Nanosheets as highly active solid acid catalysts for green chemical syntheses
Atsushi
Takagaki
a,
Caio
Tagusagawa
b,
Shigenobu
Hayashi
c,
Michikazu
Hara
d and
Kazunari
Domen
*b
aSchool of Materials Science, Japan Advanced Institute of Science and Technology (JAIST), 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan
bDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: domen@chemsys.t.u-tokyo.ac.jp; Fax: +81-3-5841-8838; Tel: +81-3-5841-1148
cResearch Institute of Instrumentation Frontier, National Institute of Advanced Industrial Science and Technology (AIST), Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan
dMaterials and Structures Laboratory, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan
First published on 4th December 2009
Abstract
Solid acid catalysts offer the opportunity to reduce environmental impact owing to such advantages as ease of product separation and recyclability of the catalyst, which contribute considerably to green chemistry. Nanosheets, crystalline two-dimensional metal oxide sheets prepared from cation-exchangeable layered metal oxides through exfoliation and aggregation, are a novel class of potential solid acid catalysts for replacing liquid acids, such as sulfuric acid. This article reviews the acid strength and acid catalysis of several types of nanosheets, which are strongly dependent on novel strong Brønsted acid sites attributed to bridged OH groups formed only on nanosheets. An efficient acid catalysis of layered protonated niobium molybdate in Friedel–Crafts alkylation, esterification and hydrolysis, owing to its unique intercalation availability, is also discussed.
 Atsushi Takagaki | Atsushi Takagaki received a BSc in chemistry from the Tokyo University of Science in 2001 and a MSc (2003) and PhD (2006) in chemistry from the Tokyo Institute of Technology supervised by Professor Kazunari Domen and Michikazu Hara. After graduating, Dr Takagaki had moved to The University of Tokyo as a postdoc in 2006–2008. In 2008, Dr Takagaki joined Japan Advanced Institute of Science and Technology (JAIST) as Assistant Professor. His research interests are the development of heterogeneous catalysts, especially solid acids for green and sustainable chemistry, including biomass utilization. |
 Caio Tagusagawa | Caio Tagusagawa received a BSc in chemistry from The University of São Paulo in Brazil in 2005. After moving to Japan, he received a MSc in chemistry from The University of Tokyo in 2007. He is currently a PhD candidate at The University of Tokyo under the supervision of Professor Kazunari Domen. His research interests focus on the development of novel classes of solid acid catalysts based on transition metal oxides. |
 Shigenobu Hayashi | Shigenobu Hayashi was employed at the National Chemical Laboratory for Industry (currently National Institute of Advanced Industrial Science and Technology) in 1978, after leaving a graduate school of science in The University of Tokyo midcourse. He obtained his PhD degree in chemistry from The University of Tokyo in 1983. He has conducted extensive research on the application of solid-state nuclear magnetic resonance to materials science, especially to hydrogen-storage materials, proton-conducting materials, and microporous and mesoporous materials. |
 Michikazu Hara | Michikazu Hara received his PhD degree (1992) in chemistry from the Tokyo Institute of Technology. After four years as a researcher at the Toshiba Corporation he joined the Tokyo Institute of Technology in 1997 and was promoted to Associate Professor in 2000. In 2006, as a Professor, he moved to the Materials and Structures Laboratory in the same university and concurrently holds the post of a project leader at Kanagawa Academy of Science and Technology. His research interests are in the development of solid catalysts for the environmentally benign production of chemicals and energy. He was awarded Scientific American 50 (2006). |
 Kazunari Domen | Kazunari Domen received BSc (1976), MSc (1979), and PhD (1982) honors in chemistry from the University of Tokyo. Dr Domen joined the Chemical Resources Laboratory, Tokyo Institute of Technology in 1982 as Assistant Professor and was subsequently promoted to Associate Professor in 1990 and Professor in 1996. Moving to the University of Tokyo in 2004, his research interests now include heterogeneous catalysis and materials chemistry, with particular focus on surface chemical reaction dynamics, photocatalysis, solid acid catalysis, and mesoporous materials. |
Broader contextMost chemicals and fuels are currently synthesized by catalytic reactions. Over 15 million tons of sulfuric acid is annually consumed for the production of industrially important chemicals. However, there are inherent drawbacks to the use of liquid acids as catalysts, such as the cost of separating the acid from the reaction solution and the generation of large amounts of waste. Solid acid catalysts, which are reusable and readily separable from the reaction product, offer the opportunity to reduce the impact on the environment and increase profits. The chemical industry is currently searching for a highly active and stable solid acid to improve the environmental safety of the production of chemicals and energy. Here, we describe metal oxide nanosheets, a novel class of potential solid acid catalysts. Exfoliated nanosheets have strong Brønsted acid sites on the two-dimensional sheets, and exhibit remarkable performances for several acid-catalyzed reactions. |
1. Introduction
Catalytic technologies are crucial for the current chemical industry. More than 90% of all chemicals are produced through at least one catalytic process.1 Acids are the most widely used catalysts for the production of a variety of fuels and chemicals. Until the first half of the 20th century, the most commonly used acids were mineral acids, such as HF and H2SO4. These acids, however, have serious drawbacks: they necessitate the costly and inefficient separation of the catalyst from homogeneous reaction mixtures, resulting in a huge waste of energy and a large amount of waste products. The use of solid acid catalysts generally offers several advantages over liquid acids.1–9 These include reduced equipment corrosion, ease of product separation and recyclability of the catalyst. The principles of “green chemistry” and “green technology” dictate that production should have a minimal adverse effect on the environment and human health,5,6 stimulating the replacement of these acid catalysts with recyclable, nontoxic solid acids with strong acid sites. From the 1940s onwards, liquid acids have been replaced by solid acids for the production of fuels and bulk chemicals in the fields of oil refining and petrochemicals on an enormous scale.4 Most of these reactions are carried out in the gas phase, and zeolites are widely used because of their excellent thermal and chemical stability, acid properties and shape selectivity.1–7 At present, more than 180 industrial processes, such as alkylation, isomerization, dehydration, condensation, cracking and etherification, employ solid acid catalysts, including zeolites, clays, metal oxides and ion-exchange resins.4However, in liquid phase reactions, most processes are still catalyzed by homogeneous acids, such as H2SO4, HF and AlCl3. This disagreeable situation is due to the lack of suitable highly active solid acid catalysts. Thus, the development of solid acids as heterogeneous catalysts is desirable from the viewpoint of environmentally sustainable chemistry. One of the most attractive classes of acid-catalyzed reactions is chemical synthesis in water. Organic synthesis using water as a solvent has several advantages, such as non-flammability, non-toxicity and safety, which make it a highly enviromentally-benign process. These reactions require the replacement of liquid acids with water-tolerant solid acids.8 Several types of solid acids, such as high-silica zeolite,10–13 niobium oxide,14–18 ion-exchange resin9,19–21 and functionalized mesoporous materials,22,23 have been reported. It should be noted that the development of water-tolerant solid acids is also an important issue in the field of biorefinery.24–30 In contrast to petroleum, biomass is already highly functionalized and condensed to form a complexed structure. As a major biomass, lignocellulose, which is composed of cellulose, hemicellulose and lignin, should be depolymerized by hydrolysis for use in the production of fuels and chemicals. Monosaccharides, such as glucose, fructose and xylose, obtained from hydrolysis of polysaccharides, are further transformed into furan derivatives, such as 5-hydroxymethylfurfural and furfural, as key intermediates for the production of fuels and chemicals by dehydration of water.24–28,31–37 Such essential biorefinery processes, including hydrolysis and dehydration, are also catalyzed by acids. In biorefinery, too, then, sulfuric acid is consumed. Thus, from the petroleum industry to biorefinery, there is a strong demand for the replacement of liquid acids by solid acid catalysts in order to ensure green chemical synthesis.
2. Exfoliated nanosheets
Ion-exchangeable layered metal oxides offer several attractive features owing to their wide variety of structural and electronic properties. Cation-exchangeable layered metal oxides consist of polyanion sheets of metal oxides with intercalated alkaline cations. The metal oxide polyanion sheets are negatively charged, and the high negative charge density of these metal oxide sheets allow them to function as host structures for ion-exchange reactions with various cations. The sheets of the material often exhibit electrical conductivity and photoresponses based on bandgap transitions beneficial to highly active photocatalysts.38–63From the viewpoint of heterogeneous catalysis, these layered materials offer several attractive features. For example, the large inner surface area is potentially applicable to various catalytic reactions if the reactant molecules can be intercalated. Another interesting property is the solid acidity, which the H+-exchanged form of the layered compound is expected to exhibit. In the case of clay minerals, such catalytic properties have already been examined extensively.64–69 However, for layered transition-metal oxides, applications have yet to be explored because of the difficulty of intercalation of reactant molecules. This resistance to intercalation is attributed to the higher charge density of the sheets. Exfoliation of the layered sheets may overcome this disadvantage and allow the use of layered materials in catalytic reactions.
