Suppression of breast xenograft growth and progression in nude mice: implications for the use of orally administered sphingolipids as chemopreventive agents against breast cancer
Kirk W.
Simon
a,
Larry
Tait
b,
Fred
Miller
b,
Chun
Cao
c,
Kevin P.
Davy
c,
Tanya
LeRoith
d and
Eva M.
Schmelz
*ce
aDepartment of Nutrition and Food Science, Wayne State University, Detroit, MI, USA
bDepartment of Karmanos Cancer Institute, Wayne State University, Detroit, MI, USA
cDepartment of Human Nutrition, Foods and Exercise (HNFE), Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA
dDepartment of Anatomic Pathology and Biomedical Sciences & Pathobiology, Virginia-Maryland Regional College of Veterinary Medicine, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA
eDepartment of Human Nutrition, Foods and Exercise (HNFE), Corporate Research Center Building 23 (0913), Room 1011, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA. E-mail: eschmelz@vt.edu; Fax: +1 540-231-2947; Tel: +1 540-231-3649
First published on 23rd September 2010
Abstract
Sphingolipids are lipid messengers involved in the regulation of many different cellular processes. Sphingolipid enzymes and bioactive metabolites have been targets of in vitro and in vivo efforts to suppress cancer growth, progression and metastasis of various cancer types. Dietary sphingomyelin effectively suppressed colon cancer in several rodent models without causing toxic side effects. In the present study, we determined if the effect of sphingolipid metabolites derived from the hydrolysis of dietary sphingomyelin is restricted to the intestinal tract or if their systemic concentrations are sufficient to suppress cancers of distant sites. For these studies, we used MCF10AT1 cells, a model for progressive breast cancer, injected into the mammary fatpad of nude mice as a single cell suspension. The mice were fed 0.1% sphingomyelin supplements in a semi-purified AIN76A control diet when the lesions were palpable. The study was terminated when the first lesions had grown to 5 mm. In the sphingomyelin-fed group, there was a trend to smaller lesion size and, importantly, a delayed progression to more malignant stages without apparent side effects. This may be the result of significantly reduced rates of proliferation and angiogenesis, while no increase of apoptosis was detected. Changes in aberrantly expressed proteins in the sphingomyelin-fed group, such as E-cadherin, VEGF and sphingosine kinase-1, may be associated with the suppression of tumor growth. These results demonstrate that diet-derived sphingolipids can efficiently suppress the growth and progression of MCF10AT1 xenografts, suggesting that dietary sphingomyelin may also be effective against cancers of other sites.
Introduction
Breast cancer incidence and mortality has remained high, despite improved diagnostic and treatment efforts to eradicate the disease. An estimated 192,370 women were newly diagnosed in 2009 and 40,170 will die from breast cancer in the United States of America.1 Early detection improves survival,2 but preventive efforts may be more effective at maintaining health and increasing survival. Many in vitro and in vivo studies have demonstrated the preventive potential of dietary compounds; these often lack toxic side effects, and together with an easy route of administration could increase the patients' compliance with and well-being during the prevention regimen.Sphingolipids are membrane-bound lipids with both structural and signaling properties. Sphingolipid metabolites are generated by growth factors, cytokines, cellular stresses and other factors, and as lipid second messengers are involved in the generation of an appropriate cellular response. These responses include the regulation of cell growth, differentiation, cell death, motility, angiogenesis, inflammation and many more processes in a concentration, structure, time and cell type-specific manner.3,4 Several enzymes in sphingolipid metabolism have been identified as targets for drug treatments to generate a sphingolipid pattern that exerts anticancer activities, such as the suppression of cell growth, motility, angiogenesis, etc. Among these enzymes are sphingomyelinases,5,6 glucosylceramide synthase,7 sphingosine kinase 1,8,9 sphingosine-1-phosphate lyase,10 sphingosine-1-phosphate phosphatase11,12 and others. However, the inhibitors are sometimes not sufficiently specific and can cause adverse side effects. Furthermore, cancer cells can exhibit an aberrant expression or activity of these enzymes, rendering the cells resistant to the regulatory stimuli. Thus, the delivery of bioactive metabolites directly to the cancer cells is an attractive alternative. We have shown in previous studies that administration of natural or synthetic complex sphingolipids via the diet suppressed chemically-induced colon cancer13–18 or cancer caused by gene mutations in mice19,20 by up to 80%. There were no side effects of this route of administration in any of our studies, and even very high doses used in other studies were tolerated well.21
Diet-derived sphingolipid metabolites are taken up in the gastrointestinal tract, and a small portion is transported to the liver via the lymphatic system, mostly as sphingosine and fatty acids.22–24 Free sphingoid bases can also be re-utilized for the synthesis of more complex sphingolipids such as sphingomyelin (SM), and can be incorporated into chylomicrons and secreted from the intestinal tract; the impact of dietary SM on the content of SM in plasma lipoproteins, however, remains unclear.25 To determine if the diet provides sufficient amounts of bioactive metabolites to prevent cancer of distant sites in the body, in the present study, we investigated if orally administered SM can suppress breast cancer growth and progression. The amounts used in this study, 0.1% by weight, were highly effective against colon cancer (see above) without causing serious side effects, and are about 5–10 times higher than the daily intake in humans, which has been estimated at 0.01 to 0.02% of the diet.26 A recent study demonstrated that in humans, a dose of 250 mg SM is sufficient to increase SM and SM metabolites in the colon.24 The focus of this study is to determine if SM supplements are sufficient to affect breast xenograft growth and progression using, in contrast to a strictly chemopreventive approach, in which the diet is supplemented before cell injection, a study design in which SM is added to the diet when the lesions are palpable.
As a model for proliferative breast disease, we used MCF10AT1 cells, pre-malignant human breast epithelial cells derived from the spontaneously immortalized, normal appearing MCF10A cells by transfection with a constitutively active Ha-ras.27 Although ras mutations are rare events in humans and only found in approximately 5% of patients, the aberrant activation of Ras and down-stream targets of Ras are seen in human breast cancer overexpressing ErbB-228 or Notch,29 and it is thought that activated Ras can contribute to transformation, unlimited proliferation,29,30 motility and invasion.31 Ha-ras alone, however, is not sufficient to induce transformation.32 Lesions formed in this system include mild to moderate hyperplasia, atypical hyperplasia, carcinoma in situ, moderately differentiated carcinoma, and undifferentiated carcinoma, as well as histologically normal ducts.32 Thus, the MCF10AT1 cell line represents a model for the slow and heterogeneous progression of preneoplastic breast cells and provides a transplantable xenograft model of human proliferative breast disease with proven neoplastic potential32,33 that is ideal to test the capacity of natural compounds to suppress breast cancer development and progression in vivo.
