 05508-090, São Paulo, SP, Brazil
First published on 29th October 2009
Abstract
We present the magnetic separation approach to facilitate the recovery of gold nanoparticle (AuNP) catalysts. The use of magnetically recoverable supports for the immobilization of AuNPs instead of traditional oxides, polymers or carbon based solids guarantees facile, clean, fast and efficient separation of the catalyst at the end of the reaction cycle. Magnetic separation can be considered an environmentally benign separation approach, since it minimizes the use of auxiliary substances and energy for achieving catalyst recovery. The catalyst preparation is based on the immobilization of Au3+ on the surface of core–shell silica-coated magnetite nanoparticles, followed by metal reduction using two different methods. AuNPs were prepared by thermal reduction in air and by hydrogen reduction at mild temperature. Interestingly, the mean particle size of the supported AuNPs was similar (ca. 5.9 nm), but the polydispersity of the samples is quite different. The catalytic activity of both catalysts in the aerobic oxidation of alcohols was investigated and a distinct selectivity for benzyl alcohol oxidation was observed.
Introduction
The development of new strategies for the recovery and recycling of catalysts, which minimizes the consumption of auxiliary substances, energy and time used in achieving separations can result in significant economical and environmental benefits.Gold in the bulk form has been regarded as being chemically inert towards chemisorption of reactive molecules such as oxygen and hydrogen. Consequently, bulk gold was considered to be an uninteresting metal from the point of view of catalysis. However, the catalytic properties of gold are revealed when the size is reduced to few nanometers, particularly with dimension less than 10 nm.1 The overall performance of gold nanoparticle (AuNP) catalysts highly depends on the size and shape of the nanoparticles, the structure and properties of the catalyst support and the gold–support interface interactions.2 A variety of interesting catalytic properties of AuNPs in both oxidation and reduction reactions has been reported.1-3 Despite this, little attention has been paid to possible difficulties arising from size reduction, and the challenges of separating the very small particles, most of the time in a colloidal equilibrium, from the products. In this regard, AuNPs have been dispersed on solid supports to increase the catalyst stability and to facilitate the catalyst recovery.3 As the size of the support decreases, separation using physical methods, such as filtration or centrifugation, becomes a difficult and time-consuming procedure. Simple filtration is inefficient to accomplish product isolation in systems comprised of nanoparticles stabilized in solution or very thin solids. In these circumstances, an alternative that has drawn attention lately is the use of magnetically recoverable solid supports. Magnetic separation is easy, fast, clean and a very promising recovery tool for isolation of thin solids from the reaction medium. It can be considered an environmentally benign separation approach, since it minimizes the use of auxiliary substances and energy for achieving catalyst recovery. Magnetic supports, mainly composed of superparamagnetic nanoparticles, can be attracted to relatively low static magnetic field strengths and are easily recovered, since no residual magnetization is observed when the magnetic field is removed. The immobilization of homogeneous and NP-based catalysts on magnetically separable solid supports has been shown to be a powerful tool for catalyst recovery and recycling.4-7
We report herein the preparation of AuNPs supported on a magnetic solid that can be easily recovered from solution by applying an external magnetic field, while keeping good control over different parameters, such as particle size, size distribution and catalytic activity. The oxidation of alcohols was used to probe the catalytic activity of the catalyst. Catalytic oxidations are green alternatives to stoichiometric oxidations with permanganate, manganese dioxide and chromium(VI) reagents, major sources of waste, particularly in fine chemicals and pharmaceuticals manufacture.8