It is well known that some clay minerals spontaneously undergo delamination in water, producing single-layer colloidal suspensions. In the last decade, it has been reported that a similar exfoliation can be artificially achieved for several classes of layered materials by a number of soft chemical processes.70–72 Exfoliated single layers are expected to behave as a new kind of inorganic macromolecule, which may exhibit unique properties different from those of bulk materials.
A variety of layered metal oxides can be exfoliated into single nanosheets. Layered metal oxides which could be exfoliated to date include CsxTi2−x/4□x/4O4 (□: vacancy),73–83 Na2Ti3O7,84 KCa2Nb3O10,85–94 KSr2Nb3O10,93,95,96 KLaNb2O7,93,97 KLa2Ti3O10,97,98 KTiNbO5,92,99 CsTi2NbO7,100 KNb3O8,99,101 K4Nb6O17,82,94,100,102–111 RbTaO3,112 Bi2SrTa2O9,113 LiNbWO6,114 LiTaWO6,114 H2W2O9,114 Cs6+xW11O36,115 manganese oxide83,116–120 and ruthenium oxide.121
Typically, layered transition-metal oxides are exfoliated by the addition of bulky quarternary alkylammonium ions (tetrabutylammonium, TBA) into aqueous solutions containing protonated layered transition-metal oxides. The resulting exfoliated nanosheets are inorganic macromolecules with a two-dimensional crystalline lattice. Exfoliated nanosheets are characterized by unique properties, such as single-crystalline quality, high crystallinity and a well-defined composition. Most exfoliated nanosheets are unilamellar and retain their high crystallinity after multiplex synthesis processes, including ion-exchange, intercalation and exfoliation, as determined by X-ray powder diffraction (XRD) and transmission electron microscopy (TEM).
Exfoliated nanosheets are used for photochemical and photocatalytic applications. Aggregated nanosheets and dye-nanosheet hybrid materials have photocatalytic activity for hydrogen production under ultraviolet and visible irradiation.86–88,90,91,93,94,102–104,109,110 They are also utilized for electrochemical energy storage.
Here, we introduce the application of exfoliated transition metal oxide nanosheets as solid acid catalysts. The exfoliated sheets are regarded as two-dimensional macropolyanions. Heteropolyanions, such as SiW12O404− and PW12O403−, are known to exhibit strong Brønsted acidity when the counter cations are protons, and these materials have already been applied to several industrial catalytic processes.122,123 A notable difference between the two systems is that macropolyanion sheets are heterogeneous catalysts, while heteropolyanions are essentially homogeneous catalysts in aqueous solution. Another important aspect of exfoliated macropolyanion sheets is their two-dimensional single-crystal structure. For most solid acid materials of transition-metal oxides, the surface structures are as spontaneously defined as in most heterogeneous catalysts. However, exfoliated sheets maintain their two-dimensional crystal structure, forming a precisely defined atomic structure. This facilitates examination of the origin of solid acidity based on an atomic scale and quantum chemical levels, which are not well understood for most solid acid materials. Moreover, a wide variety of cation-exchangeable transition metal layered oxides have been reported, which will be helpful in an analysis of the strength of the solid acidity.
3. Nanosheets as solid acid catalysts
3.1. Exfoliated aluminosilicates
Before we describe the application of exfoliated transition metal oxides as potential solid acid catalysts, it should be mentioned that delaminated aluminosilicates are highly promising materials as solid acids. Corma and co-workers have conducted extensive research on the exfoliated zeolites, such as ITQ-2,124–131 ITQ-6130,132,133 and ITQ-18,130,134 which have been successfully used for a variety of chemical syntheses, including cracking,124–126,132 acetalization,127 isomerization,128 condensation,130 and Beckmann rearrangement.1313.2. Acid properties of exfoliated transition metal oxide nanosheets
Acid properties of exfoliated transition metal oxide nanosheets depend strongly on the surface OH groups on the two-dimensional sheets. The crystal structure of the parent layered metal oxides affects the formation of acidic OH groups on exfoliated nanosheets, resulting in different acid properties.A distinct difference can be observed between HSr2Nb3O10 nanosheets and HTiNbO5 nanosheets.135Fig. 1 schematizes the structures of these two types of nanosheets, [Sr2Nb3O10]− and [TiNbO5]−. Sr2Nb3O10 nanosheets are slabs of layered perovskite. In HTiNbO5, the two-dimensional anion sheets are composed of TiO6 and NbO6 octahedra where TiO6 and NbO6 are randomly located.
![Schematic structures of [Sr2Nb3O10]− nanosheet and [TiNbO5]− nanosheet (green ball: Nb, white ball: Sr, blue ball: Ti or Nb).](/image/article/2010/EE/b918563a/b918563a-f1.gif) | ||
| Fig. 1 Schematic structures of [Sr2Nb3O10]− nanosheet and [TiNbO5]− nanosheet (green ball: Nb, white ball: Sr, blue ball: Ti or Nb). | ||
Esterification of acetic acid with ethanol on HTiNbO5 nanosheets and HSr2Nb3O10 nanosheets in the liquid phase indicated that HTiNbO5 nanosheets exhibited higher activity for the reaction than two protonated conventional zeolites (H-ZSM-5 and HMOR), whereas HSr2Nb3O10 nanosheets showed no activity. Layered HTiNbO5 and HSr2Nb3O10 also hardly catalyzed the reaction, confirming that the interlayer H+ ions in layered metal oxides are not available because the reactant molecules are unable to penetrate the interlayer space to utilize the H+ ions as catalyst. Similar results were obtained for cumene cracking and dehydration of 2-propanol in gas phase reactions.135 HTiNbO5 nanosheets exhibited high turnover frequency comparable to SiO2–Al2O3 for cumene cracking. The dehydration of 2-propanol also proceeded on HTiNbO5 nanosheets, whereas HSr2Nb3O10 nanosheets did not show activity for either reaction.
Coloration using indicator reagents and 1H magic angle spinning (MAS) NMR measurements were examined to determine the acidities of two different nanosheets. The coloration indicated that the acidity of HTiNbO5 nanosheets corresponds to 90% sulfuric acid or stronger (Hammet acidity function H0 < −8.2), whereas no coloration was observed for HSr2Nb3O10 nanosheets even in chalcone (H0 > −5.6). The properties of hydrogen species in the interlayer space and on the nanosheets were examined in detail by 1H MAS NMR spectroscopy. The 1H chemical shift reflects acidity, and hydrogen species with a large chemical shift are expected to possess strong acidity. A peak at 10.0 ppm, ascribed to strongly acidic OH groups fixed in the interlayer space, was observed for layered HSr2Nb3O10 after dehydration (Fig. 2). However, this peak disappeared after exfoliation and aggregation. For HSr2Nb3O10 nanosheets, two peaks were observed at 5.1 and 1.6 ppm. The latter main peak was ascribed to isolated OH groups without hydrogen bonds, and the former to OH groups with hydrogen bonds. CD3CN adsorption indicated that there were no strong acid sites on HSr2Nb3O10 nanosheets. In the original layered oxide, strong hydrogen bonds were formed in the narrow interlayer clearance, which made the OH groups strongly acidic. These strong hydrogen bonds disappeared with the formation of HSr2Nb3O10 nanosheets.
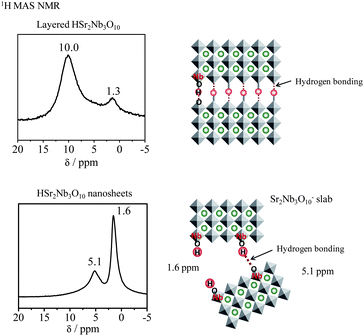 | ||
| Fig. 2 1H MAS NMR spectra for layered HSr2Nb3O10 and HSr2Nb3O10 nanosheets. | ||
In the case of HTiNbO5 nanosheets, four peaks are observed at 12, 7.6, 4.9 and 1.6 ppm (Fig. 3). The peak at 12 ppm is ascribed to OH groups in an environment similar to that of layered HTiNbO5, suggesting that some of the exfoliated sheets aggregate in a regular stacking pattern. Layered HTiNbO5 exhibits no activity for acid catalysis reactions, indicating that the OH groups are not available for these reactions in this case. Fig. 3 also schematizes the structure of the HTiNbO5 nanosheets. In [TiNbO5]− sheets obtained by the exfoliation of layered HTiNbO5, a negative charge is expected to be localized on the TiO6 octahedra. It is considered that there are two types of OH groups on the nanosheets: isolated Ti–OH and bridged Ti(OH)Nb. By analogy to isolated Nb–OH and hydrogen-bonded Nb–OH in HSr2Nb3O10 nanosheets, the peaks at 1.6 and 4.9 ppm can be assigned to isolated Ti–OH and hydrogen-bonded Ti–OH, respectively. The peak at 7.6 ppm is not observed in the 1H MAS NMR spectrum of HSr2Nb3O10 nanosheets, suggesting that this peak is due to strong acid species on HTiNbO5 nanosheets. Thus, it seems reasonable to assign the peak at 7.6 ppm to Ti(OH)Nb. CD3CN-adsorbed HTiNbO5 nanosheets are comparable to CD3CN-adsorbed strong Brønsted acid sites in zeolites,136 which is consistent with the fact that HTiNbO5 nanosheets function as a strong solid acid. Ti(OH)Nb are regarded as strong Brønsted acid sites in HTiNbO5 nanosheets.