Results and discussion
Sphingomyelin suppressed tumor development and progression.
Sphingolipids are natural components of the diet that are hydrolyzed in the intestinal tract to the bioactive metabolites ceramide and sphingosine, which are taken up by the intestinal cells and either degraded or incorporated into complex sphingolipids.22,23 The exposure to these bioactive metabolites is sufficient to suppress colon cancer in mice.13–20 Only a small fraction of the absorbed sphingolipid metabolites reaches systemic distribution;22,23 therefore, the highest dose effective in colon cancer prevention was used for the present study. Female NCR nude mice (8 weeks old) were injected with MCF10AT1 cells while on the semi-purified AIN76A diet; once the lesions were palpable, the SM group received 0.1% SM in the diet, and the development of the lesions was compared to the controls who did not receive any supplements. SM supplements did not affect the health of the mice as reflected by their general appearance and weight. The mean weight of the control group (27.8 ± 2.3 g) did not differ significantly from the SM-fed group (25.8 ± 0.5 g) (p = 0.663). This confirms earlier studies from our and other’s laboratories that neither a sphingolipid-free diet nor SM supplements to the diet (up to 1% by weight21) cause adverse side effects, and SM is therefore safe to use as a dietary supplement in our studies.Since cells were injected at two conlateral sites per mouse, the development of 30 lesions (2 per mouse) was possible in each group. In the control group, 22 lesions were detected (1.47 per mouse), and 25 in the SM-fed group (1.67 per mouse); this difference is not statistically significant (p = 0.417). There were 2 mice without lesions in each group (13.3%). This lesion development lies in the expected range for this cell line when injected into nude mice in a Matrigel solution.27 The volume of the lesions was lower in the SM-fed group, albeit not statistically significant (99.33 ± 23.99 mm3 in the control and 63.72 ± 7.89 mm3 in the SM-fed group; p = 0.190). This is especially interesting, since we added the SM supplements only after the lesions were already palpable and not as a strictly preventive regimen before cell injection.
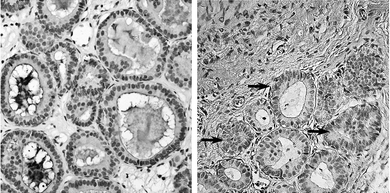 | ||
| Fig. 1 MCF10AT1 xenograft architecture. MCF10AT1 cells are organized either as tightly packed breast-like ducts with little (A) or high (B, arrows) amounts of stromal tissue with no apparent difference between the control group fed the AIN76A diet alone and the SM supplemented group (0.1%, by weight). | ||
MCF10AT1 cells were organized as ducts rather than as cell clusters; this confirms previous findings demonstrating that these cells form structures that resemble normal human breast ducts when injected into nude mice in a Matrigel solution.27 The lesions were solid and without a necrotic core. It is important to note the difference in the amount of stromal tissue in individual xenografts. Some lesions consisted mostly of epithelial cells, and there was very little stromal tissue visible (Fig. 1A), while it was more abundant in others, and the epithelial cells appeared as small islands of ducts (Fig. 1B, arrows). Stromal tissue has been shown to generate a microenvironment that regulates breast cell growth, migration, morphology, differentiation, migration and survival.34 In cancer, the cancer cells signal to alter protein secretion of the stroma but stroma activated by inflammatory signals is involved in tumor development and progression (see recent review34). Thus, the interactions of tumor stroma and epithelial cells can have a profound effect on the progression of the epithelial cells, and the tumor size per se may not truly reflect the preventive potential of the tested compound; the epithelial and stroma geno- and phenotype may be more critical. In this study, the amount of stroma was not the same in sections of different areas of a single xenograft or among the lesions found in one mouse; thus, an association of SM supplements and the epithelial cell to stroma ratio could not be made by immunohistochemical methods but changes in gene expression levels in the stroma as a result of SM treatment will be considered in future studies using real-time PCR.
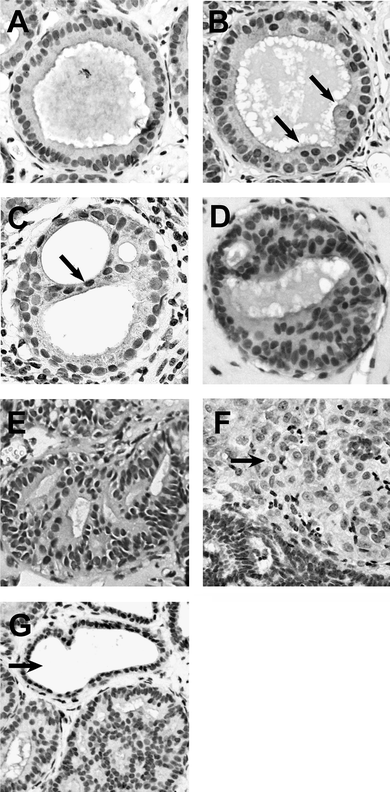 | ||
| Fig. 2 MCF10AT1 xenograft lesions. The lesions were graded using the criteria outlined in the Experimental to approximate the scoring system in place for human surgical specimen: A, stage 0, normal ducts; B, stage 1, mild hyperplasia with multiple layers (arrows); C, stage 2, moderate hyperplasia with bridging (arrow); D, stage 3, atypical hyperplasia; E, stage 4, carcinoma in situ; F, stage 5, invasive carcinoma (arrow); G, fibrocystic structure (arrow) and hyperplastic duct. | ||
We next determined the progression of the MCF10AT1 cells using the criteria outlined in the Experimental section to define the different disease stages, as shown in Fig. 2. Most of the xenografts in the SM-fed group progressed to stage 2 (22 or 88%), characterized by moderate hyperplasia, bridging and papillary hyperplasia. Only two areas of stage 3 (8%) and one of stage 5 (4%) were detected in this group. In contrast, half of the xenografts in control animals progressed further and exhibited areas of stage 3 (6 or 27.3%), 4 (2 or 9.1%) and 5 (3 or 13.6%) (different from the SM-fed group at p = 0.0212) (Fig. 3). This lies closely in the range of the progression rate reported for this cell line.27 These results indicate that SM supplements suppressed or delayed the progression of these lesions, confirming our hypothesis that diet-derived SM metabolites are sufficient to exhibit a systemic effect and suppress breast cancer progression without causing side effects. Thus, dietary sphingolipids are not only effective against colon (our previous studies) and liver cancer35 but can successfully target cancer of distant sites in the body. Whether dietary sphingolipids are as effective against more aggressive breast cancer lesions is currently being investigated in our laboratory.