Experimental
Synthesis of silica-coated magnetite nanoparticles
The synthesis of oleic acid-coated Fe3O4 nanoparticles and the core–shell Fe3O4@SiO2 composite was reported elsewhere.7 The solid was modified with amino groups by reaction with 3-aminopropyltriethoxysilane (APTES) in dry toluene under N2, to give the amino-functionalized support (Fe3O4@SiO2/NH2). The final solid was washed with toluene and dried at 100 °C for 20 h.Synthesis of supported Au(0) nanoparticles
Supported AuNPs were prepared as follows: 50 mg of Fe3O4@SiO2/NH2 was added to 10 mL of an aqueous HAuCl4 solution (1 g L−1, pH∼6). The mixture was stirred at 25 °C for 2 h. The solid was then magnetically collected from the solution and washed twice with hot distilled water. The Au(0)NPs were obtained by reduction of the Au(III) precursor either by thermal treatment (oven, 150 °C (constant), 8 h, in air) or by H2 reduction (glass reactor, 650 mg 2, 3 mL cyclohexane, 80 °C, 4 atm H2, 3 h). The color of the solid changes from brown to dark purple after formation of Au NPs.Characterization methods
High-resolution transmission electron microscopy (HRTEM) and X-ray energy dispersive spectroscopy (EDS) were performed at the Laboratório Nacional de Luz Síncrotron (LNLS, Campinas, SP) using a Jeol-3010 ARP microscope, and conventional TEM images were performed using a Philips CM 200 microscope (IF-USP). Samples for TEM observations were prepared by placing a drop containing the nanoparticles in a carbon-coated copper grid. The histograms of the gold nanoparticles size distribution, assuming a spherical shape, were obtained from the measurement of the diameter of about 300 particles found in arbitrarily chosen areas of enlarged micrographs. The samples were observed in different regions of the Cu grid.Catalytic experiments
All reactions were performed using a modified Fischer–Porter glass reactor loaded under a positive pressure of dry N2 from a Schlenk line. Liquids and solutions were transferred via a syringe. In a typical reaction, the glass reactor was loaded with the supported Au(0)NPs catalyst (50 mg, 1.8 μmol Au), the substrate (1.5 mmol), and 2 mL of toluene. The reactor, immersed in an oil bath, was loaded with O2 to the desired pressure. The temperature was maintained by an oil bath placed in a hot-stirring plate connected to a digital controller (ETS-D4 IKA). The reactions were conducted under magnetic stirring, using a Teflon-coated magnetic stir bar, for the desired time. The catalyst was magnetically recovered by placing a permanent magnet in the reactor wall and the products were collected with a syringe and analysed by gas chromatography (GC) and GC-MS. The isolated catalyst could be reused when a new amount of substrate was added.Results and discussion
A schematic illustration of the step-by-step approach used for the preparation of gold catalysts composed of supported AuNPs on a magnetically recoverable solid is shown in Scheme 1. The magnetically recoverable support used in this study consists of core–shell silica-coated magnetite nanoparticles (Fe3O4@SiO2) prepared following the procedure described recently.6 The approach to prepare AuNPs is based on the uptake of Au3+ ions from HAuCl4 aqueous solution by the silica-coated magnetic nanoparticles, previously functionalized with –NH2 groups, followed by metal reduction under mild conditions. The amino-functionalized support (1) was loaded with 0.72 wt% of gold, as determined by atomic absorption analysis (ICP OES). It is interesting to notice that the non-functionalized support, when submitted to a similar metal solution, do not adsorb gold ions. The Au3+-loaded magnetic solid (2) was used as precursor for the preparation of supported AuNPs either by thermal treatment (catalyst 3) or by H2 reduction (catalyst 4). The formation of the AuNPs could be evidenced by the SPR bands at 567 nm (catalyst 3) and 522 nm (catalyst 4) observed in the UV-Vis spectra shown in Fig. 1.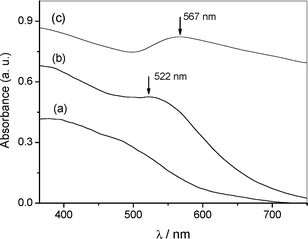 | ||
| Fig. 1 UV-Vis spectra of ethanolic suspensions of Fe3O4@SiO2-NH21 (a), Au(0) catalyst 4 (b) and Au(0) catalyst 3 (c). | ||
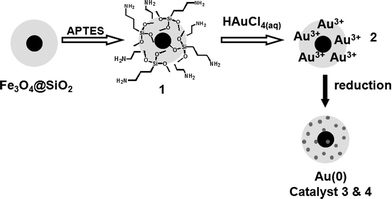 | ||
| Scheme 1 Step-by-step preparation of magnetically recoverable AuNPs. | ||