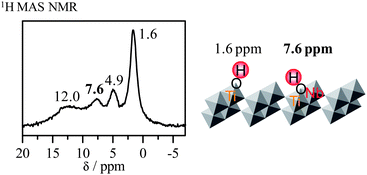 | ||
| Fig. 3 1H MAS NMR spectrum for HTiNbO5 nanosheets. | ||
The acid catalytic activity of HTiNbO5 nanosheets is attributable to the strong Brønsted acid sites, Ti(OH)Nb. In contrast, HSr2Nb3O10 nanosheets exhibited no or slight acid catalytic activity for these reactions. These nanosheets host only isolated and hydrogen-bonded OH groups, which do not have strong acidity.
3.3. Exfoliated metal oxide nanosheets with strong Brønsted acid sites
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Nb5+ = 1:1) (an amorphous oxide and a mixture of crystalline TiO2 (rutile), Nb2O5 and TiNb2O7). Neither sample exhibited catalytic activity for the reaction, whereas both titanium niobate nanosheets (HTiNbO5 and HTi2NbO7) gave high activity. It was confirmed that the peak due to Ti(OH)Nb was absent in the 1H MAS NMR spectrum for the bulk samples. Thus, it appears that Ti(OH)Nb cannot be formed on bulky metal oxides prepared simply by mixing Ti4+ and Nb5+, suggesting that nanosheets of TiO6 and NbO6 octahedra mixed at an atomic level are essential for the generation of such strong acid sites.
:Nb5+ = 1:1) (an amorphous oxide and a mixture of crystalline TiO2 (rutile), Nb2O5 and TiNb2O7). Neither sample exhibited catalytic activity for the reaction, whereas both titanium niobate nanosheets (HTiNbO5 and HTi2NbO7) gave high activity. It was confirmed that the peak due to Ti(OH)Nb was absent in the 1H MAS NMR spectrum for the bulk samples. Thus, it appears that Ti(OH)Nb cannot be formed on bulky metal oxides prepared simply by mixing Ti4+ and Nb5+, suggesting that nanosheets of TiO6 and NbO6 octahedra mixed at an atomic level are essential for the generation of such strong acid sites.
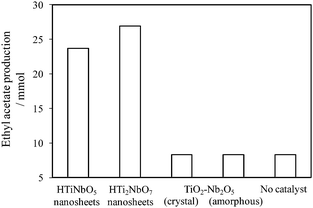 | ||
| Fig. 4 Production of ethyl acetate on titanium niobate nanosheets and Ti–Nb mixed oxides (343 K, catalyst: 0.2 g, acetic acid: 0.10 mol, ethanol: 0.10 mol, reaction time: 6 h). | ||
The formation of this kind of strong Brønsted acid site on pristine niobium oxide nanosheets would be helpful for understanding the unique acid properties of niobic acid.138 Niobic acid, which is amorphous niobium oxide with incorporated water molecules, Nb2O5·nH2O, is sufficiently stable and remains active for acid catalysis in the presence of water. Nb2O5·nH2O is already being used for the production of industrially important chemicals, such as methyl tertiary butyl ether (MTBE), methyl methacrylate and 2,5-dimethyl-2,4-hexadiene.4,15 However, the properties of Nb2O5·nH2O have yet to be examined thoroughly, and although it has been reported that Nb2O5·nH2O hosts several types of acid sites,14,139–142 the details remain unclear, presenting an obstacle to the further development of active Nb2O5·nH2O catalysts. The lack of investigation can largely be attributed to the complexity of amorphous Nb2O5·nH2O.140–144 Niobium oxide nanosheets (HNb3O8) can be readily synthesized from layered KNb3O8 through exfoliation and aggregation. The two-dimensional single-crystal nanosheets enable a simpler characterization of the surface structure and surface functional groups. The hydrolysis of ethyl acetate in the presence of a large amount of water showed that the HNb3O8 nanosheets functioned as a water-tolerant solid acid catalyst and exhibited high catalytic performance, reaching a level twice that of niobic acid. The HNb3O8 nanosheets were also superior to niobic acid for other acid-catalyzed reactions, including esterification and Friedel–Crafts alkylation. As mentioned above, HSr2Nb3O10 nanosheets did not function as a solid acid. Although Nb2O5·nH2O, HNb3O8 nanosheets and HSr2Nb3O10 nanosheets are all Nb5+-containing oxides, there appears to be a definite distinction in catalysis and acidity between these materials, suggesting that the appearance of strong Brønsted acid sites is largely structure-dependent. Fig. 5 shows 1H MAS NMR spectra for HSr2Nb3O10 nanosheets, HNb3O8 nanosheets and niobic acid, along with the schematic structures of the sheets. On HSr2Nb3O10 sheets, which consist of corner-sharing NbO6 octahedra,145–147 H is located only on oxygen atoms at the vertices of the NbO6 octahedra. The two peaks at 5.1 and 1.6 ppm in the NMR spectrum for the HSr2Nb3O10 nanosheets can be assigned to Nb–OH groups with and without hydrogen bonds, respectively.124 In the NMR spectrum for HNb3O8 nanosheets, three peaks appear at 1.3, 5.5 and 8.5 ppm. As the HNb3O8 sheets are composed of edge-sharing NbO6 octahedra,148–150 it is thought that there are OH groups shared by two Nb5+ (Nb(OH)Nb) in addition to isolated Nb–OH groups. As a result, the chemical shift at 8.5 ppm can be attributed to Nb(OH)Nb. This peak has a large chemical shift and is not observed in the 1H MAS NMR spectrum of HSr2Nb3O10 nanosheets (not an acid catalyst), indicating that Nb(OH)Nb functions as a strong Brønsted acid site. The 1H MAS NMR spectrum for dehydrated Nb2O5·nH2O reveals a broad peak centered around 6.9 ppm with a shoulder peak at 1.7 ppm. Nb2O5·nH2O is formed by the random condensation of H8Nb6O19 clusters,141,142 resulting in a large variety of hydroxyl groups; hence the broad peak in the 1H MAS NMR spectrum. Thus, Nb(OH)Nb functions as a strong Brønsted acid site in Nb2O5·nH2O as well.
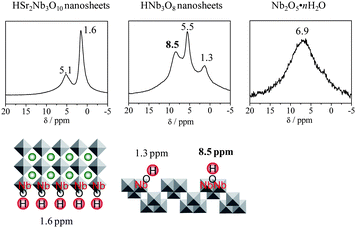 | ||
| Fig. 5 1H MAS NMR spectra for HSr2Nb3O10 nanosheets, HNb3O8 nanosheets and niobic acid. | ||
Fig. 6 shows SEM images of the aggregated HTiNbO5, HNb3O8 and HNbWO6 nanosheets. The addition of H+ to solutions containing colloidal nanosheets causes the nanosheets to aggregate randomly as precipitates with the expected composition ratios, as confirmed by energy dispersive X-ray spectroscopy, and the resultant precipitates are free of the tabular particles observed in the original layered compounds.151 The Brunauer-Emmett-Teller (BET) surface areas of the aggregated nanosheet precipitates are 153 m2 g−1 for HTiNbO5, 101 m2 g−1 for HNb3O8 and 66 m2 g−1 for HNbWO6, all much higher than that of the original protonated layered oxides (about 1 m2 g−1). This indicates that the exfoliation-aggregation process provides access to the surface of the nanosheets.
 | ||
| Fig. 6 SEM images of aggregated HTiNbO5, HNb3O8 and HNbWO6 nanosheets. | ||
The XRD pattern of HTiNbO5 nanosheets shows a weaker (002) diffraction peak and a complete absence of (004) and (008) peaks, in contrast to the original sample, indicating a much poorer periodic layer structure than that of the original HTiNbO5 (Fig. 7). However, diffraction peaks such as (200) and (020), corresponding to in-plane diffraction, are preserved in the XRD pattern of HTiNbO5 nanosheets, which means the in-plane structure is maintained after precipitation. The HNb3O8 precipitate also has a weaker diffraction peak at low angle (2θ < 10°), and the in-plane diffraction peaks (e.g., (101)) are similarly retained upon exfoliation-aggregation. HNbWO6 nanosheets form the same poorly periodic structure with a partial absence of diffraction peaks at low angle (<10°) and the preservation of in-plane diffraction peaks ((110) and (200)). For these nanosheets, the XRD patterns displayed much weaker diffraction peaks compared to those of the original layered compounds, while the in-plane diffraction peaks such as (410), (200) and (210) persisted even after exfoliation-aggregation.