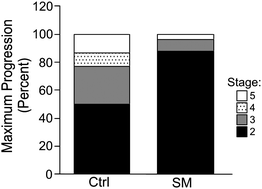 | ||
| Fig. 3 Suppression of progression. Mice (n = 15 per group) were injected with MCF10AT1 cells while being fed the AIN76A diet. After the lesions were palpable (after 4 weeks), the diet of the SM group was supplemented with 0.1% of SM. Using the criteria in the Experimental, the area of the highest disease stage found in one lesion determined the overall category of the lesion. (different at p < 0.05). | ||
Sphingomyelin reduced cell proliferation but did not induce apoptosis
Sphingolipid metabolites regulate cell growth both in vitro and in vivo. Since unlimited proliferation is considered a driving force for progression, the effect of the SM supplements on the rate of proliferation in the breast xenografts was determined using Ki-67 expression as a marker. As shown in Fig. 4, SM supplements significantly suppressed the proliferation of MCF10AT1 xenografts (22.9 ± 2.1% Ki-67 positive nuclei in the control group vs. 11.6 ± 1.7% in the SM-fed group; p < 0.001).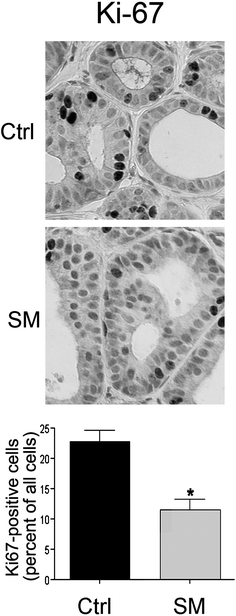 | ||
| Fig. 4 Inhibition of proliferation by orally administered sphingomyelin. Sections from the control (Ctrl) or sphingomyelin-fed (SM) group (n = 15 per group, 5 images per section) were immunostained for Ki-67 as a marker for proliferation and expressed as percent Ki-67 positive cells. *p < 0.001. | ||
One objective of many anticancer treatments is the induction of apoptosis in cancer cells to eradicate tumors. Sphingolipid metabolites induce apoptosis in many cancer cell lines in vitro; therefore, we also evaluated the rate of apoptosis in the MCF10AT1 xenografts. There were very few epithelial cells in either group that were stained positive for activated caspase-3, with most sections being completely negative (not shown). The lack of apoptosis was verified by evaluating the shape of the nuclei in H&E stained sections—condensed or fragmented nuclei of epithelial cells were rarely detected, indicating that orally administered SM did not induce apoptosis in the xenografts. This low rate of apoptosis in MCF10AT1 xenografts is not specific for our study but has been reported before in MCF10AT1 cells grown in three-dimensional cultures36 or as xenografts37,38 These results confirm our previous observations and the reports of other groups that dietary sphingolipids do not induce apoptosis above normal levels in the colon,16,17 liver35,39,40 or spleen.40 Instead, we have previously observed a suppression of aberrant proliferation in the colonic mucosa.16,17 Thus, dietary sphingolipids may exert their antitumor effect against colon and breast cancer in part by reducing the rate of proliferation. This is in contrast to many other natural compounds that clearly show an association of tumor suppression and the induction of apoptosis by many different pathways41 but may be associated with the lack of toxic side effects of dietary sphingolipids, which was apparent by the general appearance of the mice, a comparable weight gain and blood parameters we have reported previously (normal blood urea nitrogen, creatinine, albumin, serum glutamate pyruvate transaminase, alkaline phosphatase, etc.14). This may be the result of a limited digestion of complex sphingolipids, which prevents the generation of toxic levels of bioactive sphingolipid metabolites in the intestinal tract.42 Even the feeding of SM over two generations did not affect the health of rats,21 suggesting that the supplementation of the diet with SM is a safe route of administration.
Sphingomyelin reduced VEGF expression and the number of blood vessels in the xenografts
The growth and progression of tumors are dependent on a sufficient supply of nutrients and oxygen, and thus on the establishment of blood vessels. In addition to the vascular endothelial growth factor (VEGF) secretion by endothelial and stromal cells, breast cancer cells also secrete VEGF to stimulate angiogenesis by enhancing endothelial migration, proliferation and survival,43 but also to attract mesenchymal stem cells that differentiate into stroma.44 To evaluate if the suppression of growth and progression of MCF10AT1 xenografts by dietary SM is associated with a reduced angiogenesis, the expression of VEGF in the xenografts was determined by immunohistochemistry. VEGF was expressed in endothelial cells without apparent differences among the treatment groups, but there was also significant immunostaining of the epithelial cells. The control group expressed moderate to high levels of VEGF in lower grade ducts (Fig. 5A) but higher levels in more advanced stages. Undetectable/very low to moderate levels of VEGF were observed in the SM-fed group (Fig. 5B) (luminal staining is unspecific). Higher grade lesions (stage 5) in either group showed substantial VEGF staining. These results confirm previous findings, demonstrating a reduction of VEGF mRNA expression levels in the colonic mucosa of Min mice after feeding sphingolipids for four weeks.20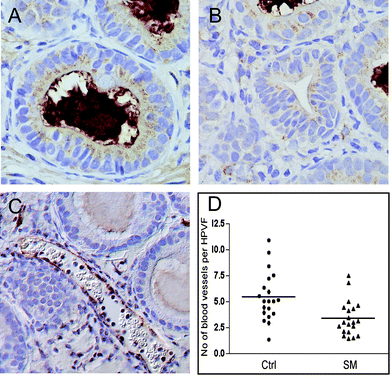 | ||
| Fig. 5 Regulation of angiogenesis by dietary sphingomyelin. Sections of tumors from the control group (Ctrl) and SM-fed group (SM) were immunostained with VEGF (A, B) or Pecam-1 (C). A moderate to high expression of VEGF was detected in the lower grade ducts of the control group (A) with high levels in higher grades (unspecific luminal staining). Low to moderate levels were seen in the SM-fed group (B). PECAM-1 staining was used to visualize endothelial cells (C), and blood vessels were counted in 10 high-powered viewing fields (HPVF) per section (D). The number of blood vessels per HPVF was significantly higher in the control (Ctrl) group than in the SM-fed group (p < 0.001). | ||
Immunostaining for Pecam-1 allowed for the visualization of blood vessels (Fig. 5C) in the xenografts. In the control group, there were 5.41 ± 0.26 blood vessels per high-powered viewing field (Fig. 5D); SM significantly reduced the number of blood vessels to 3.39 ± 0.18 (p < 0.001). There was no consistent difference in the size of these blood vessels between the groups, but it appeared that there were generally more and larger vessels associated with the stromal tissue than with areas of tightly packed epithelial tissue. These results demonstrate the suppression of angiogenesis by SM, possibly via suppression of VEGF expression. It has been shown that stromal cells directly adjacent of tumor cells promote angiogenesis via the activation of gene expression;45 the secretion of VEGF by stromal cells has been suggested to be a contributing factor.46 Thus, the phenotype of the stroma appears to be critical for tumor growth and progression, and the effect of dietary SM on tumor-associated stroma and its secretion of growth factors and other tumor-supporting proteins needs to be evaluated in more detail.
Expression levels of gene products associated with progression of breast cancer
The promotion and progression of tumor cells is associated with changes in the expression levels of specific proteins, usually identified as tumor suppressors or promoters. In heterogenic diseases such as breast cancer, this protein expression pattern can be employed to predict the progression of the tumor cells and their response to treatment, and therefore these proteins are potential biomarkers for in vivo efficacy determinations. To confirm the suppression of progression by orally administered SM and to identify targets that are altered by the treatment, we used immunohistochemistry to evaluate changes in gene products that have been associated with breast cancer progression and may be a potential biomarker for SM efficacy against breast cancer in vivo.The loss of E-cadherin is a frequent early event in cancers of many organs such as colon, the ovaries and also breast.47 This has been associated with changes in cellular architecture that may permit the epithelial–mesenchymal transition (EMT), cancer progression and invasion.48,49 In our study, there was weak to no expression of E-cadherin in the control group, even in lower grade lesions (Fig. 6A). Higher magnification showed an expression of E-cadherin in some ducts, predominantly located in the cytoplasm (Fig. 6B). The staining in these cells was sometimes diffuse but also appeared granulated, sometimes localized in the perinuclear area. In contrast, most of the ducts in the SM-fed group exhibited E-cadherin positive staining in at least some cells of each duct (Fig. 6C). Here, E-cadherin was often localized at the membranes (Fig. 6D), but diffuse or sometimes granular cytoplasmic expression was often seen in ducts lacking membranous E-cadherin. The appearance of cytosolic E-cadherin in the early stages has been described in pre-malignant breast lesions as a result of increased endocytosis, leading to a loss of membrane anchoring that weakens adhesion, and altered cell dissociation, motility and invasive potential.49,50 Later stages of breast cancer, even ductal carcinoma in situ, often demonstrate a complete loss of E-cadherin protein expression.51 Overexpression of E-cadherin in MDA-MB-231 cells partially restored the epithelial morphology with increased cell–cell contacts and adhesion, and reduced migration properties.52 It is not known if there is a different function of the cytosolic and membrane-bound E-cadherin since the cells increased the expression levels in both cellular fractions,52 but the expression and recruitment of tumor suppressors such as PTEN are dependent on the E-cadherin/β-catenin complex at the adherens junctions.53 Natural compounds in the diet can restore the expression of E-cadherin and suppress invasion.54 Thus, the regulation of E-cadherin expression and localization may affect several signaling intermediates restoring cellular differentiation and suppressing proliferation.
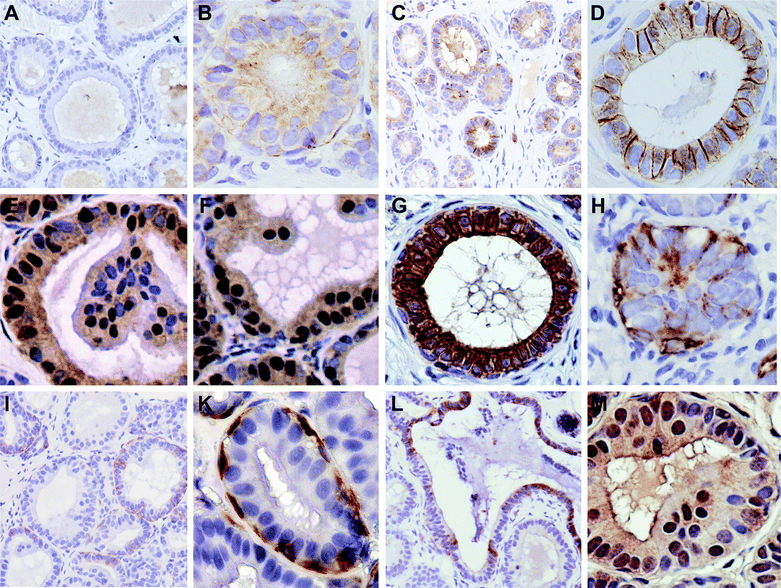 | ||
| Fig. 6 Expression levels and the localization of proteins associated with breast cancer progression are altered with dietary SM. E-cadherin expression in the control group (A low, B high magnification) and the SM-group (C low, D high magnification); H4K16ac in control (E) and SM-group (F); (G) cytokeratin expression confirms the epithelial origin of the ducts; (H) Vimentin expression in the control group; (I, K) Vimentin-positive myoepithelial cells; (L) SK1 expression; (M) ceramide kinase expression (the luminal staining of the duct-like structure is unspecific). | ||
Since E-cadherin expression is often down-regulated via epigenetic silencing—also reported in breast cancer55—we investigated the global methylation of histones as a marker for transcriptional regulation; the loss of histone 4 lysine 16 acetylation (H4K16ac) has been suggested to be a hallmark of human cancer cells56 and has been found to be associated with larger breast tumors.57 H4K16 hypoacetylation also has been observed in the silencing of E-cadherin.58 As shown in Fig. 6E and 6F, SM feeding leads to an increase in H4K16 acetylation, which followed the stage distribution of the tissue in most cases; in SM-fed mice, positive nuclei were also seen in stage 3 (Fig. 6F). The cytosolic staining seen with this antibody is unspecific since it was still visible in the sections treated with blocking peptides. While these results need to be confirmed in a larger study, and further corroborated by chromatin immunoprecipitation and quantitative PCR analysis for specific epigenetically regulated genes, they suggest that dietary SM indeed affects histone marks involved in epigenetic silencing and thereby potentially influences the expression of genes such as E-cadherin. The mechanisms for gene expression regulation are currently a research focus in our laboratory.
Next, we investigated the expression of vimentin, expressed after EMT, and thus indicative of progression. All ducts expressed cytokeratin, confirming their epithelial nature (Fig. 6G). There was no vimentin expression in the lower grade lesions of either group, but a diffuse staining in higher grade lesions in both groups (Fig. 6H). Since there were less higher grade lesions in the sphingomyelin-fed group, there was also less vimentin staining. Myoepithelial cells also stained strongly positive for vimentin, as has been reported previously.59 These cells closely surround the ducts formed by the MCF10AT1 cells (Fig. 6I). There was no apparent difference in the number of myoepithelial cells between the groups. However, myoepithelial cells have been shown to regulate breast epithelial polarity, morphogenesis, differentiation, etc. via secretion of regulatory factors such as laminin-1, fibronectin, matrix metalloproteinases, ephrin receptor B4, pleiotrophin and others, and are thought to suppress breast cancer progression.60 While there is a reported loss of myoepithelial cells during breast cancer progression,60 the most drastic and consistent changes during this progression occur in myoepithelial cells, specifically in genes encoding secretory and cell surface proteins.61 Since this can alter their function and interaction with breast epithelial cells and affect their role in cancer progression, the changes in their gene expression levels—and functions—in response to sphingolipid metabolites is therefore an important question.
We have investigated the expression of other proteins often associated with breast cancer progression but did not find either a consistent or reproducible change in their expression (CXCL4, SDF-1, Her-2, pHer-2 EGFR, pEGFR), or their expression levels were low or not changed by dietary SM (Cyclin D1, N-cadherin, F4/80). However, as reported above, several molecular markers for breast cancer progression were aberrantly expressed in advanced stages and were affected by the dietary SM, suggesting that these proteins may be involved in the suppression of breast cancer progression by dietary SM.
Expression of sphingosine kinase-1 and ceramide kinase
Changes in the expression of enzymes regulating the sphingolipid metabolism that result in changes in the expression pattern of sphingolipid metabolites have been reported in several cancers, including breast cancer. Sphingosine kinase-1 (SK1) generates sphingosine-1-phosphate, which, in contrast to sphingosine or ceramide, mostly promotes cell growth, motility, inflammation, tumorigenesis, angiogenesis and invasion, and therefore has been implicated in the uncontrolled growth of cancer cells.8,62 SK1 exhibits higher expression levels in estrogen receptor-negative breast tumors and has been associated with a higher rate of metastasis and a worse outcome.63 SK1 expression was detected in the cytosol of approximately half of the fibrocystic structures (depicted in Fig.2G) in the control lesions (Fig. 6L) and only occasionally in the duct-like structures. There was no SK1 expression, even in the higher-grade ductal structures in the SM-fed group, and only an infrequent staining of the fibrocystic structures. Nuclear SK1 was only detected in small areas of one lesion per group, and therefore appears not to be important in the progression of MCF10AT1 lesions.Ceramide kinase is a less well-described member of the sphingolipid metabolic enzyme family that generates ceramide-1-phosphate (C1P). In lung cancer cells, C1P increased survival and stimulated proliferation.64 We did not detect drastic differences between the controls and the sphingomyelin-fed group in both groups: ceramide kinase was weakly to strongly expressed in the cytosol of few epithelial cells, and often also in the nucleus (Fig. 6M). A high ceramide kinase expression, however, has been shown in estrogen receptor negative breast cancers and was associated with a worse prognosis.65 It has been reported that C1P in macrophages and other immune cells plays a role in inflammatory processes (see the recent review66). Thus, the cancer cells may not be the only critical cell population expressing ceramide kinase; this needs to be investigated in an immune competent model.
Experimental
Cell culture
MCF10AT1 cells were obtained from the Cell Core at the Karmanos Cancer Institute at Wayne State University, Detroit, MI. Cells were routinely cultured at 37 °C in DMEM/Ham's F12 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Gibco, Invitrogen), supplemented with 5% horse serum (Gibco), 10 μg ml−1 insulin (Sigma, St. Louis, MO), 0.02 μg ml−1 EGF (Calbiochem, San Diego, CA), 0.5 μg ml−1 hydrocortisone (Sigma) and 0.1 μg ml−1 cholera toxin (Calbiochem) in 5% CO2 in a humidified atmosphere.
:1) (Gibco, Invitrogen), supplemented with 5% horse serum (Gibco), 10 μg ml−1 insulin (Sigma, St. Louis, MO), 0.02 μg ml−1 EGF (Calbiochem, San Diego, CA), 0.5 μg ml−1 hydrocortisone (Sigma) and 0.1 μg ml−1 cholera toxin (Calbiochem) in 5% CO2 in a humidified atmosphere.
Animals and diets
Female NCR nude mice (seven weeks old) were purchased from Taconic (Germantown, NY). Upon arrival, they were randomly divided into two experimental groups (n = 15 per group) and weighed to ensure an equal weight. After acclimatization for one week, they were placed on the semi-purified AIN 76A diet (Dyets, Bethlehem, PA). This diet is essentially sphingolipid-free,67 in contrast to the newer formulations that contain soy oil and may contain significant amounts of glucosylceramide. Since sphingolipids are not essential nutrients, placing animals on sphingolipid-free diets is not harmful and has neither caused adverse effects in any of our studies, nor those published by others.Xenografts and treatment
Cells were grown to approximately 70% confluence and harvested by gentle trypsination. 1 × 107 cells were mixed into 100 μl of cold Matrigel (BD, Franklin Lakes, NJ) and subcutaneously injected into the area of two contralateral mammary fatpads per mouse. When the xenografts were palpable (approximately 1–2 mm in diameter, 4 weeks later), one group of mice was placed on the AIN 76A diet containing 0.1% sphingomyelin (by weight) (Avanti, Alabaster, AL), while the control group did not receive any dietary supplements. After the xenografts reached a size of about 5 mm (16 weeks after injection), the mice were killed, the xenografts excised, measured, fixed overnight in 10% neutral buffered formalin, embedded in paraffin and sectioned for immunohistochemical analyses at 4–5μm. The tumor volume was calculated by the formula: V = {W2 × L} ÷ 2, where V is the tumor volume, W is the width and L is the length.Determination of progression
Hematoxylin and eosin-stained sections of each animal were graded in a blinded manner for progression according to the criteria of Dawson et al. (1996). The lesions were classified according to the highest grade found in the xenograft. The categories were defined as follows:0 simple small tubules with 1–2 cell layers, no nuclear enlargement.
1 mild hyperplasia, simple small tubules with >2 cell layers but no bridging or architectural complexity, variable nuclear contours.
2 moderate hyperplasia, mildly distended ducts, 4 or more layers of epithelial cells, bridging, irregularly shaped lumen, papillary hyperplasia.
3 atypical hyperplasia, grossly distended ducts, marked cellular proliferation often forming luminal mass, some loss of polarity, cells become monotonous, tendency to clear cytoplasm with distinct borders, enlarged, non-round nuclei, small nucleoli, occasional mitoses.
4 carcinoma in situ, distended ducts filled with uniform cells, rigid intraluminal bridges forming round spaces, occasional central necrosis, distinct cell boundaries, uniform round, hyperchromatic, enlarged nuclei, frequent mitoses.
5 invasive carcinoma, glandular, squamous or undifferentiated.
Immunohistochemical analyses
Tissue sections were immunostained using our established procedures.19 Briefly, sections were deparaffinized in xylene, re-hydrated through graded alcohol and steamed in citrate buffer (Target Retrieval Solution, Dako, Carpinteria, CA) for antigen retrieval if suggested for the specific antibody by the manufacturer. Endogenous peroxidase activity was blocked with Peroxoblock (Zymed, Carlsbad, CA). Blocking of the sections for 1 h was followed by overnight incubation at 4 °C with the indicated antibodies, without primary antibody or with antibody plus blocking peptide as negative or antibody specificity controls. The sections were rinsed, incubated with the appropriate secondary antibodies for 1 h at room temperature, and treated with the ABC staining systems (Vector, Burlingame, CA) according to the manufacturer's instructions. The immunocomplex was visualized with diaminobenzidine (Dako).The sections were briefly counterstained with hematoxylin (Zymed) and permanently mounted with Histomount® (Zymed). All images were digitally captured on a Nikon Eclipse 80i epifluorescence microscope, equipped with DIC, digital cameras, and acquisition and analysis software (NIS Elements). Images were processed with Adobe Photoshop®.
Antibodies used in this study were generated against Ki-67 (Dako), activated Caspase-3 (Cell Signaling, Danvers, MA), ceramide kinase (Abgent, San Diego, CA), CRCX4 (R&D), cytokeratin (Sigma), SDF-1 (R&D), E-cadherin (R&D Systems), H4K14ac (abcam), N-cadherin (abcam), EGFR (Upstate Biolabs, Chicago, IL), F4/80 (abcam), Her-2 and phospho-Her-2 (Cell Signaling), VEGF (Santa Cruz), SK1 (Imgenex, San Diego, CA), Vimentin (Dako) and Pecam1 (Santa Cruz). The quantitation of proliferation was achieved by immunostaining for Ki-67. A red filter was used to obscure staining and pictures were taken from 5 randomly selected areas per slide in a blinded manner. The nuclei stained positively for Ki67, all unstained epithelial nuclei were counted, and the rate of proliferation was expressed Ki67 positive as a percentage of all nuclei. Data are expressed as mean ± SEM.
Determination of angiogenesis
Sections were immunostained (as described above) for Pecam1 to visualize blood vessels in the xenografts. 10 high-powered viewing fields per section were randomly selected by a person unaware of the treatment at 40× magnification, and all the visible blood vessels that stained positive for Pecam-1 and/or showed blood cells were counted. Other vessels that might have been lymphatic vessels and microvessels without Pecam1 staining were not counted in any section. Furthermore, the diameter of the blood vessels were not measured since the random angle of sectioning the vessels did not allow for the quantitation of this parameter. 20 tumors per group were analyzed by this method. Data are expressed as the mean number of blood vessels per high-powered viewing field ± SEM.Statistical analyses
Experimental groups were compared using the unpaired t-test after ANOVA for groups that had values sampled from Gaussian distributions. An unpaired t-test followed by Welch's correction was used when the standard deviations differed significantly among the groups, and the non-parametric Mann–Whitney test was used in groups that did not follow a Gaussian distribution. These statistical analyses were performed with Instat 3.0a (GraphPad Software, Inc.). The association of tumor progression and dietary supplementation was calculated using the one-sided Mantel–Haenszel chi square test.Conclusion
The present study demonstrates for the first time that dietary SM in amounts that are approximately 2.5 to 10 times higher than the estimated human intake of complex sphingolipids26 suppressed the growth and progression of MCF10AT1 xenografts without causing adverse side effects. This was associated with a significantly reduced rate of angiogenesis. Proteins that are aberrantly expressed in breast cancer progression, i.e., E-cadherin, VEGF and enzymes of the sphingolipid metabolism that could generate a pro-tumorigenic microenvironment, i.e., SK1, were targeted by the diet-derived sphingolipid metabolites and exhibited more normal expression levels or protein localization in the SM-fed group, suggesting the possibility that their functions may be restored. Whether these gene products can be used as molecular markers for sphingolipid efficacy in vivo remains to be determined. However, our results demonstrate systemic, in addition to the previously described local, effects of diet-derived sphingolipid metabolites. Whether the amounts of SM that were effective in this pre-malignant breast cancer model can also prevent tumor growth and progression in more advanced models is currently under investigation in our laboratory.Acknowledgements
This study was supported by NCI grant R03 CA101125, The Susan G. Komen Race for the Cure grant BCT0503453 and funds from the Prevention Program, Karmanos Cancer Institute, Wayne State University, Detroit, MI, USA.References
- M. J. Horner, L. A. G. Ries, M. Krapcho, M. Neyman, R. Aminou, N. Howlader, S. F. Altekruse, E. J. Feuer, L. Huang, A. Mariotto, B. A. Miller, D. R. Lewis, M. P. Eisner, D. G. Stinchcomb, B. K. Edwards (eds). SEER Cancer Statistics Review, 1975–2006, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2006/, based on November 2008 SEER data submission, posted to the SEER web site, 2009 Search PubMed.
- H. D. Nelson, K. Tyne, A. Naik, C. Bougatsos, B. K. Chan and L. Humphrey, Ann. Intern. Med., 2009, 151, 727–737 (W237–W742).
- W. Zheng, J. Kollmeyer, H. Symolon, A. Momin, E. Munter, E. Wang, S. Kelly, J. C. Allegood, Y. Liu, Q. Peng, H. Ramaraju, M. C. Sullards, M. Cabot and A. H. Merrill, Jr, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1864–1884 CrossRef CAS.
- N. Bartke and Y. A. Hannun, J. Lipid Res., 2009, 50 Suppl., S91–96.
- G. Grammatikos, V. Teichgraber, A. Carpinteiro, T. Trarbach, M. Weller, U. R. Hengge and E. Gulbins, Antioxid. Redox Signaling, 2007, 9, 1449–1456 Search PubMed.
- E. L. Smith and E. H. Schuchman, Mol. Ther., 2008, 16, 1565–1571 CrossRef CAS.
- Y. Y. Liu, T. Y. Han, A. E. Giuliano and M. C. Cabot, J. Biol. Chem., 1999, 274, 1140–1146 CrossRef CAS.
- V. E. Nava, J. P. Hobson, S. Murthy, S. Milstien and S. Spiegel, Exp. Cell Res., 2002, 281, 115–127 CrossRef CAS.
- M. Kohno, M. Momoi, M. L. Oo, J. H. Paik, Y. M. Lee, K. Venkataraman, Y. Ai, A. P. Ristimaki, H. Fyrst, H. Sano, D. Rosenberg, J. D. Saba, R. L. Proia and T. Hla, Mol. Cell. Biol., 2006, 26, 7211–7223 CrossRef CAS.
- M. Serra and J. D. Saba, Adv. Enzyme Regul., 2009 Search PubMed.
- W. I. Leong and J. D. Saba, Biochimie, 2010, 92, 716–723 CrossRef CAS.
- N. J. Pyne, J. S. Long, S. C. Lee, C. Loveridge, L. Gillies and S. Pyne, Adv. Enzyme Regul., 2009 Search PubMed.
- D. L. Dillehay, S. K. Webb, E. M. Schmelz and A. H. Merrill, Jr, J. Nutr., 1994, 124, 615–620 CAS.
- E. M. Schmelz, D. L. Dillehay, S. K. Webb, A. Reiter, J. Adams and A. H. Merrill, Jr, Cancer Res., 1996, 56, 4936–4941 CAS.
- E. M. Schmelz, A. S. Bushnev, D. L. Dillehay, D. C. Liotta and A. H. Merrill, Jr, Nutr. Cancer, 1997, 28, 81–85 Search PubMed.
- E. M. Schmelz, M. C. Sullards, D. L. Dillehay and A. H. Merrill, Jr, J. Nutr., 2000, 130, 522–527 CAS.
- L. A. Lemonnier, D. L. Dillehay, M. J. Vespremi, J. Abrams, E. Brody and E. M. Schmelz, Arch. Biochem. Biophys., 2003, 419, 129–138 CrossRef CAS.
- K. W. Simon, P. C. Roberts, M. J. Vespremi, S. Manchen and E. M. Schmelz, Mol. Nutr. Food Res., 2009, 53, 332–340 CrossRef CAS.
- E. M. Schmelz, P. C. Roberts, E. M. Kustin, L. A. Lemonnier, M. C. Sullards, D. L. Dillehay and A. H. Merrill, Jr, Cancer Res., 2001, 61, 6723–6729 CAS.
- H. Symolon, E. M. Schmelz, D. L. Dillehay and A. H. Merrill, Jr, J. Nutr., 2004, 134, 1157–1161 CAS.
- T. Kobayashi, T. Shimizugawa, T. Osakabe, S. Watanabe and H. Okuyama, Nutr. Res., 1997, 17, 111–123 CrossRef CAS.
- A. Nilsson, Biochim. Biophys. Acta, Lipids Lipid Metab., 1969, 187, 113–121 Search PubMed.
- E. M. Schmelz, K. J. Crall, R. Larocque, D. L. Dillehay and A. H. Merrill, Jr, J. Nutr., 1994, 124, 702–712 CAS.
- L. Ohlsson, E. Hertervig, B. A. Jonsson, R. D. Duan, L. Nyberg, R. Svernlov and A. Nilsson, Am. J. Clin. Nutr., 2010 Search PubMed.
- A. Nilsson and R. D. Duan, J. Lipid Res., 2005, 47, 154–171.
- H. Vesper, E. M. Schmelz, M. N. Nikolova-Karakashian, D. L. Dillehay, D. V. Lynch and A. H. Merrill, Jr, J. Nutr., 1999, 129, 1239–1250 CAS.
- P. J. Dawson, S. R. Wolman, L. Tait, G. H. Heppner and F. R. Miller, Am. J. Pathol., 1996, 148, 313–319 Search PubMed.
- F. C. von Lintig, A. D. Dreilinger, N. M. Varki, A. M. Wallace, D. E. Casteel and G. R. Boss, Breast Cancer Res. Treat., 2000, 62, 51–62 CrossRef CAS.
- S. Mittal, D. Subramanyam, D. Dey, R. V. Kumar and A. Rangarajan, Mol. Cancer, 2009, 8, 128 CrossRef.
- G. J. Clark and C. J. Der, Breast Cancer Res. Treat., 1995, 35, 133–144 CrossRef CAS.
- H. Y. Yong, I. Y. Kim, J. S. Kim and A. Moon, Int. J. Oncol., 36, 501–507 Search PubMed.
- B. Wang, H. D. Soule and F. R. Miller, Anticancer Res., 1997, 17, 4387–4394 CAS.
- F. R. Miller, R. J. Pauley and B. Wang, Anticancer Res., 1996, 16, 1765–1769 CAS.
- E. S. Radisky and D. C. Radisky, Rev. Endocr. Metab. Disord., 2007, 8, 279–287 Search PubMed.
- I. Silins, J. Hogberg and U. Stenius, Food Chem. Toxicol., 2006, 44, 1552–1561 CrossRef CAS.
- A. Sadlonova, S. Mukherjee, D. B. Bowe, S. R. Gault, N. A. Dumas, B. A. Van Tine, N. Frolova, G. P. Page, D. R. Welch, L. Novak and A. R. Frost, Am. J. Pathol., 2007, 170, 1064–1076 Search PubMed.
- S. Iravani, L. Mora, F. R. Miller and P. J. Dawson, Int. J. Oncol., 1998, 12, 369–375 CAS.
- A. Sadlonova, Z. Novak, M. R. Johnson, D. B. Bowe, S. R. Gault, G. P. Page, J. V. Thottassery, D. R. Welch and A. R. Frost, Breast Cancer Res., 2005, 7, R46–R59 CrossRef.
- I. Silins, M. Nordstrand, J. Hogberg and U. Stenius, Carcinogenesis, 2003, 24, 1077–1083 CrossRef CAS.
- H. Furuya, S. Ohkawara, K. Nagashima, N. Asanuma and T. Hino, Int. J. Vitam. Nutr. Res., 2008, 78, 41–49 Search PubMed.
- N. Khan, V. M. Adhami and H. Mukhtar, Biochem. Pharmacol., 2008, 76, 1333–1339 CrossRef CAS.
- L. Nyberg, A. Nilsson, P. Lundgren and R. D. Duan, J. Nutr. Biochem., 1997, 8, 112–118 CrossRef CAS.
- H. F. Dvorak, T. M. Sioussat, L. F. Brown, B. Berse, J. A. Nagy, A. Sotrel, E. J. Manseau, L. Van de Water and D. R. Senger, J. Exp. Med., 1991, 174, 1275–1278 CrossRef CAS.
- E. Ritter, A. Perry, J. Yu, T. Wang, L. Tang and E. Bieberich, Ann. Surg., 2008, 247, 310–314 CrossRef.
- J. A. Tuxhorn, S. J. McAlhany, T. D. Dang, G. E. Ayala and D. R. Rowley, Cancer Res., 2002, 62, 3298–3307 CAS.
- A. C. Levine, X. H. Liu, P. D. Greenberg, M. Eliashvili, J. D. Schiff, S. A. Aaronson, J. F. Holland and A. Kirschenbaum, Endocrinology, 1998, 139, 4672–4678 CrossRef CAS.
- B. E. Gould Rothberg and M. B. Bracken, Breast Cancer Res. Treat., 2006, 100, 139–148 CrossRef CAS.
- D. Sarrio, S. M. Rodriguez-Pinilla, D. Hardisson, A. Cano, G. Moreno-Bueno and J. Palacios, Cancer Res., 2008, 68, 989–997 CrossRef CAS.
- J. Y. Yang, C. S. Zong, W. Xia, Y. Wei, M. Ali-Seyed, Z. Li, K. Broglio, D. A. Berry and M. C. Hung, Mol. Cell. Biol., 2006, 26, 7269–7282 CrossRef CAS.
- H. Zhang, L. C. Stephens and R. Kumar, Clin. Cancer Res., 2006, 12, 1479–1486 CrossRef CAS.
- K. Suzuki and K. Takahashi, Biochem. Biophys. Res. Commun., 2006, 349, 255–260 CrossRef CAS.
- Y. L. Chao, C. R. Shepard and A. Wells, Mol. Cancer, 2010, 9, 179 CrossRef.
- M. V. Fournier, J. E. Fata, K. J. Martin, P. Yaswen and M. J. Bissell, Cancer Res., 2009, 69, 4545–4552 CrossRef CAS.
- Q. Chu, M. T. Ling, H. Feng, H. W. Cheung, S. W. Tsao, X. Wang and Y. C. Wong, Carcinogenesis, 2006, 27, 2180–2189 CrossRef CAS.
- W. C. Reinhold, M. A. Reimers, P. Lorenzi, J. Ho, U. T. Shankavaram, M. S. Ziegler, K. J. Bussey, S. Nishizuka, O. Ikediobi, Y. G. Pommier and J. N. Weinstein, Mol. Cancer Ther., 2010, 9, 1–16 CrossRef CAS.
- M. F. Fraga, E. Ballestar, A. Villar-Garea, M. Boix-Chornet, J. Espada, G. Schotta, T. Bonaldi, C. Haydon, S. Ropero, K. Petrie, N. G. Iyer, A. Perez-Rosado, E. Calvo, J. A. Lopez, A. Cano, M. J. Calasanz, D. Colomer, M. A. Piris, N. Ahn, A. Imhof, C. Caldas, T. Jenuwein and M. Esteller, Nat. Genet., 2005, 37, 391–400 CrossRef CAS.
- S. E. Elsheikh, A. R. Green, E. A. Rakha, D. G. Powe, R. A. Ahmed, H. M. Collins, D. Soria, J. M. Garibaldi, C. E. Paish, A. A. Ammar, M. J. Grainge, G. R. Ball, M. K. Abdelghany, L. Martinez-Pomares, D. M. Heery and I. O. Ellis, Cancer Res., 2009, 69, 3802–3809 CrossRef CAS.
- H. M. O'Hagan, H. P. Mohammad and S. B. Baylin, PLoS Genet., 2008, 4, e1000155 Search PubMed.
- C. Mark, B. van Deurs and O. W. Petersen, Differentiation, 1990, 43, 146–156 CrossRef.
- M. C. Adriance, J. L. Inman, O. W. Petersen and M. J. Bissell, Breast Cancer Res., 2005, 7, 190–197 CrossRef CAS.
- M. Allinen, R. Beroukhim, L. Cai, C. Brennan, J. Lahti-Domenici, H. Huang, D. Porter, M. Hu, L. Chin, A. Richardson, S. Schnitt, W. R. Sellers and K. Polyak, Cancer Cell, 2004, 6, 17–32 CrossRef CAS.
- O. Cuvillier, I. Ader, P. Bouquerel, L. Brizuela, B. Malavaud, C. Mazerolles and P. Rischmann, Curr. Mol. Pharmacol., 2010, 3, 53–65 Search PubMed.
- E. Ruckhäberle, A. Rody, K. Engels, R. Gaetje, G. von Minckwitz, S. Schiffmann, S. Grosch, G. Geisslinger, U. Holtrich, T. Karn and M. Kaufmann, Breast Cancer Res. Treat., 2008, 112, 41–52 CrossRef.
- P. Mitra, M. Maceyka, S. G. Payne, N. Lamour, S. Milstien, C. E. Chalfant and S. Spiegel, FEBS Lett., 2007, 581, 735–740 CrossRef CAS.
- E. Ruckhäberle, T. Karn, A. Rody, L. Hanker, R. Gatje, D. Metzler, U. Holtrich and M. Kaufmann, J. Cancer Res. Clin. Oncol., 2009, 135, 1005–1013 CrossRef.
- N. F. Lamour and C. E. Chalfant, Curr. Drug Targets, 2008, 9, 674–682 Search PubMed.
- A. Von der Decken, J. Nutr., 1977, 107, 1340–1348.
| This journal is © The Royal Society of Chemistry 2010 |