The morphology of the supported AuNPs were examined by transmission electron microscopy (TEM), shown in Fig. 2a and b. The support is comprised of magnetic cores (Fe3O4∼ 10 nm) spherically coated with silica (spheres of ∼ 40 nm in diameter). The mean diameter of the supported AuNPs in both catalysts was estimated at 5.9 nm with polydispersity of 18% for catalyst 4 and 32% for catalyst 3. The AuNPs are fairly well-distributed over the support in both catalysts; however, the particle size distribution of catalyst 4 is sharper if compared to catalyst 3, as can be seen in the size distribution histograms adjusted to lognormal functions presented in Fig. 2c–d. The SPR band of catalyst 4 appears at λmaxca. 520 nm, which agrees with the higher monodispersity of the AuNPs. Moreover, the λmax red shift of about 45 nm and the broadened optical spectrum of catalyst 3 can be realized by aggregation of small particles, light scattering and lower overall quality of the supported nanoparticles.9
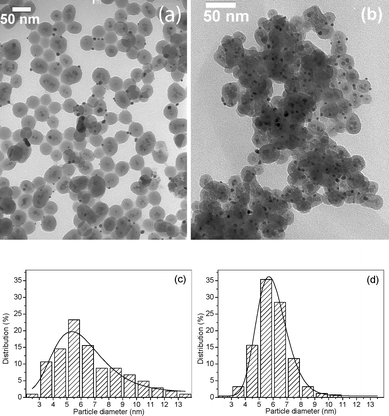 | ||
| Fig. 2 TEM images of catalyst 3 (a) and catalyst 4 (b). Histogram showing particle size distribution and lognormal fitting of the AuNPs of catalyst 3 (c) and the AuNPs of catalyst 4 (d). | ||
The distribution and composition of each nanoparticle of the nanocomposite was revealed by energy dispersive spectroscopy (EDS) analysis of the image shown in Fig. 3. The core nanoparticles were found to be exclusively composed of Fe atoms (Fig. 3b), and the darker nanoparticles of Au atoms (Fig. 3c), through a detailed EDS analysis of the image areas shown in box (b) and box (c), respectively. The EDS analysis shown in Fig. 3a corresponds to the whole nanocomposite which contains Fe, Au and Si (Cu from sample holder).
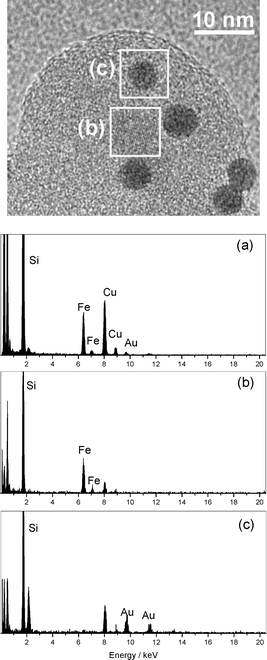 | ||
| Fig. 3 HRTEM image and EDS analysis of the AuNP magnetically recoverable catalyst 4 in different areas of the image: (a) corresponds to the whole area, (b) corresponds to the iron oxide particle shown in box b and (c) corresponds to the gold particle shown in box c. | ||
The magnetic properties of the Fe3O4@SiO2 nanocomposite were investigated elsewhere.7 It displays excellent magnetic properties characterized by negligible coercivity and remanence, and very high saturation magnetization MS at room temperature (MS∼ 69 emu g−1 at 70 kOe). These magnetic properties describe a superparamagnetic material that responds to an external magnetic field but does not remain magnetized when the magnetic field is removed. Such materials can be easily concentrated from the solution with a magnet and immediately redispersed after removing the magnetic field, since the absence of remanence prevents aggregation of particles.
The catalytic oxidation of benzyl alcohol was used to probe the activity and recovery of the supported AuNPs in catalysts 3 and 4. The first attempt to catalyze the oxidation of benzyl alcohol using our supported AuNPs was performed under atmospheric pressure of O2 and no products (aldehyde or ester) were observed (Table 1, entry 1). It was also observed that in the absence of base, the catalyst was not efficient in catalyzing the selective oxidation of alcohols, even at 100 °C and 5 atm O2 (Table 1, entry 2). The conversion increased to 75% (95% selectivity) as long as a small amount of K2CO3 was present (Table 1, entry 3). As it can be seen in Fig. 4, there is a strong dependence of the oxidation reaction with the O2 pressure for both catalysts 3 and 4. Catalyst 3 is less active and requires higher pressure for the substrate conversion, but the selectivity was maintained in the pressure range studied. On the other hand, catalyst 4 is more active, reaching 100% conversion at 3 atm of O2, but the high activity is accompained by the drop of selectivity to benzaldehyde to ca. 50%. The oxidation reaction catalyzed by catalyst 3 is highly selective to benzaldehyde, found as the main product. It is also known that benzaldehyde can be further oxidized into benzoic acid, and, finally, benzyl alcohol and benzoic acid would interact and produce benzyl benzoate.10 In fact, benzyl benzoate was found as the only by-product in the oxidation of benzyl alcohol by catalysts 3 and 4.