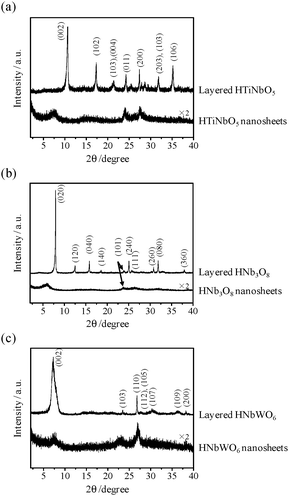 | ||
| Fig. 7 XRD patterns for layered- and nanosheets metal oxides. (a) HTiNbO5, (b) HNb3O8, and (c) HNbWO6. | ||
NH3 temperature programmed desorption (NH3-TPD) is a basic method for measuring the acid amount and acid strength from the desorbed NH3 quantity and desorbed temperature. Fig. 8 shows the NH3-TPD (m/z = 16) results for metal-oxide aggregated nanosheets. The NH3-TPD profiles for HTiNbO5 and HNb3O8 nanosheets exhibit three and two distinct peaks, respectively, at 430, 482 and 535 K for HTiNbO5, and at 440 and 550 K for HNb3O8. HNbWO6 nanosheets exhibit similar peaks at 455 K and 560 K. The peaks at 535–560 K are attributable to strong acid sites. The order of acid strength in these solid acids increases in the order HTiNbO5 < HNb3O8 ≤ HNbWO6. The strong acid amount calculated from the NH3-TPD results for the aggregated nanosheets is 0.24 mmol g−1 for HTiNbO5, 0.28 mmol g−1 for HNb3O8 and 0.34 mmol g−1 for HNbWO6.
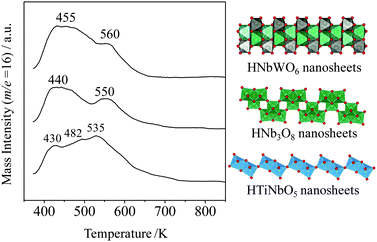 | ||
| Fig. 8 NH3-TPD (m/z 16) spectra for HTiNbO5, HNb3O8, and HNbWO6 nanosheets. | ||
The acid properties of the aggregated nanosheets were also evaluated by 31P MAS NMR, using trimethylphosphine oxide (TMPO) as a probe molecule. As the 31P chemical shifts of protonated TMPO (i.e., TMPOH+) tend to move downfield, higher chemical shifts indicate higher protonic acid strength. Fig. 9 shows 31P MAS NMR spectra for 50 mol% TMPO adsorbed aggregated nanosheets. The peaks below 50 ppm are ascribed to physisorbed or crystalline TMPO, which are excluded from the following discussion. The HNb3O8 nanosheet catalyst exhibits a main broad 31P peak centered at ca. 70 ppm, while the HTiNbO5 nanosheet catalyst exhibits a main peak at ca. 63 ppm. The HNbWO6 nanosheet catalyst exhibits a main broad peak at ca. 71 ppm, with a shoulder peak at 65 ppm. The larger chemical shift corresponds to the stronger acidity. The results in Fig. 9 demonstrate that stronger acidic sites are present in the order HNbWO6 ≥ HNb3O8 > HTiNbO5, which correlates with the trend in maximum temperature of the NH3-TPD curves assigned to M(OH)M′ acid sites. The peak positions in the 31P MAS NMR spectra indicate that the strong acid sites in the HNbWO6 and HNb3O8 nanosheet precipitates (70–71 ppm) are comparable in strength to those in HY zeolites (65 ppm).152
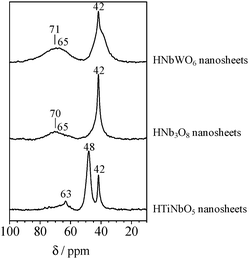 | ||
| Fig. 9 31P MAS NMR spectra for trimethylphosphine oxide adsorbed HTiNbO5, HNb3O8, and HNbWO6 nanosheets. | ||
The acid catalytic activity of the aggregated nanosheets is examined through liquid-phase Friedel–Crafts alkylation (Table 1). The original layered metal oxides (HTiNbO5, HNb3O8 and HNbWO6) do not catalyze these reactions, with alkylated products absent or afforded at yields of less than 1%. However, the exfoliated and aggregated materials, with the exception of HTiNbO5, exhibit significant activity for this reaction. The HNb3O8 aggregated nanosheets have a relatively low activity for this reaction at 373 K, but exhibit remarkable activity at 423 K. The catalytic activity of the nanosheet precipitates decreases in the order HNbWO6 > HNb3O8 ≫ HTiNbO5, which is consistent with the acid strength determined by NH3-TPD. The nanosheet materials consisting of the group-5 element (Nb5+) and group-6 element (W6+) afford higher benzylanisole yields and higher NH3 desorption temperatures than the nanosheet precipitate bearing the group-4 element (Ti4+). Catalysts with the aggregated nanosheet structure have higher acid strength, depending on the combination of metal oxides, decreasing in the order Nb5+(OH)W6+ > Nb5+(OH)Nb5+ > Ti4+(OH)Nb5+.
| Catalyst | Yield (%)b | Selectivity (%)b | Turnover rate /h−1 |
|---|---|---|---|
| a Reaction conditions: anisole (100 mmol), benzyl alcohol (10 mmol), catalyst (0.2 g), 373 K, 2 h. b benzylanisole yield and selectivity. c 423 K, 4 h. | |||
| HTiNbO5 nanosheets | 0 (0.3 c) | — | 0 (0.2) |
| HNb3O8 nanosheets | 1.8 (94c) | 23 | 1.6 (42) |
| HNbWO6 nanosheets | 8.6 | 42 | 6.3 |
| HTaWO6 nanosheets | 58.4 | 66 | 104 |
| H-ZSM5 | 10.5 | 37 | 13.1 |
| H-Beta | 36.6 | 56 | 9.2 |
| Amberlyst-15 | 57.9 | 58 | 3.0 |
| Nafion NR50 | 63.0 | 63 | 17.5 |
Substitution of niobium by tantalum is also effective in generating strong acid sites on nanosheets.137,151 HTiTaO5 and HTaWO6 are known to form layered structures similar to HTiNbO5 and HNbWO6. The BET surface areas of these tantalum oxide nanosheets are 92 m2 g−1 for HTiTaO5 nanosheets and 47 m2 g−1 for HTaWO6 nanosheets, lower than that of niobium-based oxides (153 m2 g−1: HTiNbO5 nanosheets; 66 m2 g−1: HNbWO6 nanosheets). NH3-TPD measurement revealed that tantalum-based nanosheets possess much stronger acid sites than the corresponding niobium-based nanosheets. HTaWO6 nanosheets exhibit a broad TPD profile with peaks at 455 K, 555–595 K, and 667 K (Fig. 10). The highest peak at 667 K is due to preserved acid sites in the layered structure that cannot contribute to acid catalysis. The peaks at 555–595 K for HTaWO6 nanosheets are assigned to strong acid sites, higher than that for HNbWO6 nanosheets (560 K). The difference of acidity between tantalum- and niobium-based nanosheets strongly affects acid catalysis. HTaWO6 nanosheets exhibit remarkable activity for Friedel–Crafts alkylation of anisole with benzyl alcohol, giving a yield six times higher than that for HNbWO6 nanosheets and comparable to that for Amberlyst-15 and Nafion NR50 (Table 1). The turnover rate for the HTaWO6 nanosheets are estimated to be 104 h−1, higher than that for Nafion NR50 (17.5 h−1) and H-ZSM5 (13.1 h−1). A similar behavior was observed for HTiTaO5 and HTiNbO5 nanosheets. For hydrolysis of ethyl acetate and esterification of acetic acid, HTiTaO5 nanosheets function as a water-tolerant solid acid superior to HTiNbO5 nanosheets. The catalytic activity of HTiTaO5 nanosheets reached about twice that of HTiNbO5 nanosheets and Nb2O5·nH2O.
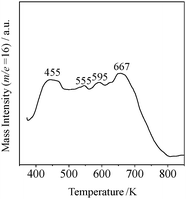 | ||
| Fig. 10 NH3-TPD (m/z 16) spectrum for HTaWO6 nanosheets. | ||
3.4. Layered niobium molybdate
During our investigation, we have discovered that protonated niobium molybdate, HNbMoO6, exhibits a significantly high catalytic activity.153–156 Layered HNbMoO6·nH2O can be obtained easily from the Li form by a proton-exchange reaction. The resultant material consists of layers formed of randomly sited MO6 (M = Nb and Mo) octahedra with H2O in the interlayer. This layered protonated niobium molybdate is highly active for several acid-catalyzed reactions, including Friedel–Crafts alkylation, acetalization, esterification, hydrolysis, and hydration. Fig. 11 shows the catalytic performance results for the Friedel–Crafts alkylation of toluene with benzyl alcohol. HNbMoO6 exhibits remarkable activity for the reaction, much higher than that of the other solid acids tested. HNbMoO6 also catalyzes the alkylation of anisole or benzene, achieving high yields: the yield of benzyl anisole reached 99% after 30 min, whereas those for ion-exchange resins only reached ca. 42% even after 1 h. The results of acetalization of cyclohexanone, esterification of lactic acid, hydrolysis of ethyl lactate, and hydration of 2,3-dimethyl-2-butene over HNbMoO6 are summarized in Table 2. For these reactions, HNbMoO6 showed remarkable activity, comparable or superior to conventional ion-exchange resins (Nafion NR50 and Amberlyst-15). Even in the presence of water (hydrolysis and hydration), HNbMoO6 functioned as an efficient catalyst. The HNbMoO6 was recoverable by filtration and washing with water and acetone to remove the residue within the interlayer, and the material was confirmed to be reusable with no change in crystal structure or activity after three reuse cycles. | ||
| Fig. 11 Yield of benzyltoluene for Friedel–Crafts alkylation over layered HNbMoO6. Reaction conditions: toluene (100 mmol), benzyl alcohol (10 mmol), catalyst (0.2 g), 353 K, 4 h. | ||
| Catalyst | Acetalizationa reaction rate /mmol h−1 | Esterificationb reaction rate /mmol h−1 | Hydrolysisc reaction rate /mmol h−1 | Hydrationd Yield (%) |
|---|---|---|---|---|
| a Cyclohexanone (20 mmol), methanol (5 mL), catalyst (0.1 g), 323 K. b Lactic acid (100 mmol), ethanol (1 mol), catalyst (0.2 g), 343 K. c Ethyl lactate (50 mmol), H2O (9 mL), catalyst (0.1 g), 343 K. d 2,3-dimethyl-2-butene (12.5 mmol), H2O (7.6 mL), catalyst (0.2 g), 343 K, 5 h. | ||||
| HNbMoO6 | 158 | 5.7 | 0.9 | 3.4 |
| Amberlyst-15 | 66 | 4.5 | 1.22 | 3.7 |
| Nafion NR50 | 58 | 6.0 | 1.06 | 2.2 |
| — | 0 | 1.4 | 0 | 0.4 |
This high catalytic activity of layered HNbMoO6 is attributed to the efficient intercalation of substrates within the interlayer with strong acidity. Substrates such as benzyl alcohol are readily intercalated into HNbMoO6. In order to validate this process, the adsorption of benzyl alcohol onto HNbMoO6 was investigated in isolation by immersing HNbMoO6·nH2O (n = 1.23) in benzyl alcohol solution under constant shaking for 30 min. Fig. 12 shows the XRD pattern of the resultant material after drying at room temperature. Immersion in benzyl alcohol shifts the (001) peak due to HNbMoO6 to lower angles, corresponding to an increase in basal spacing from 13.7 to 16.3 Å, which confirms the intercalation of benzyl alcohol into the layered HNbMoO6. Given the interlayer spacing of the fully dehydrated sample (10.8 Å),157 the total expansion is estimated to be 5.5 Å. In the Friedel–Crafts alkylation of toluene in the presence of benzyl alcohol, the interlayer spacing of the layered HNbMoO6 was increased to 16.6 Å. A preliminary benzyl alcohol-intercalated sample, placed in anisole solution, then heated at 373 K for 1 h, indicated that alkylated products (benzyl anisole) were detected, and the basal spacing of HNbMoO6 was found to decrease to 14.1 Å. These results indicate that intercalated benzyl alcohol is consumed in the reaction and that the interlayer sites of HNbMoO6 function as active sites. Other substrates, including n-alkyl alcohols and ketones, could also be successfully intercalated into the interlayer gallery of HNbMoO6.
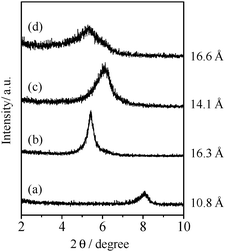 | ||
| Fig. 12 XRD patterns of HNbMoO6: (a) dehydrated, (b) after immersion in benzyl alcohol for 30 min, (c) after reaction of anisole, (d) during Friedel–Crafts alkylation. | ||
The intercalation mechanism greatly affects the catalytic activity of this layered catalyst. A difference in intercalation behavior was observed between carboxylic acid and hydroxycarboxylic acid. The latter could easily intercalate, whereas the former did not, resulting in a unique behavior of acid catalysis for esterification.154 Layered HNbMoO6 exhibits negligible activity for the esterification of carboxylic acid, and remarkable activity for the esterification of hydroxycarboxylic acid. Fig. 13 shows the results of the esterification of three carboxylic acids (acetic, propionic, butyric) and lactic acid with n-butanol. Amberlyst-15, Nafion NR50 and layered HNbMoO6 were compared. For the three carboxylic acids, the ester yield over the three catalysts examined decreases in the order Amberlyst-15 > Nafion NR50 ≫ HNbMoO6, and the activity decreases with increasing carbon number of the carboxylic acid. The two ion-exchange resins exhibit similar yields for propionic acid and lactic acid, which have the same carbon number. However, layered HNbMoO6 exhibits a very high activity for lactic acid, with a yield of 35.1%, as compared to the 6.2% for propionic acid. The catalytic activity of ion-exchange resins is influenced only by the carbon number of the carboxylic acid, and is unaffected by the presence of OH groups in lactic acid. In contrast, the OH groups of lactic acid appear essential for allowing intercalation into the layered HNbMoO6. Given that activation of carboxylic acid by a proton is necessary for the reaction, esterification of the three carboxylic acids did not occur due to the difficulty of intercalation. However, lactic acid, which has an OH group adjacent to the carboxyl group, could intercalate into the oxide, resulting in a high catalytic activity for the reaction.
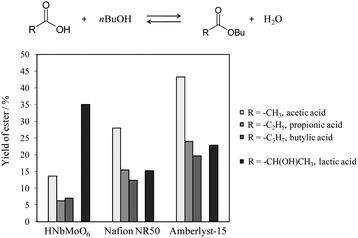 | ||
| Fig. 13 The results of esterification of carboxylic acids (acetic, propionic, butyric) and lactic acid with n-butanol over HNbMoO6, Amberlyst-15 and Nafion NR50. Reaction condition: carboxylic acid (0.05 mol), n-butanol (0.25 mol), catalyst (0.1 g), 343 K, 5 h. | ||
The acid properties of HNbMoO6 were determined by 31P MAS NMR using TMPO as a probe molecule.155 HNbMoO6 exhibits distinct peaks at 86.4 and 81 ppm, attributable to strong acid sites. The peak positions indicate that the acid sites of layered HNbMoO6 are stronger than those of both H-type zeolites (65 ppm for HY,152 78 ppm for H-Beta158) and ion-exchange resin (81 ppm for Amberlyst-15), and comparable in strength to those on strongly acidic zeolites (86 ppm for HZSM-5159 and HMOR158). It was also confirmed by XRD that TMPO intercalates into HNbMoO6, indicating that the strong acid sites of HNbMoO6, detectable in the NMR spectrum, are present in the interlayer.
3.5. Biorefinery applications
Exfoliated nanosheets and layered HNbMoO6 function as efficient solid acid catalysts even in the presence of water. This remarkable feature can be exploited in biorefinery applications. Valente and co-workers reported that exfoliated metal oxide nanosheets could dehydrate D-xylose to produce furfural in a water–toluene solvent mixture.160 HTiNbO5 nanosheets catalyzed this reaction to achieve a furfural yield of 55% at 92% conversion, whereas the furfural yield in the presence of niobic acid (Nb2O5·nH2O) was 12%. Other nanosheets, including HTi2NbO7, H2Ti3O7, HNb3O8, and H4Nb6O17, also gave a high furfural yield. These nanosheets could be reused at least three times without loss of activity.Layered HNbMoO6 exhibits remarkable performance for hydrolysis of saccharides.156 Polysaccharides such as starch and cellulose can be converted into monosaccharides such as glucose, a major platform for the synthesis of a variety of chemicals, by a homogeneous acid-catalyzed reaction using sulfuric acid and/or by enzymatic reactions. The development of a reusable and readily separable solid acid catalyst for the hydrolysis of saccharides is considered essential for converting biomass into bioethanol and useful chemicals with the lowest environmental impact. Fig. 14 shows the results of the hydrolysis of sucrose and cellobiose. The hydrolysis of cellobiose was carried out at 373 K using 0.2 g of catalyst, 1.0 g of cellobiose, and 10 mL of water. The catalytic performance of these catalysts are compared with that of a range of conventional solid acids, including ion-exchange resins, H-type zeolite and niobic acid. For the hydrolysis of sucrose, an equivalent amount of glucose and fructose was produced. H-ZSM5 zeolite was inactive for this reaction, and niobic acid, although known to be water-tolerant, did not achieve appreciable activity for this reaction. The activity of layered HNbMoO6, however, substantially exceeded the maximum performance of any of the other solid materials tested. This high activity is also found for hydrolysis of cellobiose. Cellobiose, a subunit of cellulose, consists of β-1,4-glycosidic bonds, which are much more stable than the α-1,2-glycosidic bonds comprising sucrose, and thus is more resistant to hydrolysis. The layered HNbMoO6 catalyst also exhibited the highest performance in this hydrolysis reaction, producing glucose at twice the rate of the ion-exchange resins and achieving a turnover rate comparable to that of Nafion NR50. It should be noted that the turnover rate of HNbMoO6 was much higher than that of Amberlyst-15 and twice that of sulfuric acid. This layered oxide could produce glucose from starch and cellulose in the presence of water.