| Entry | Catalyst | Substrate | P O2 (atm) | Conversion (%)b | Selectivity (%)b | |
|---|---|---|---|---|---|---|
| Aldehyde or ketone | Ester (homocoupling) | |||||
| a Reaction conditions: 1.5 mmol of substrate, 2 mL of toluene, 50 mg of catalyst (1.8 μmol Au), 70 mg K2CO3, 100 °C, reaction time 6 h.b Determined by GC-MS.c Bubbling O2.d Without base. | ||||||
| 1 | 3 | Benzyl alcohol | —c | 0.0 | — | — |
| 2d | 3 | Benzyl alcohol | 5 | 2.2 | 99 | 1 |
| 3 | 3 | Benzyl alcohol | 5 | 75 | 95 | 5 |
| 4 | 3 | Benzyl alcohol | 6 | 84.3 | 95 | 5 |
| 5 | 4 | Benzyl alcohol | 3 | 100 | 47 | 53 |
| 6 | 3 | 1-Phenyl ethanol | 6 | 100 | 100 | 0 |
| 7 | 4 | 1-Phenyl ethanol | 3 | 100 | 100 | 0 |
| 8 | 3 | Cyclohexanol | 6 | 31 | 100 | 0 |
| 9 | 4 | Cyclohexanol | 6 | 42 | 100 | 0 |
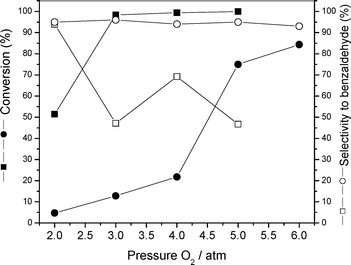 | ||
| Fig. 4 Pressure dependence of the catalytic performance of supported AuNPs: catalyst 3 (circle) and catalyst 4 (square). Reaction conditions: 1.5 mmol of benzyl alcohol, 50 mg catalyst, 70 mg K2CO3, 2 mL toluene, 100 °C, 6 h. | ||
Secondary alcohols such as 1-phenyl ethanol and cyclohexanol (Table 1, entries 6–9) were also oxidized to the corresponding ketones with 100% selectivity. In all experiments, the catalyst was recovered by using a permanent magnet and the products were collected with a syringe and analysed by GC-MS.
Recycling experiments were examined for the catalytic oxidation of benzyl alcohol at 5 atm of O2 and 100 °C using catalysts 3 and 4. After the first reaction, the catalyst was recovered in the reactor wall by using a permanent magnet, the products were collected with a syringe, and the recovered catalyst was washed with ethanol (2 mL) inside the reactor, and dried under vaccum. The catalyst was reused in successive oxidation reactions by adding new portions of solvent, base and substrate. This procedure was repeated four times, however the supported AuNP catalyst 3 lost 50% of the initial conversion, considering the same reaction time. After the 4th cycle, the conversion of benzyl alcohol was 35% but the selectivity to aldehyde was maintained as shown in Fig. 5. The deactivation process of catalyst 4 is more severe than catalyst 3 under similar experimental conditions. The activity of 100% in the 1st run drops to less than 10% in the 4th run.
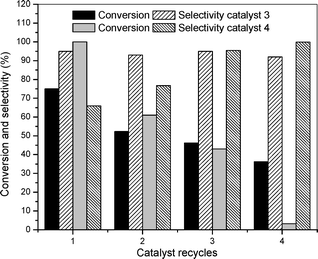 | ||
| Fig. 5 Conversion and selectivity of the catalytic oxidation of benzyl alcohol to benzaldehyde using the magnetically recovered AuNP catalysts 3 and 4 (conditions similar to entry 3, Table 1). | ||
The low reusability of the catalyst is a point of concern. One of the key factors that must be considered is the possibility that the active metal would leach into the reaction mixture, especially in liquid phase oxidation reactions, because of the possible dissolution of metals by the reactants and, particularly, the carboxylic acid-type (by)products, thereby leading to catalyst deactivation. To address this possibility, the isolated products obtained after magnetic separation were analysed by atomic absorption analysis (ICP OES). Results have shown that negligible Au leaching of our catalysts occurs (less than the limit of quantitation of 2.5 ppb of Au). This demonstrates the high affinity of gold and our magnetic support, the high performance of the magnetic separation in the present catalytic system and, as a first conclusion, indicates that the catalyst deactivation is not related to metal loss. Another key factor to be considered is the stability of the catalyst support and nanoparticles when subjected to the reaction conditions. TEM images and EDS analysis of the spent catalysts (see ESI†, Fig. S1) and TEM image of the catalyst support subjected to basic reaction conditions (see ESI†, Fig. S2) provided supporting evidence for the catalyst deactivation under reuse. First, the morphology of the catalyst support (core–shell Fe3O4@SiO2) has changed; the silica layer was strongly affected by the presence of base in the reaction medium and the catalyst support lost the spherical morphology, which caused aggregation. Second, large Au particles (∼80 nm) were revealed in the TEM image of spent catalyst, which suggests the coalescence of the small, active gold particles into larger, less active or inactive particles. The growth of the Au particles, above the catalytic limit of about 10 nm,1 can also be a consequence of the changes occurring in the catalyst support during the reaction. Therefore, the use of base associated with a silica-based catalyst support should be the main reason for the catalyst deactivation. Another point that might be relevant concerns the presence of potassium as detected by EDS analysis. This is also a consequence of the use of base that can cause metal surface contamination.
It is also important to mention that some catalytic activity still remains even after the 4th reuse (ca. 50% for catalyst 3), most probably because small particles are still present, as indicated by the TEM image.
Conclusions
We successfuly prepared supported AuNP catalysts by the reduction of gold salts previously immobilized on a magnetically recoverable support. The method used in the reduction step for metal nanoparticle formation can be crucial in determining particle dispersion and distribution over the solid support and, consequently, the catalytic activity of the metal nanoparticles. In this work, two reduction methods were compared. Supported AuNPs were prepared by thermal reduction in air and by hydrogen reduction at mild temperature. Interestingly, the mean particle size was similar ca. 5.9 nm, but the polydispersity of the samples is quite different. The UV-Vis and TEM data of the AuNPs prepared by hydrogen reduction, catalyst 4, suggest that a higher control over the particle dispersion was achieved by this reduction method. With respect to the catalytic activity, catalyst 4 is more active in the aerobic oxidation reactions, but less selective for the aldehyde product.The catalysts reported here are very efficiently recovered by magnetic separation, with negligible Au leaching to the solution. This separation procedure is an efficient and green alternative to the conventional separation techniques used for the recovery of solids, such as filtration and centrifugation, since it is fast, clean, easy to scale-up and constitutes a waste minimization and low energy consumption procedure.
The use of base, K2CO3, was necessary to attain good catalytic activities in the oxidation of alcohols by our AuNPs catalysts, but seems to be responsible for the catalyst deactivation under reuse due to degradation of the catalyst support, which causes metal particles to grow beyond the catalytically active limit. The literature contains alternatives for the use of base in such reactions, such as the use of basic and redox supports3q,10 or bimetallic Au–Pd alloys.2f Additional experiments are in progress in our laboratory in order to synthesize and charaterize magnetic catalysts comprised of supports and/or metal particles more resistant to base or, hopefully, active in the absence of base.
Acknowledgements
The authors are grateful to the Brazilian agencies FAPESP and CNPq for financial support and indebted to LNLS-Laboratório de Microscopia Eletrônica (Brazil) for HRTEM images and EDS analysis. The authors also acknowledge the Instituto Nacional de Ciência e Tecnologia de Catálise em Sistemas Moleculares e Nanoestruturados (INCT-CMN).Notes and references
- (a) T. V. Choudhary and D. W. Goodman, Top. Catal., 2002, 21, 25 CrossRef CAS; (b) T. V. Choudhary and D. W. Goodman, Appl. Catal., A, 2005, 291, 32 CrossRef CAS; (c) G. C. Bond and D. T. Thompson, Catal. Rev. Sci. Eng., 1999, 41, 319 CrossRef CAS; (d) G. C. Bond and D. T. Thompson, Appl. Catal., A, 2006, 302, 1 CrossRef CAS; (e) G. J. Hutchings, Catal. Today, 2005, 100, 55 CrossRef CAS; (f) G. J. Hutchings and M. Haruta, Appl. Catal., A, 2005, 291, 2 CrossRef CAS; (g) M. Haruta, Gold Bull., 2004, 37, 27 CAS; (h) F. L. Didier Astruc and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS and references therein; (i) G. J. Hutchings, Chem. Commun., 2008, 1148 RSC; (j) A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef.