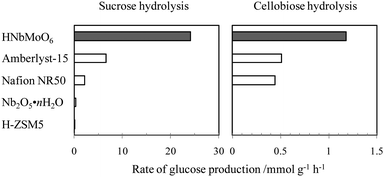 | ||
| Fig. 14 Hydrolysis of disaccharides (sucrose and cellobiose) over solid acid catalysts. Sucrose hydrolysis: sucrose (2.92 mmol), H2O (20 mL), catalyst (0.2 g), 353 K. Cellobiose hydrolysis: cellobiose (2.92 mmol), H2O (10 mL), 373 K. | ||
Conclusions
Strong solid acid catalysts based on nanosheets obtained through protonation, exfoliation and aggregation of cation-exchangeable layered metal oxides have been developed. Exfoliation-aggregation of protonated layered oxides does not always result in strong solid acids, and the formation of bridging hydroxyl groups, M(OH)M′, which are intrinsic to the crystal structure of the two-dimensional sheets, is essential for the preparation of nanosheet catalysts with strong acidity. The acid strength of the strong Brønsted acid sites is significantly influenced by metal cations M and M′ in the order (Ti4+, Nb5+) < (Nb5+, Nb5+) < (Nb5+, W6+). In addition, a unique method has been developed for the acid catalysis of layered niobium molybdate, exploiting its facile intercalation availability for substrate within the interlayer gallery with strong acidity. Exfoliated nanosheets and layered HNbMoO6 functioned as efficient solid acid catalysts even in the presence of water, and were exploited in biorefinery applications, including the formation of monosaccharides by hydrolysis and furan-derivatives by dehydration.Notes and references
- W. F. Hölderich, J. Röseler, G. Heitmann and A. T. Leibens, Catal. Today, 1997, 37, 353 CrossRef CAS.
- A. Corma, Chem. Rev., 1995, 95, 559 CrossRef CAS.
- A. Corma, Chem. Rev., 1997, 97, 2373 CrossRef CAS.
- K. Tanabe and W. F. Hölderich, Appl. Catal., A, 1999, 181, 399 CrossRef CAS.
- J. H. Clark, Acc. Chem. Res., 2002, 35, 791 CrossRef CAS.
- J. H. Clark, Green Chem., 1999, 1, 1 RSC.
- R. A. Sheldon and R. S. Downing, Appl. Catal., A, 1999, 189, 163 CrossRef CAS.
- T. Okuhara, Chem. Rev., 2002, 102, 3641 CrossRef CAS.
- A. M. Harmer and Q. Sun, Appl. Catal., A, 2001, 221, 45 CrossRef CAS.
- S. Namba, N. Hosonuma and T. Yashima, J. Catal., 1981, 72, 16 CrossRef CAS.
- H. Ishida, Catal. Surv. Jpn., 1997, 1, 241 CrossRef CAS.
- H. Ogawa, T. Koh, K. Taya and T. Chihara, J. Catal., 1994, 148, 493 CrossRef CAS.
- P. Botella, A. Corma, J. M. Lopez Nieto, S. Valencia, M. E. Lucas and M. Sergio, Appl. Catal., A, 2000, 203, 251 CrossRef CAS.
- I. Nowak and M. Ziolek, Chem. Rev., 1999, 99, 3603 CrossRef CAS.
- K. Tanabe and S. Okazaki, Appl. Catal., A, 1995, 133, 191 CrossRef CAS.
- K. Ogasawara, T. Iizuka and K. Tanabe, Chem. Lett., 1984, 645 CrossRef CAS.
- O. Okazaki and H. Harada, Chem. Lett., 1988, 1313 CrossRef.
- K. Hanaoka, T. Takeuchi, T. Matsuzaki and Y. Sugi, Catal. Today, 1990, 8, 123 CrossRef CAS; T. Hanaoka, K. Takeuchi, T. Matsuzaki and Y. Sugi, Catal. Lett., 1990, 5, 13 CrossRef CAS.
- G. A. Olah, P. S. Lyer and G. K. S. Prakasn, Synthesis, 1986, 513 CrossRef CAS.
- M. A. Harmer, Q. Sun and W. E. Farneth, J. Am. Chem. Soc., 1996, 118, 7708 CrossRef CAS.
- K. Okuyama, X. Chen, K. Takata, D. Odawara, T. Suzuki, S. Nakata and T. Okuhara, Appl. Catal., A, 2000, 190, 253 CrossRef CAS.
- R. L. Dhepe, M. Ohashi, S. Inagaki, M. Ichikawa and A. Fukuoka, Catal. Lett., 2005, 102, 163 CrossRef CAS.
- J. A. Bootsma and B. H. Shanks, Appl. Catal., A, 2007, 327, 44 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164 CrossRef CAS.
- G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184 CrossRef CAS.
- R. Rinaldi and F. Schüth, Energy Environ. Sci., 2009, 2, 610 RSC.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033 RSC.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008, 130, 12787 CrossRef CAS.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933 CrossRef CAS; J. N. Chheda, Y. Román-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342 RSC.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597 CrossRef CAS; Y. Su, H. M. Brown, X. Huang, X.-D. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 361, 117 CrossRef CAS.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979 CrossRef CAS.
- D. Mercadier, L. Rigal, A. Gaset and J.-P. Gorrichon, J. Chem. Technol. Biotechnol., 1981, 31, 489 Search PubMed.
- Y. Nakamura and S. Morikawa, Bull. Chem. Soc. Jpn., 1980, 53, 3705 CrossRef CAS.
- C. Moreau, R. Durand, S. Razigade, J. Duhamet, P. Faugeras, P. Rivalier, P. Ros and G. Avignon, Appl. Catal., A, 1996, 145, 211 CrossRef CAS.
- C. Carlini, M. Giuttari, A. M. R. Galletti, G. Sbrana, T. Armaroli and G. Busca, Appl. Catal., A, 1999, 183, 295 CrossRef CAS.
- K. Domen, A. Kudo, A. Shinozaki, A. Tanaka, K.-I. Maruya and T. Onishi, J. Chem. Soc., Chem. Commun., 1986, 356 RSC.
- K. Domen, A. Kudo, M. Shibata, A. Tanaka, K.-I. Maruya and T. Onishi, J. Chem. Soc., Chem. Commun., 1986, 1706 RSC.
- A. Kudo, A. Tanaka, K. Domen, K.-I. Maruya and T. Onishi, J. Catal., 1988, 111, 67 CrossRef CAS.
- A. Kudo, K. Sayama, A. Tanaka, K. Asakura, K. Domen, K. Maruya and T. Onishi, J. Catal., 1989, 120, 337 CrossRef CAS.
- K. Sayama, A. Tanaka, K. Domen, K. Maruya and T. Onishi, Catal. Lett., 1990, 4, 217 CrossRef CAS.
- K. Sayama, A. Tanaka, K. Domen, K. Maruya and T. Onishi, J. Catal., 1990, 124, 541 CrossRef CAS.
- K. Sayama, A. Tanaka, K. Domen, K. Maruya and T. Onishi, J. Phys. Chem., 1991, 95, 1345 CrossRef CAS.
- J. Yoshimura, A. Tanaka, J. N. Kondo and K. Domen, Bull. Chem. Soc. Jpn., 1995, 68, 2439 CAS.
- T. Takata, Y. Furumi, K. Shinohara, A. Tanaka, M. Hara, J. N. Kondo and K. Domen, Chem. Mater., 1997, 9, 1063 CrossRef CAS.
- S. Ikeda, M. Hara, J. N. Kondo, K. Domen, H. Takahashi, T. Okubo and M. Kakihana, Chem. Mater., 1998, 10, 72 CrossRef CAS.
- S. Ikeda, M. Hara, J. N. Kondo, K. Domen, H. Takahashi, T. Okubo and M. Kakihana, J. Mater. Res., 1998, 13, 852 CrossRef CAS.
- J. Yoshimura, Y. Ebina, J. Kondo, K. Domen and A. Tanaka, J. Phys. Chem., 1993, 97, 1970 CrossRef CAS.
- K. Domen, Y. Ebina, T. Sekine, A. Tanaka, J. Kondo and C. Hirose, Catal. Today, 1993, 16, 479 CrossRef CAS.
- Y. Ebina, A. Tanaka, J. N. Kondo and K. Domen, Chem. Mater., 1996, 8, 2534 CrossRef CAS.
- Y. Yamashita, K. Hyuga, V. Petrykin, M. Kakihana, M. Yoshimura, K. Domen and A. Kudo, J. Ceram. Soc. Jpn., 2007, 115, 511 CrossRef CAS.
- H. Hata, Y. Kobayashi, V. Bojan, W. J. Youngblood and T. E. Mallouk, Nano Lett., 2008, 8, 794 CrossRef CAS.
- T. Mitsuyama, A. Tsutsumi, T. Hata, K. Ikeue and M. Machida, Bull. Chem. Soc. Jpn., 2008, 81, 401 CrossRef CAS.
- K. Shimizu, Y. Tsuji, M. Kawakami, K. Toda, T. Komada, M. Sato and Y. Kitayama, Chem. Lett., 2002, 1158 CrossRef CAS.
- K. Shimizu, S. Itoh, T. Hatamachi, T. Komada, M. Sato and K. Toda, Chem. Mater., 2005, 17, 5161 CrossRef CAS.
- H. Takahashi, M. Kakihana, Y. Yamashita, K. Yoshida, S. Ikeda, M. Hara and K. Domen, J. Alloys Compd., 1999, 285, 77 CrossRef CAS.
- H. Takahashi, M. Kakihana, Y. Yamashita, K. Yoshida, S. Ikeda, M. Hara and K. Domen, Phys. Chem. Chem. Phys., 2000, 2, 4461 RSC.
- M. Machida, J. Yabunaka and T. Kijima, Chem. Commun., 1999, 1939 RSC.
- M. Machida, J. Yabunaka and T. Kijima, Chem. Mater., 2000, 12, 812 CrossRef CAS.
- M. Machida, K. Miyazaki, S. Matsushima and M. Arai, J. Mater. Chem., 2003, 13, 1433 RSC.
- Y. I. Kim, S. J. Atherton, E. S. Brigham and T. E. Mallouk, J. Phys. Chem., 1993, 97, 11802 CrossRef CAS.
- T. Nakato, K. Kusunoki, K. Yoshizawa, K. Kuroda and M. Kaneko, J. Phys. Chem., 1995, 99, 17896 CrossRef CAS.
- T. J. Pinnavaia, Science, 1983, 220, 365 CAS.
- P. Laszlo, Acc. Chem. Res., 1986, 19, 121 CrossRef CAS.
- Y. Izumi and M. Onaka, Adv. Catal., 1992, 38, 245 CAS.
- K. Motokura, N. Fujita, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Angew. Chem., Int. Ed., 2006, 45, 2605 CrossRef CAS.
- K. Motokura, N. Nakagiri, K. Mori, T. Mizugaki, K. Ebitani, K. Jitsukawa and K. Kaneda, Org. Lett., 2006, 8, 4617 CrossRef CAS.
- K. Motokura, N. Nakagiri, T. Mizugaki, K. Ebitani and K. Kaneda, J. Org. Chem., 2007, 72, 6006 CrossRef CAS.
- A. J. Jacobson, Mater. Sci. Forum, 1994, 152–153, 1 CAS.
- R. E. Schaak and T. E. Mallouk, Chem. Mater., 2002, 14, 1455 CrossRef CAS.
- M. Osada and T. Sasaki, J. Mater. Chem., 2009, 19, 2503 RSC.
- T. Sasaki, M. Watanabe, H. Hashizume, H. Yamada and H. Nakazawa, J. Am. Chem. Soc., 1996, 118, 8329 CrossRef CAS.
- T. Sasaki and M. Watanabe, J. Am. Chem. Soc., 1998, 120, 4682 CrossRef CAS.
- R. Abe, K. Shinohara, A. Tanaka, M. Hara, J. N. Kondo and K. Domen, Chem. Mater., 1998, 10, 329 CrossRef CAS.
- F. Kooli, T. Sasaki, V. Rives and M. Watanabe, J. Mater. Chem., 2000, 10, 497 RSC.
- T. Sasaki, Y. Ebina, T. Tanaka, M. Harada, M. Watanabe and G. Decher, Chem. Mater., 2001, 13, 4661 CrossRef CAS.
- N. Sakai, Y. Ebina, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2004, 126, 5851 CrossRef CAS.
- M. Osada, Y. Ebina, K. Takada and T. Sasaki, Adv. Mater., 2006, 18, 295 CrossRef CAS.
- M. Osada, Y. Ebina, H. Funakubo, S. Yokoyama, T. Kiguchi, K. Takada and T. Sasaki, Adv. Mater., 2006, 18, 1023 CrossRef CAS.
- T. Shibata, K. Fukuda, Y. Ebina, T. Kogure and T. Sasaki, Adv. Mater., 2008, 20, 231 CrossRef CAS.
- S. Ida, C. Ogata, D. Shiga, K. Izawa, K. Ikeue and Y. Matsumoto, Angew. Chem., Int. Ed., 2008, 47, 2480 CrossRef CAS.
- N. Sakai, K. Fukuda, Y. Omomo, Y. Ebina, K. Takada and T. Sasaki, J. Phys. Chem. C, 2008, 112, 5197 CrossRef CAS.
- N. Miyamoto, K. Kuroda and M. Ogawa, J. Mater. Chem., 2004, 14, 165 RSC.
- Y. S. Han, I. Park and J. H. Choy, J. Mater. Chem., 2001, 11, 1277 RSC.
- Y. Ebina, T. Sasaki, M. Harada and M. Watanabe, Chem. Mater., 2002, 14, 4390 CrossRef CAS.
- Y. Ebina, N. Sakai and T. Sasaki, J. Phys. Chem. B, 2005, 109, 17212 CrossRef CAS.
- O. C. Compton, E. C. Carroll, J. Y. Kim, D. S. Larsen and F. E. Osterloh, J. Phys. Chem. C, 2007, 111, 14589 CrossRef CAS.
- E. C. Carroll, O. C. Compton, D. Madsen, F. E. Osterloh and D. S. Larsen, J. Phys. Chem. C, 2008, 112, 2394 CrossRef CAS.
- O. C. Compton, C. H. Mullet, S. Chiang and F. E. Osterloh, J. Phys. Chem. C, 2008, 112, 6202 CrossRef CAS.
- O. C. Compton and F. E. Osterloh, J. Phys. Chem. C, 2009, 113, 479 CrossRef CAS.
- M. Fang, C. H. Kim, G. B. Saupe, H.-N. Kim, C. C. Waraksa, T. Miwa, A. Fujishima and T. E. Mallouk, Chem. Mater., 1999, 11, 1526 CrossRef CAS.
- K. Maeda and T. E. Mallouk, J. Mater. Chem., 2009, 19, 4813 RSC.
- K. Maeda, M. Eguchi, S. A. Lee, W. J. Youngblood, H. Hata and T. E. Mallouk, J. Phys. Chem. C, 2009, 113, 7962 CrossRef CAS.
- K. Saruwatari, H. Sato, T. Idei, J. Kameda, A. Yamagishi, A. Takagaki and K. Domen, J. Phys. Chem. B, 2005, 109, 12410 CrossRef CAS.
- K. Okamoto, H. Sato, K. Saruwatari, K. Tamura, J. Kameda, T. Kogure, Y. Umemura and A. Yamagishi, J. Phys. Chem. C, 2007, 111, 12827 CrossRef CAS.
- S. Ida, C. Ogata, M. Eguchi, W. J. Youngblood, T. E. Mallouk and Y. Matsumoto, J. Am. Chem. Soc., 2008, 130, 7052 CrossRef CAS.
- R. E. Schaak and T. E. Mallouk, Chem. Mater., 2000, 12, 3427 CrossRef CAS.
- T. Nakato, N. Miyamoto and A. Harada, Chem. Commun., 2004, 78 RSC.
- S. W. Keller, H.-N. Kim and T. E. Mallouk, J. Am. Chem. Soc., 1994, 116, 8817 CrossRef CAS.
- K. Akatsuka, G. Takanashi, Y. Ebina, N. Sakai, M. Haga and T. Sasaki, J. Phys. Chem. Solids, 2008, 69, 1288 CrossRef CAS.
- K. Domen, Y. Ebina, S. Ikeda, A. Tanaka, J. N. Kondo and K. Maruya, Catal. Today, 1996, 28, 167 CrossRef CAS.
- R. Abe, K. Shinohara, A. Tanaka, M. Hara, J. N. Kondo and K. Domen, Chem. Mater., 1997, 9, 2179 CrossRef CAS.