- (a) N. Zheng and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 14278 CrossRef CAS; (b) N. Zheng and G. D. Stucky, Chem. Commun., 2007, 3862 RSC; (c) N. S. Patil, R. Jha, B. S. Uphade, S. K. Bhargava and V. R. Choudhary, Appl. Catal., A, 2004, 275, 87 CrossRef CAS; (d) A. Abad, P. Concepción, A. Corma and H. Gracía, Angew. Chem., Int. Ed., 2005, 44, 4066 CrossRef CAS; (e) H. Tsunoyama, H. Sakurai and T. Tsukuda, Chem. Phys. Lett., 2006, 429, 528 CrossRef CAS; (f) D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362 CrossRef CAS; (g) D. I. Enache, D. W. Knight and G. J. Hutchings, Catal. Lett., 2005, 103, 43 CrossRef CAS.
- (a) M. Haruta, CATTECH, 2002, 6, 102–115 CrossRef CAS; (b) M. Haruta, Catal. Today, 1997, 36, 153 CrossRef CAS; (c) R. Zanella, S. Giorgio, C. R. Henry and C. Louis, J. Phys. Chem. B, 2002, 106, 7634 CrossRef; (d) C. Aprile, A. Abad, H. García and A. Corma, J. Mater. Chem., 2005, 15, 4408 RSC; (e) E. Taaring, Anders Theilgarard, J. M. Marchetti, K. Egeblad and C. H. Chrristensen, Green Chem., 2008, 10, 408 RSC; (f) M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812 CrossRef CAS; (g) R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2009, 11, 1366 RSC; (h) S. Biella, G. L. Castiglioni, C. Fumagalli, L. Patri and M. Rossi, Catal. Today, 2002, 72, 43 CrossRef CAS; (i) V. R. Choudhary, R. Jha and P. Jana, Green Chem., 2007, 9, 267 RSC; (j) H. Tsunoyama, T. Tsukuda and H. Sakurai, Chem. Lett., 2007, 36, 212 CrossRef CAS; (k) S. Carrettin, P. McMorn, P. Jonhnston, K. Griffin and G. J. Hutchings, Chem. Commun., 2002, 696 RSC; (l) S. Kim, S. W. Bae, J. S. Lee and J. Park, Tetrahedron, 2009, 65, 1461 CrossRef CAS; (m) C. Raptis, H. Garcia and M. Stratakis, Angew. Chem., Int. Ed., 2009, 48, 3133 CrossRef CAS; (n) A. Abad, A. Corma and H. Garcia, Chem.–Eur. J., 2008, 14, 212 CrossRef CAS; (o) J. Hu, L. Chen, K. Zhu, A. Suchopar and R. Richards, Catal. Today, 2007, 122, 277 CrossRef CAS; (p) J. Yang, Y. Guan, T. Verhoeven, R. van Santen, C. Li and E. J. M. Hensen, Green Chem., 2009, 11, 322 RSC; (q) S. Demirel, M. Lucas, J. Waerna, T. Salmi, D. Murzin and P. Claus, Top. Catal., 2007, 44, 299 CrossRef CAS; (r) P. Haider and A. Baiker, J. Catal., 2007, 248, 175 CrossRef CAS; (s) S. Biella and M. Rossi, Chem. Commun., 2003, 378 RSC; (t) L. Patri and M. Rossi, J. Catal., 1998, 176, 552 CrossRef CAS; (u) N. S. Patil, B. S. Uphade, P. Jana, R. S. Sonawane, S. K. Bhargava and V. R. Choudhary, Catal. Lett., 2004, 94, 89 CrossRef CAS; (v) T. A. Nijhuis, E. Sacaliuc-Parvulescu, N. S. Govender, J. C. Schouten and B. M. Weckhuysen, J. Catal., 2009, 265, 161 CrossRef CAS; (w) C. Aprile, A. Corma, M. E. Domine, H. Garcia and C. Mitchell, J. Catal., 2009, 264, 44 CrossRef CAS; (x) V. R. Choudhary, N. S. Patil and S. K. Bhargava, Catal. Lett., 2003, 89, 55 CrossRef CAS.
- For examples, see: (a) T.-J. Yoon, W. Lee, T.-S. Oh and J. K. Lee, New J. Chem., 2003, 27, 227 RSC; (b) P. D. Stevens, G. Li, J. Fan, M. Yen and Y. Gao, Chem. Commun., 2005, 4435 RSC; (c) P. D. Stevens, G. Li, J. Fan, H. M. R. Gardimalla, M. Yen and Y. Gao, Org. Lett., 2005, 7, 2085 CrossRef CAS; (d) A. Hu, G. T. Yee and W. Lin, J. Am. Chem. Soc., 2005, 127, 12486 CrossRef CAS; (e) M. Kotani, T. Koike, K. Yamaguchi and N. Mizuno, Green Chem., 2006, 8, 735 RSC; (f) R. Abu-Reziq, H. Alper, D. Wang; and M. L. Post, J. Am. Chem. Soc., 2006, 128, 5279 CrossRef; (g) D. K. Yi, S. S. Lee and J. Y. Ying, Chem. Mater., 2006, 18, 2459 CrossRef CAS; (h) M. Kawamura and K. Sato, Chem. Commun., 2006, 4718 RSC; (i) Y. Zheng, P. D. Stevens and Y. Gao, J. Org. Chem., 2006, 71, 537 CrossRef CAS; (j) M. Kawamura and K. Sato, Chem. Commun., 2007, 3404 RSC; (k) D. Guin, B. Baruwati and S. V. Manorama, Org. Lett., 2007, 9, 1419 CrossRef CAS; (l) Z. Yinghuai, S. C. Peng, C. Emi, A. Zhenshun and R. A. Kemp, Adv. Synth. Catal., 2007, 349, 1917 CrossRef; (m) B. Baruwati, D. Guin and S. Manorama, Org. Lett., 2007, 9, 5377 CrossRef CAS; (n) C. O. Dalaigh, S. A. Corr, Y. Gunko and S. J. Connon, Angew. Chem., Int. Ed., 2007, 46, 4329 CrossRef CAS; (o) S. Luo, X. Zheng, H. Xu, X. Mi, L. Zhang and J.-P. Cheng, Adv. Synth. Catal., 2007, 349, 2431 CrossRef CAS; (p) M. Shokouhimehr, Y. Piau, J. Kim and Y. Jang, Angew. Chem., Int. Ed., 2007, 46, 7039 CrossRef CAS; (q) S. Z. Luo, X. X. Zheng and J.-P. Cheng, Chem. Commun., 2008, 5719 RSC; (r) X. Zheng, S. Luo, L. Zhang and J.-P. Cheng, Green Chem., 2009, 11, 455 RSC; (s) B. Panella, A. Vargas and A. Baiker, J. Catal., 2009, 261, 88 CrossRef CAS.
- L. M. Rossi, F. P. Silva, L. L. R. Vono, P. K. Kiyohara, E. L. Duarte, R. Itri, R. Landers and G. Machado, Green Chem., 2007, 9, 379 RSC.
- M. J. Jacinto, P. K. Kiyohara, S. H. Masunaga, R. F. Jardim and L. M. Rossi, Appl. Catal., A, 2008, 338, 52 CrossRef CAS.
- M. J. Jacinto, O. H. C. F. Santos, R. F. Jardim, R. Landers and L. M. Rossi, Appl. Catal., A, 2009, 360, 177 CrossRef CAS.
- (a) R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC; (b) S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943 CrossRef CAS.
- K. R. Brown, D. G. Walter and M. J. Natan, Chem. Mater., 2000, 12, 306 CrossRef CAS.
- V. R. Choudhary, A. Dhar, P. Jana, R. Jha and B. S. Uphade, Green Chem., 2005, 7, 768 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: TEM and EDS analyses of the spent catalyst. See DOI: 10.1039/b916825g |
| This journal is © The Royal Society of Chemistry 2010 |