- R. Abe, K. Shinohara, A. Tanaka, M. Hara, J. N. Kondo and K. Domen, J. Mater. Res., 1998, 13, 861 CrossRef CAS.
- R. Abe, M. Hara, J. N. Kondo, K. Domen, K. Shinohara and A. Tanaka, Chem. Mater., 1998, 10, 1647 CrossRef CAS.
- G. B. Saupe, C. C. Waraksa, H. N. Kim, Y. J. Han, D. M. Kaschak, D. M. Skinner and T. E. Mallouk, Chem. Mater., 2000, 12, 1556 CrossRef CAS.
- N. Miyamoto, H. Yamamoto, R. Kaito and K. Kuroda, Chem. Commun., 2002, 2378 RSC.
- G. H. Du, Q. Chen, Y. Yut, S. Zhang, W. Z. Zhou and K. M. Peng, J. Mater. Chem., 2004, 14, 1437 RSC.
- K. Maeda, M. Eguchi, W. J. Yongblood and T. E. Mallouk, Chem. Mater., 2008, 20, 6770 CrossRef CAS.
- R. Z. Ma, Y. Kobayashi, W. J. Youngblood and T. E. Mallouk, J. Mater. Chem., 2008, 18, 5982 RSC.
- N. Miyamoto, Y. Yamada, S. Koizumi and T. Nakato, Angew. Chem., Int. Ed., 2007, 46, 4123 CrossRef CAS.
- K. Fukuda, I. Nakai, Y. Ebina, R. Ma and T. Sasaki, Inorg. Chem., 2007, 46, 4787 CrossRef CAS.
- S. Ida, C. Ogata, U. Unal, K. Izawa, T. Inoue, O. Altuntasoglu and Y. Matsumoto, J. Am. Chem. Soc., 2007, 129, 8956 CrossRef CAS.
- R. E. Schaak and T. E. Mallouk, Chem. Commun., 2002, 706 RSC.
- K. Fukuda, K. Akatsuka, Y. Ebina, R. Ma, K. Takada, I. Nakai and T. Sasaki, ACS Nano, 2008, 2, 1689 CrossRef CAS.
- Y. Omomo, T. Sasaki, L. Z. Wang and M. Watanabe, J. Am. Chem. Soc., 2003, 125, 3568 CrossRef CAS.
- L. Z. Wang, Y. Omomo, N. Sakai, K. Fukuda, I. Nakai, Y. Ebina, K. Takada, M. Watanabe and T. Sasaki, Chem. Mater., 2003, 15, 2873 CrossRef CAS.
- X. Yang, Y. Makita, Z. Liu, K. Sakane and K. Ooi, Chem. Mater., 2004, 16, 5581 CrossRef CAS.
- N. Sakai, Y. Ebina, K. Takada and T. Sasaki, J. Phys. Chem. B, 2005, 109, 9651 CrossRef CAS.
- K. Kai, Y. Yoshida, H. Kageyama, G. Saito, T. Ishigaki, Y. Furukawa and J. Kawamata, J. Am. Chem. Soc., 2008, 130, 15938 CrossRef CAS.
- W. Sugimoto, H. Iwata, Y. Yasunaga, Y. Murakami and Y. Takasu, Angew. Chem., Int. Ed., 2003, 42, 4092 CrossRef CAS.
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199 CrossRef CAS.
- T. Okuhara, N. Mizuno and M. Misono, Appl. Catal., A, 2001, 222, 63 CrossRef CAS.
- A. Corma, V. Fornes, S. B. Pergher, Th. L. M. Maesen and J. G. Buglass, Nature, 1998, 396, 353 CrossRef CAS.
- A. Corma, V. Fornés, J. Martínez-Triguero and S. B. Pergher, J. Catal., 1999, 186, 57 CrossRef CAS.
- A. Corma, U. Diaz, V. Fornés, J. M. Guil, J. Martínez-Triguero and E. J. Creyghton, J. Catal., 2000, 191, 218 CrossRef CAS.
- I. Rodriguez, M. J. Climent, S. Iborra, V. Fornés and A. Corma, J. Catal., 2000, 192, 441 CrossRef CAS.
- A. Corma, V. Fornés, J. M. Guil, S. Pergher, Th. L. M. Maesen and J. G. Buglass, Microporous Mesoporous Mater., 2000, 38, 301 CrossRef CAS.
- A. Corma, A. Martínez and V. Martínez-Soria, J. Catal., 2001, 200, 259 CrossRef CAS.
- A. Corma, P. Botella and C. Mitchell, Chem. Commun., 2004, 2008 RSC.
- P. Botella, A. Corma, S. Iborra, R. Monton, I. Rodriguez and V. Costa, J. Catal., 2007, 250, 161 CrossRef CAS.
- A. Corma, U. Diaz, M. E. Domine and V. Fornés, Angew. Chem., Int. Ed., 2000, 39, 1499 CrossRef CAS.
- A. Corma, U. Diaz, M. E. Domine and V. Fornés, J. Am. Chem. Soc., 2000, 122, 2804 CrossRef CAS.
- A. Corma, V. Fornés and U. Díaz, Chem. Commun., 2001, 2642 RSC.
- A. Takagaki, M. Sugisawa, D. Lu, J. N. Kondo, M. Hara, K. Domen and S. Hayashi, J. Am. Chem. Soc., 2003, 125, 5479 CrossRef.
- C. Pazé, A. Zecchina, S. Spera, G. Spano and F. Rivetti, Phys. Chem. Chem. Phys., 2000, 2, 5756 RSC.
- A. Takagaki, T. Yoshida, D. Lu, J. N. Kondo, M. Hara, K. Domen and S. Hayashi, J. Phys. Chem. B, 2004, 108, 11549 CrossRef CAS.
- A. Takagaki, D. Lu, J. N. Kondo, M. Hara, S. Hayashi and K. Domen, Chem. Mater., 2005, 17, 2487 CrossRef CAS.
- Z. Chen, T. Iizuka and K. Tanabe, Chem. Lett., 1984, 1085 CAS.
- T. Ushikubo, Y. Koike, K. Wada, L. Xie, D. Wang and X. Guo, Catal. Today, 1996, 28, 59 CrossRef CAS.
- B. K. Sen, A. V. Saha and N. Chatterjee, Mater. Res. Bull., 1981, 16, 923 CrossRef CAS.
- T. Ikeya and M. Senna, J. Non-Cryst. Solids, 1988, 105, 243 CAS.
- J. M. Jehng and I. E. Wachs, Chem. Mater., 1991, 3, 100 CrossRef CAS.
- L. J. Burcham, J. Datka and I. E. Wachs, J. Phys. Chem. B, 1999, 103, 6015 CrossRef CAS.
- M. Dion, M. Ganne and M. Tournoux, Mater. Res. Bull., 1981, 16, 1429 CrossRef CAS.
- A. J. Jacobson, J. T. Lewandowski and J. W. Johnson, J. Less Common Met., 1986, 116, 137 CrossRef CAS.
- X. Wang, J. Adhikari and L. J. Smith, J. Phys. Chem. C, 2009, 113, 17548 CrossRef CAS.
- P. M. Gasperin, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 2024 CrossRef.
- K. Nassau, J. W. Shiever and J. L. Bernstein, J. Electrochem. Soc., 1969, 116, 348 CAS.
- R. Nedjar, M. M. Borel and B. Raveau, Mater. Res. Bull., 1985, 20, 1291 CrossRef CAS.
- C. Tagusagawa, A. Takagaki, S. Hayashi and K. Domen, J. Phys. Chem. C, 2009, 113, 7831 CrossRef CAS.
- E. F. Rakiewicz, A. W. Peters and R. F. Wormsbecher, J. Phys. Chem. B, 1998, 102, 2890 CrossRef CAS.
- C. Tagusagawa, A. Takagaki, S. Hayashi and K. Domen, J. Am. Chem. Soc., 2008, 130, 7230 CrossRef CAS.
- A. Takagaki, R. Sasaki, C. Tagusagawa and K. Domen, Top. Catal., 2009, 52, 592 CrossRef CAS.
- C. Tagusagawa, A. Takagaki, S. Hayashi and K. Domen, Catal. Today, 2009, 142, 267 CrossRef CAS.
- A. Takagaki, C. Tagusagawa and K. Domen, Chem. Commun., 2008, 5363 RSC.
- N. S. P. Bhuvanesh and J. Gopalakrishinan, Inorg. Chem., 1995, 34, 3760 CrossRef CAS.
- H. M. Kao, C. Y. Yu and M. C. Yeh, Microporous Mesoporous Mater., 2002, 53, 1 CAS.
- Q. Zhao, W. H. Chen, S. J. Huang, Y. C. Wu, H. K. Lee and S. B. Liu, J. Phys. Chem. B, 2002, 106, 4462 CrossRef CAS.
- A. S. Dias, S. Lima, D. Carriazo, V. Rives, M. Pillinger and A. A. Valente, J. Catal., 2006, 244, 230 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |