The teraton challenge. A review of fixation and transformation of carbon dioxide
Mette
Mikkelsen
,
Mikkel
Jørgensen
and
Frederik C.
Krebs
*
Risø National Laboratory for Sustainable Energy, Technical University of Denmark, Frederiksborgvej 399, DK-4000, Roskilde, Denmark
First published on 24th November 2009
Abstract
The increase in atmospheric carbon dioxide is linked to climate changes; hence there is an urgent need to reduce the accumulation of CO2 in the atmosphere. The utilization of CO2 as a raw material in the synthesis of chemicals and liquid energy carriers offers a way to mitigate the increasing CO2 buildup. This review covers six important CO2 transformations namely: chemical transformations, photochemical reductions, chemical and electrochemical reductions, biological conversions, reforming and inorganic transformations. Furthermore, the vast research area of carbon capture and storage is reviewed briefly. This review is intended as an introduction to CO2, its synthetic reactions and their possible role in future CO2 mitigation schemes that has to match the scale of man-made CO2 in the atmosphere, which rapidly approaches 1 teraton.
 Mette Mikkelsen | Mette Mikkelsen did her Master of Science in Chemistry from the Technical University of Denmark (DTU) with a specialty in organic chemistry (1997–2003). She then worked in industry as an organic chemist at LiPlasome Pharma A/S (2003–2004) and as a synthetic chemist at H. Lundbeck A/S (2004–2006) before pursuing PhD studies at Risø National Laboratory, Technical University of Denmark. The topic of her PhD work has been fixation of carbon dioxide from the atmosphere with the purpose of transforming it into a storable and combustible fuel by use of solar energy. Her main scientific interests are synthetic organic chemistry, structural characterization of organic compounds, solar energy, crystallography. |
 Mikkel Jørgensen | Mikkel Jørgensen did his Master of Science in chemistry from the University of Copenhagen and a PhD in organic chemistry from the University of Copenhagen (1990). He worked as an industrial chemist at NycoMed (1987–1990) during his PhD studies and later as an industrial chemist at PNA Diagnostics (1990–1993). He then became employed as a senior scientist at Risø National Laboratory, DTU, Denmark (1994-present). His scientific interests include synthetic chemistry, nuclear magnetic resonance (NMR), chemistry of materials, carrier mobilities in organic materials, energy levels and energy level alignment in organic materials by UPS studies, solar cells, polymers, fluorine chemistry and supramolecular chemistry. |
 Frederik C. Krebs | Frederik Christian Krebs did a BSc in chemistry (1993) and a BSc In biochemistry/immunology (1994) from University of Aberdeen, Scotland, DEA in solid state chemistry from the Université de Nantes, France (1995), Master of Science in chemistry from the University of Copenhagen (1996), PhD in chemistry from the Technical University of Denmark (DTU) (2000). He did a postdoc (2001–2002) and then became employed as senior scientist at Risø National Laboratory, DTU (2002–present). His scientific interests include all aspects of chemistry, physics and engineering. He is currently associate editor for the international journal Solar Energy Materials and Solar Cells and has published more than 200 peer reviewed papers, conference proceedings, editorials, book reviews, patents and reports. |
Broader contextThe level of carbon dioxide in the atmosphere has risen significantly since pre-industrial times and today there is an excess of 1 teraton of carbon dioxide in the atmosphere. The contribution of this extra teraton toward global warming have resulted in a considerable effort towards mitigating carbon dioxide. Various approaches ranging from reduction of emission, a change to renewable energy sources, and methods to safely capture and store carbon dioxide have been investigated. The good news is that there is plenty of space to store captured carbon dioxide and the capacity underground and in the deep sea is vast compared to the problem at hand. The more challenging part of the problem is that the 24 gigaton annual increase in atmospheric carbon dioxide is man-made and unlikely to reduce significantly in the next decades. This naturally raises the problem of how to actively remove carbon dioxide from the atmosphere. Currently the 120 megaton scale at which we are able to industrially convert carbon dioxide is significantly lower than the annual emission. We review the problem of getting a carbon-dioxide-emission-free source of carbon dioxide and try to identify the currently available chemistry that could possibly be upscaled and thus enable handling carbon dioxide on an annual multi gigaton scale such that annual emission can be matched by annual capture and conversion. |
1. Introduction
Climate change is considered to be one of the greatest environmental threats of our times.1 The atmospheric concentration of green house gases can roughly be divided into carbon dioxide (CO2), nitrous oxide (N2O), methane (CH4), fluorocarbons (CFs) and chlorofluorocarbons (CFCs). The atmospheric concentrations of these gases have increased steadily over the past century.2 Current research shows that there is an excess of approximately 3.9% CO2 with respect to the natural “carbon cycle”. The natural carbon cycle is the carbon-flow between the atmosphere and oceans and the fixation of CO2 by plants and microorganisms, which is balanced by emission of CO2 from plants, animals and volcanoes. Human activities therefore produce an annual excess of 3.9% CO2 to the carbon cycle.3 The largest source of CO2 emitters comes from power generation, public electricity and heat production from fossil fuel combustion.This increase in CO2 emission, which is not balanced by CO2 fixation mainly due to deforestation, has resulted in an increase in atmospheric CO2 during the last 200 years from approximately 270 ppm to 385 ppm. This increase is thought to cause atmospheric warming, due to the prevention of infrared re-emission. The atmospheric warming is associated with a global climate change and a planetary temperature increase.3 Furthermore, as atmospheric CO2 increases, the global mean temperature increases, and this will put more water vapor into the atmosphere. Water vapor is also a very effective greenhouse gas and this will increase the earth's temperature even further.4 The International Panel on Climate Change (IPCC) predicts that, by the year 2100, the atmosphere may contain up to 570 ppm CO2, causing a rise in the mean global temperature of around 1.9 °C. IPCC predicts that this will give an increase in the mean sea level of up to 1 m by 2100, increased desert formation and the extinction of species.5
There is an ongoing research in finding ways to reduce CO2 emission into the atmosphere and there are, in principle, three possible strategies for reducing the CO2 buildup in the atmosphere: reduction of the amount of CO2 produced; usage of CO2; and storage of CO2.5,6
The first strategy can be addressed by increasing the energy efficiency or a change in the primary energy source to decrease the amount of CO2 emitted. The replacement of a C-rich energy carrier (coal) by other less C-rich fossil fuels (oil or natural gas) is an option that with relative ease leads to a reduction in CO2 emission. However the largest reduction in CO2 would be gained by switching to non-fossil fuels such as hydrogen and renewable energy. The second strategy involves the use of CO2 as a chemical feedstock in different applications. The third strategy involves the development of new technologies for capture and sequestration of CO2.5,6 There is an excess of 115 ppm by volume of CO2 in the atmosphere with respect to the pre-industrial value of 270 ppm that amounts to approximately 900 Gt CO2. In order to bring the CO2 level back to where it was, we need to develop processes, techniques and applications capable of handling CO2 on the scale of 1 teraton. Handling CO2 at this scale implies significant challenges in terms of how we extract CO2 from the atmosphere, how we transform it and how we either use it or store it safely.
1.1 Scope of this review
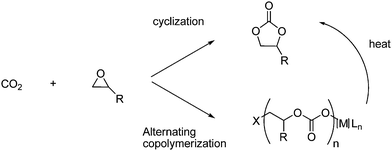
A lot of research has gone into the field of CO2 transformations, where CO2 is used as raw material in reactions. In Fig. 1 the transformations are divided into six categories and the typical product for these transformations are listed.7 This review will cover all of the categories listed, but due to the vastness of this area some of the transformations are only described briefly. However in the reference list recent reviews and/or original literature can be found for all the categories. | ||
| Fig. 1 CO2 transformations covered in this review.7 | ||
Another vast research area, namely carbon capture and storage (CCS), will also be covered briefly in this review. The synthetic reactions and their possible role in future CO2 mitigation schemes will also be evaluated.
1.2 The carbon dioxide molecule
The molecular geometry of CO2 is linear in its ground state; therefore it is apolar even though it has two polar C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. The molecule is a bi-functional catalyst due to its two different reaction sites. The carbon atom is an electrophile, while the oxygen atoms are nucleophiles.
O bonds. The molecule is a bi-functional catalyst due to its two different reaction sites. The carbon atom is an electrophile, while the oxygen atoms are nucleophiles.
The physical state of CO2 varies with temperature and pressure. CO2 is a solid at low temperatures, and will, on warming below 5.1 bar, sublime directly into the vapor state. Above the critical point (31.1 °C, 73.9 bar), which means at higher temperature and/or pressure, CO2 is said to be in a supercritical state, where it behaves like a gas while its density is approaching or even exceeding the density of liquid water.2
Infrared (IR) and nuclear magnetic resonance (NMR) techniques are used as diagnostic tools of the state of the CO2 molecule or for its quantitative determination.
Even though CO2 is an abundant and renewable carbon source only a few industrial processes utilize CO2 as a raw material. The reason for this is that the carbon atom in CO2 is in its most oxidized form and is therefore relatively unreactive. A large input of energy is required to transform CO2 into other chemicals. There are four way of altering this:
(1) By using high energy starting materials such as hydrogen and organometallics.
(2) Choosing low energy synthetic targets.
(3) Removing a compound on the product side, and thereby forcing the equilibrium to the right.
(4) Supplying physical energy, i.e., light or electricity.
Choosing the right conditions for the CO2 transformation is crucial in order to achieve a negative Gibbs energy for the reaction.5
2. Chemical transformations
CO2 has a strong affinity toward nucleophiles and electron-donation reagents; therefore CO2 can be classified as “anhydrous carbonic acid”, which rapidly reacts with basic compounds.However CO2 is not used extensively as a source of carbon in current laboratory and industrial practices. This can, in part, be ascribed to thermodynamic aspects. Thus the carbon atom in CO2 is as stated above electrophilic and a chemical reaction necessitates a reductive supply of energy in the form of electrons.8
2.1 Industrial use of carbon dioxide
The industry uses approximately 120 Mt CO2 per year, excluding use for enhanced oil recovery. In Fig. 2 a bar chart shows the amount CO2 fixed annually for different chemical applications.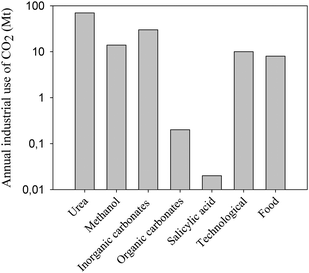 | ||
| Fig. 2 The annual industrial use of CO2 in megatons. Note the logarithmic scale on the y-axis. Urea accounts for more than 50% of the annual usage. | ||
The industrial use amounts to only 0.5% of the total anthropogenic CO2 emissions, which is about 24 Gt CO2 annually.9 The usage can be divided into two groups: those using its physical aspects and those using its chemical aspects. The physical properties of CO2 are used in the beverage industry, in enhanced oil recovery and in its supercritical state as a technological fluid for applications in reactions as solvents and in nano-particle or composite production. As an inert and safe gas it is also used as a protective gas (in chemical or steel industries, in food preservation, in welding etc.) and as a fire extinguisher. CO2 is also used in its solid state for refrigeration especially in refrigerated railcars and trailers to substitute the use of CFCs that are harmful to the atmosphere. CO2 can also be used chemically as a reactant and can be converted into chemicals such as urea, salicylic acid, inorganic carbonates, pigments, cyclic organic carbonates or used as an additive in the synthesis of methanol.
2.2 Industrial syntheses with carbon dioxide
The industrial use of CO2 as a source of chemical carbon is very limited. CO2 is used in carboxylation reactions and in certain transformation reactions, which are represented in Scheme 1.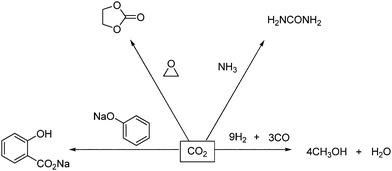 | ||
| Scheme 1 Industrial syntheses with CO2.8 | ||
One of the most important acids prepared by this synthesis is salicylic acid (intermediate in the synthesis of acetylsalicyclic acid (aspirin)) as shown in Scheme 2. The reaction has been used in the industrial synthesis of salicylic acid since 1874. The yield is increased from ca. 50% to 90%, when CO2 is introduced under pressure (5–7 bars).6,8
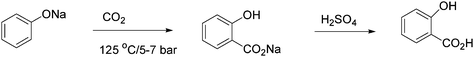 | ||
| Scheme 2 Industrial synthesis of salicyclic acid. | ||
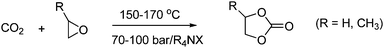 | ||
| Scheme 3 Synthesis of carbonates from CO2 and epoxides. | ||
Cyclic organic carbonates have a high boiling point and have therefore found many applications as solvents. Furthermore they can react with ammonia or amines to form carbamates. The carbamates can then be converted into polyurethane, a versatile material with a high commercial value.6,10,11
The alkene carbonates are in general excellent solvents for the production of various polymers: polyacrylonitrile, nylon, terylene and polyvinylchloride.8
The production of polycarbonates, which is also an industrialized process, comprises the same reactants as the synthesis of five-membered cyclic carbonates. The polymerization is often catalyzed by zinc complexes.7
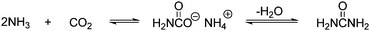
Different processes have been designed for the production of urea. The only difference between them is the techniques used for the recovery and recycling of the unreacted ammonium carbamate.8 Urea is used as a chemical fertilizer, urea resins, urea–melamine resins and as an animal feed additive.
 | ||
| Scheme 5 Methanol production from synthesis gas along with a small amount of CO2. | ||
The annual production of methanol is around 40 Mt and it is used as an intermediate for the production of a variety of chemicals including formaldehyde, methyl tert-butyl ether and acetic acid.13
The current and estimated industrial use of CO2 is summarized in Table 1.3,14
| Chemical product or application | Industrial volume/Mt y−1 | Industrial CO2 use/Mt y−1 | Future expectations in the use of CO2 | Endothermic or exothermic reaction |
|---|---|---|---|---|
| Urea | 100 | 70 | 102 Mt | Exothermic |
| Methanol (additive to CO) | 40 | 14 | Gt | Exothermic |
| Inorganic carbonates | 80 | 30 | — | Exothermic |
| Organic carbonates | 2.6 | 0.2 | 102 Mt | Exothermic |
| Salicylic acid | 0.06 | 0.02 | 102 kt | Exothermic |
| Technological | 10 | 10 | ||
| Food | 8 | 8 |
2.3 Chemical reactions with carbon dioxide
 | ||
| Scheme 6 Formation of carbonates from alcohols by a dehydrative condensation with CO2. | ||
These reactions do not proceed in high yields, due to hydrolysis of the esters and decomposition of the catalyst from the byproduct water.
The organic carbonates can be divided into: (1) acyclic carbonates (dimethyl carbonate (DMC) and diethyl carbonate (DEC)), (2) polycarbonates and (3) cyclic carbonates (ethylene carbonate and propylene carbonate). The formation of carbonates require energy either brought in externally or via the reactants with high free energy content.10 Important for these reactions are the development of new heterogeneous catalysts to replace the homogeneous catalyst used in this reaction for reasons of product separation, catalyst recovery and cost.10,15
2.3.3.1 Acyclic carbonates. DMC is used for the production of polycarbonate, polyurethane and other chemicals. Some of the best results obtained for the formation of DMC from methanol and CO2, which suffers only from thermodynamic limitations, is a 11% methanol conversion in supercritical CO2 (9.3 MPa).10,15 However the reaction can be improved significantly by employing a dehydrating agent, e.g., molecular sieves as exemplified in Scheme 7. The reaction gives under these optimized conditions 50% methanol conversion.16
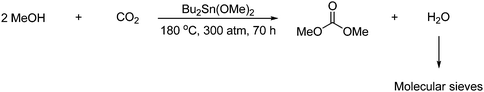 | ||
| Scheme 7 Employing molecular sieves to the reaction between alcohols and CO2 improves the yield significantly. | ||
Other dehydrating agents which could be employed are non-recyclable agents: dicyclohexyl carbodiimide (DCC), orthoesters,17 (e.g., orthoacetate, Si(OMe)4) and Mitsunobu's reagent, and recyclable agents: acetals and molecular sieves.18
An example with the synthesis of DMC using an orthoester as starting material and dehydrating agent is illustrated in Scheme 8.17
 | ||
| Scheme 8 Synthesis of DMC from trimethyl orthoacetate and CO2. | ||
Trimethyl orthoacetate which acts as both the starting material and dehydrating agent captures water to produce two molecules of methanol and one molecule of methyl acetate. Surprisingly the reaction proceeds without the addition of methanol to give DMC in 70% yield (based on the orthoester).17 A downfall to this synthesis method is that the orthoesters are relative expensive as starting materials and are difficult to regenerate from esters and alcohols.17
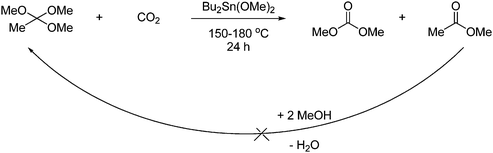
DMC can also be obtained in high yield by reaction with 2,2-dimethoxypropane and supercritical CO2 under tin catalysis as shown in Scheme 9. The reaction gives acetone as a byproduct, which can be utilized by reaction with methanol, which regenerates the starting material.19
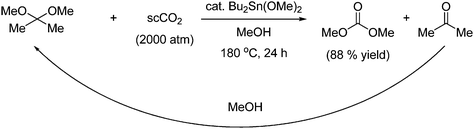 | ||
| Scheme 9 Synthesis of DMC from an acetal by reaction with supercritical CO2 under tin catalysis. | ||
Increased pressure and concentration of methanol will increase the yield. The reaction is therefore carried out in a solvent mixture of methanol and supercritical CO2. Another benefit of using supercritical CO2 is the efficient product/catalyst separation, which can be done by phase separation without having to completely depressurize the reaction mixture.18 Methanol is in fact a prerequisite for this reaction since the reaction in Scheme 9 proceeds by an alcohol reaction and a dehydration by the acetal as shown in Scheme 10.
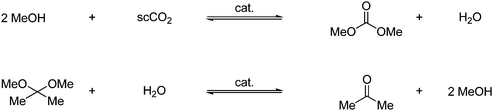 | ||
| Scheme 10 The reaction in Scheme 9 can be divided into these two reactions, which overall gives DMC. | ||
The DMC synthesis using acetals as dehydrating agents in the presence of a weakly basic tin catalyst can be significantly accelerated by employing small amounts of acid catalyst, (e.g., Ph2NH2OTf, Sc(OTf)3, etc.).18,20
This reaction type, however, has some downfalls compared to the direct synthesis of DMC from alcohols, since molecular sieves are recyclable and only some acetals are recyclable. Furthermore, there are ketone byproducts in the synthesis of DMC from acetals, while water is the byproduct in the DMC synthesis from alcohols.
DMC can also be produced by a transesterification of cyclic carbonates as shown in Scheme 11. The synthesis of cyclic carbonates from CO2 and oxiranes is described later in this section.21
 | ||
| Scheme 11 Synthesis of DMC and ethylene glycol from ethylene carbonate and methanol. | ||
A near 100% conversion of methanol with propylene carbonate to DMC (CaO/C as catalyst) has been reported.10,11b,15
The reaction of DMC from cyclic carbonates is more favorable than the direct synthesis of DMC from CO2 and methanol, since the equilibrium naturally is more to the right. However the starting material for ethylene carbonate, ethylene oxide, is highly flammable and also highly toxic, which makes the synthesis of DMC from methanol and CO2 more favorable.18,22
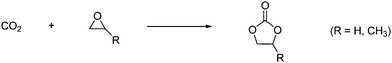
2.3.3.2 Cyclic carbonates. The reaction between oxiranes and CO2 produces cyclic or polymeric carbonates as shown in Scheme 12.
 | ||
| Scheme 12 Reaction with oxiranes and CO2 can lead to cyclic or polymeric carbonates. | ||
The reaction takes place easily due to the high steric energy of the oxiranes. It should be noted that cyclic carbonates are thermodynamically more stable than the linear carbonates.18
The current industrial synthesis of cyclic carbonates is typically catalyzed by halide salts such as Et4NBr and KI. The halide salts are also suitable in view of catalyst recycling, since they are soluble in cyclic carbonates and do not precipitate upon concentration.
Ethylene carbonate (boiling point: 521 K) and propylene carbonate (boiling point: 513 K), are as previously described produced industrially from a reaction between CO2 and ethylene oxide and propylene oxide respectively as shown in Scheme 13.
 | ||
| Scheme 13 Synthesis of ethylene and propylene carbonate from their respective oxiranes and CO2. | ||
These two cyclic carbonates are used as high-boiling solvents for natural and synthetic polymers such as lignin, cellulose ester, nylon, and PVC. Some of the best results for the synthesis of ethylene- and propylene-carbonate are obtained with a heterogeneous KI-based catalyst giving 99% selectivity and a 100% epoxide conversion.6,10,23 Excellent results for the synthesis of propylene carbonate have also been obtained by applying polyfluoroalkylphosphonium iodides ((C6F13C2H4)3MePI) as catalyst to propylene oxide in supercritical CO2. The reaction gives a high yield (93%) and high selectivity (99%). The catalyst is furthermore soluble in supercritical CO2 while the resulting product is not, which gives a facile catalyst/product separation.18,24
Cyclic carbonates can also be obtained by other pathways than the reaction between oxiranes and CO2, e.g., olefins react with CO2 in the presence of an oxidizing agent to give cyclic carbamates as shown in Scheme 14.25
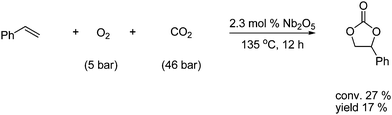 | ||
| Scheme 14 Synthesis of cyclic carbonates from a Nb-catalyzed oxidative carboxylation with olefins and CO2. | ||
Another example is the dehydrative condensation of 1,2-diols and CO2 under CeO2-ZrO2 or Bu2SnO catalysis to give cyclic carbonates, however in a very poor yield as shown in Scheme 15.26
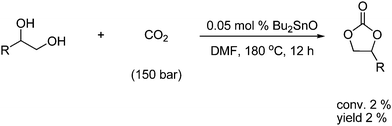 | ||
| Scheme 15 Synthesis of cyclic carbonates from a reaction with diols and CO2 under tin catalysis. | ||
Yet another example is an iron- or copper-catalyzed reaction of a cyclic ketal with supercritical CO2 to give ethylene carbonate in an excellent yield as shown in Scheme 16.27
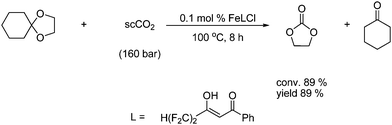 | ||
| Scheme 16 Synthesis of cyclic carbonates from acetals reaction with supercritical CO2 under iron catalysis. | ||
The reaction of propargyl alcohols with methyl iodide and CO2 proceeds in the presence of a palladium catalyst to give a substituted cyclic carbonate in a good yield as shown in Scheme 17.28
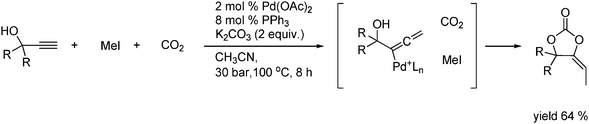 | ||
| Scheme 17 Synthesis of alkene carbonates from the reaction with propargyl alcohols. | ||
A recent review on the formation of carbonates from CO2 by Sakakura et al. describes recent progress in new reaction types, reaction conditions and catalysts.18
2.3.3.3 Polycarbonates. Polymeric carbonates produced from alternating copolymerization comprise the same reactants as the synthesis of five-membered cyclic carbonates; polymers are kinetic products, while cyclic carbonates are thermodynamic ones.29
Polycarbonates process excellent properties, which include strength, lightness, durability, high transparency, heat resistance and good electrical insulation. Hence these materials have found a wide variety of applications from soft drink bottles to building materials, automobile parts and electrical components.30 Inoue and co-workers first discovered that a mixture of ZnEt2 and H2O catalyzed the alternating copolymerization of propylene oxide and CO2 to give poly(propylene carbonate) as shown in Scheme 18.31,32
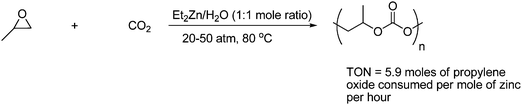 | ||
| Scheme 18 Alternative copolymerization of CO2 with propylene oxide. | ||
Zinc complexes were the first generation of catalysts for this reaction and are typified by a mixture of diethylzinc and an equimolar amount of a compound having two active hydrogen atoms, such as water, a primary amine, an aromatic dicarboxylic acid and an aromatic diol.31a In general it is found that polycarbonates are formed when di- or tri-protic sources and ZnEt2 catalyze the reaction, while monoprotic sources such as alcohols and secondary amines and ZnEt2 catalyze the formation of cyclic carbonates.29,33 Other zinc complexes have been developed and the zinc iminate complexes are the most intensively studied.34 The highly active zinc iminate catalysts give, under optimized conditions, a high reaction rate, a high molecular weight, and a narrow polydipersity. Other metal catalyst complexes have been developed for the catalysis of the aliphatic polycarbonate synthesis, e.g., cobalt,35 chromium,36 lanthanide37 and manganese complexes.38
An aromatic polycarbonate, which is based on bisphenol A is widely employed as an engineering plastic in various applications. About 2.7 million tons of this polycarbonate is produced annually and has until recently been produced from phenol, phosgene and bisphenol A. However complications with using phosgene led to the development of an alternative route where diphenyl carbonate is produced from phenol and DMC. Currently this alternative route amounts to 15% of global production capacity. The alternative route, which is carried out in four production steps, was industrialized by Asahi Kasei Chemicals.39 The first step is the formation of ethylene carbonate form ethylene oxide and CO2 as shown in Scheme 13. The second step is the formation of DMC and ethylene glycol from the transesterification of ethylene carbonate by methanol as shown in Scheme 11. The third and fourth step is shown in Scheme 19. In the third step diphenyl carbonate is formed by reaction between DMC and phenol under Pb(OPh)2 catalysis, which gives methylphenyl carbonate, which is in equilibrium with diphenyl carbonate and DMC. The fourth step is the polymerization step where diphenyl carbonate reacts with bisphenol A to produce a clear amorphous prepolymer. The polymerization consists of multiple steps where the final polymer has an average molecular weight of 11.700.21,40
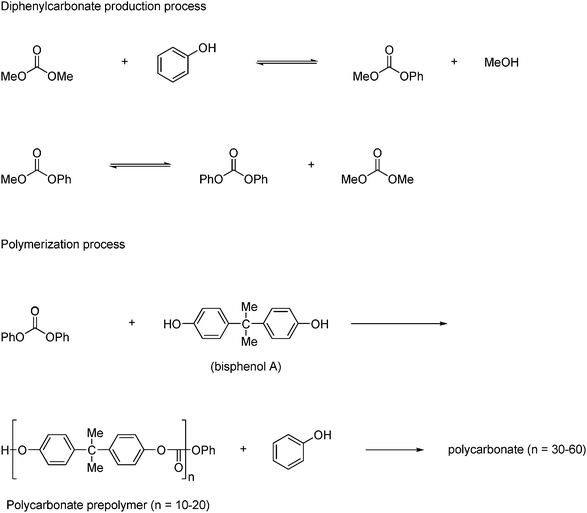 | ||
| Scheme 19 Asahi Chemical Industry's production of bisphenol A polycarbonate | ||
Current research focus on the development of a route to produce diphenyl carbonate from phenol and CO2, circumventing the use of DMC.10
Oxetanes, four-membered cyclic ethers, have a considerably lower reactivity than oxiranes. Nonetheless they react in the presence of organotin iodide with CO2 forming aliphatic poly(propyl carbonate) in excellent yield as shown in Scheme 20. By employing a different catalyst, Bu3SnI–hexamethylenephosphoric triamide (HMPA), the oxetanes undergo a cycloaddition with CO2 to give a six-membered ring carbonate in quantitative yield.41
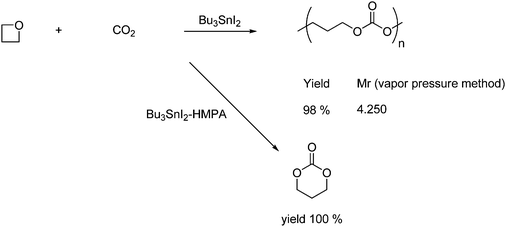 | ||
| Scheme 20 Polycarbonate or trimethylene carbonate synthesis from oxetane and CO2. | ||
Recent reviews on alternating copolymerization and its catalysts to produce polymeric carbonates have been comprehensively covered by Coates and Moore29 and more recent Darensbourg.30
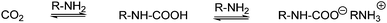 | ||
| Scheme 21 Formation of alkylammonium alkylcarbamate from two molecules of amine and CO2. | ||
Carbamates can be formed by reaction with an in situ generated carbamate ion and with electrophiles such as organic halides. These carbamates are synthesized in high yields in the presence of K2CO3 and a tetraalkylammonium salt as shown in Scheme 22.43
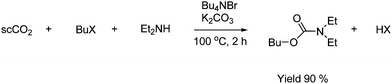 | ||
| Scheme 22 Formation of carbamates from the in situ generated carbamate ion and an organic halide. | ||
The yields for this reaction are improved by using different basic reagents, which provide stabilization to the intermediate alkylammonium alkylcarbamate ionic species.44 This synthesis has also been achieved by using a phase-transfer catalyst (18-crown-6 ether) instead of a base.45 Good to excellent yields have also been reported for the use of metallic carbonates (caesium carbonate) as basic reagents together with a phase transfer catalyst.46
Carbamates can also be formed from primary amines and alcohols in the presence of an organotin catalyst and an acetal as a dehydrating agent under high CO2 pressure, thereby replacing the organic halide with an alcohol as shown in Scheme 23.47
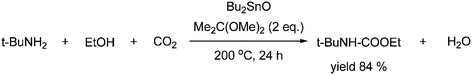
This means that the byproduct is water instead of H–X. The yield for this reaction is limited due to thermodynamic limitations and catalyst deactivation caused by water. The key to achieve high yields is therefore to add acetals, which act as dehydrating agents.7, 19a,42,47 The CO2 pressure is kept high for this reaction to avoid side reactions: (a) imine formation by reaction of the amine and the formed carbonyl compound and (b) alkylation of the amine by the alcohol.18
Substituted urea can be formed by using a dehydrating agent. The synthesis of N,N′-dialkylurea has been achieved by using hydrophilic ionic liquids as reaction media and dehydrating agent as shown in Scheme 24.48
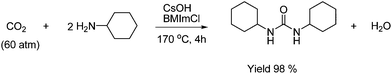 | ||
| Scheme 24 The synthesis of substituted ureas from CO2, primary amines, base and an ionic liquid BMImCl (1-n-butyl-3-methyl imidazolium chloride). | ||
O-Allyl carbamates can also be obtained through nucleophilic addition of carbamate ion to alkenes in 66–100% yield. The preformed carbamate ion is generated from primary or secondary amines, CO2 and base as shown in Scheme 25. The carbamate ion is added to a solution of allylic chlorides under palladium/phosphine catalysis.5,49
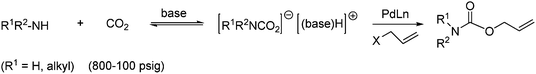 | ||
| Scheme 25 Synthesis of O-allyl carbamates from a carbamate ion and allylic chlorides. | ||
Isocyanates can be obtained from thermolysis of carbamates as shown in Scheme 26. This is an important reaction since it is a non-phosgene route to isocyanates.7,50
 | ||
| Scheme 26 Thermolysis of carbamates to give isocyanates. | ||
A mild and efficient procedure has been developed for the synthesis of isocyanates from carbamates employing chlorocatecholborane as illustrated in Scheme 27. Chlorocatecholborane intercepts the formed alcohol and the recombination of the formed isocyanate and alcohol is avoided.50
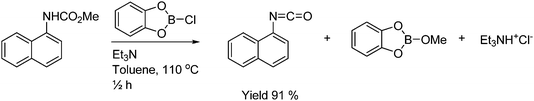 | ||
| Scheme 27 Formation of isocyanates from carbamates, chlorocatecholborane and base. | ||
Yet another way of synthesizing isocyanates without employing phosgene is from carbamic acids, which as described above are in equilibrium with amine and CO2. By dehydrating carbamic acids with, e.g., POCl3 or P4O10 in the presence of tertiary amines isocyanates can be synthesized as shown in Scheme 28.51
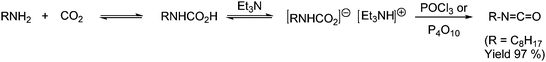 | ||
| Scheme 28 Synthesis of isocyanates by dehydrating a carbamic acid salt. | ||
Epoxides react with CO2 and the same applies for nitrogen containing three-membered rings (aziridine) which reacts with CO2 to form a five membered ring (oxazolidinone).52 Typical reaction conditions are listed in Scheme 29. Various compounds are used to promote the reaction. Typical promoters are quaternary ammonium salts, chromium salen complexes or as exemplified in Scheme 29 the alkali metal salts.
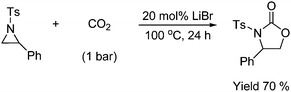 | ||
| Scheme 29 Typical reaction procedure for formation of oxazolidinones from aziridines. | ||
The regioselectivity in this ring-opening reaction leads to only one final product.52c
Another way to produce oxazolidinones in good yields is by an electrochemical procedure. The reaction is catalyzed by a Ni(II) complex (10 mol %) and performed in a single compartment cell fitted with a consumable magnesium anode and an inert cathode as shown in Scheme 30. The two regioisomers are obtained in ratios from 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 up to 86:14 mixtures depending on the substitution and reaction conditions. The major isomer corresponds to the incorporation of CO2 at the less hindered side of the mono-substituted aziridine.53
:50 up to 86:14 mixtures depending on the substitution and reaction conditions. The major isomer corresponds to the incorporation of CO2 at the less hindered side of the mono-substituted aziridine.53
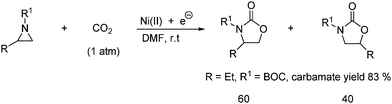 | ||
| Scheme 30 Subsituted oxazolidinones obtained from a mono-substituted aziridine and CO2 by an electrochemical procedure. | ||
2-Oxazolidinones can also be formed in good to excellent yields from substituted 1,2-aminoalcohols and CO2 under dibutyl tin oxide catalysis with 1-methyl-2-pyrrolidinone (NMP) as solvent as shown in Scheme 31. 2-Oxazolidiones were obtained in 53–94% yields depending on the substitution and degree of substitution.54
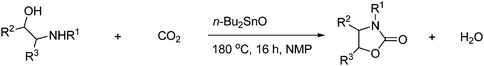 | ||
| Scheme 31 Oxazolidinone can be obtained from substituted 1,2 aminoalcohols and CO2. | ||
It is found that the amino alcohols react with CO2 to give a carbamic acid intermediate with the unexpected stereoselectivity of the Mitsunobu transformation.51b,55 The stereochemical course of the Mitsunobu reaction depends on whether N in the carbamic acid intermediate is substituted with two hydrogen atoms or a carbon atom. The former gives retention of configuration, while the latter gives inversion of configuration as illustrated in Scheme 32.56
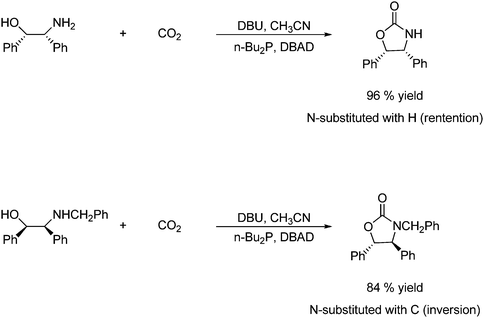 | ||
| Scheme 32 The stereochemical outcome of a reaction between a primary amino alcohol and CO2 and the outcome between a secondary amino alcohol and CO2 is illustrated. | ||
Unsaturated compounds such as acetylenes and olefins are able to react directly with CO2 and amines to afford carbamates using ruthenium catalysis. An example of this is shown in Scheme 33.
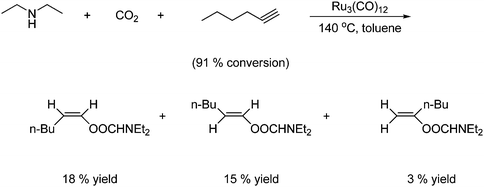
A secondary amine, CO2 and hex-1-yne react under ruthenium catalysis to give three vinylcarbamates. However, in a low overall yield.57
Cyclic carbamates can also be obtained in good yield by the reaction of CO2 with N-substituted terminal propargylamines in the presence of a ruthenium and tertiary phosphine catalyst as shown in Scheme 34.58
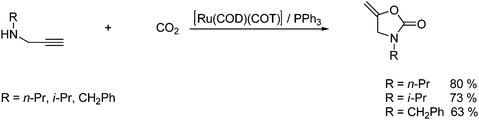 | ||
| Scheme 34 Synthesis of a cyclic carbamate from CO2 reaction with a N-substituted terminal propargylamine. | ||
The reaction between aziridine and CO2 will lead to ring-opening polymerization, when the reaction is performed under supercritical conditions as shown in Scheme 35.59
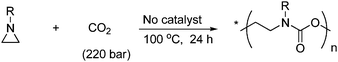 | ||
| Scheme 35 Polyurethane is formed when aziridine reacts with supercritical CO2. | ||
Amines (primary and secondary) and CO2 reacts with epoxides to afford hydroxycarbamates and aminoalcohols, and/or an oligomer of the epoxide, depending upon the nature of the amine and epoxide, and reaction conditions as shown in Scheme 36.60
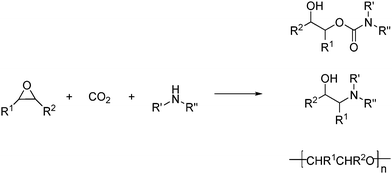 | ||
| Scheme 36 CO2 reacts with amines (primary and secondary) and epoxides to afford hydroxycarbamates and amino alcohols and/or oligomers of the epoxides. | ||
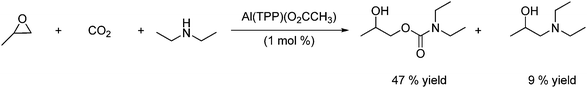
It can be seen from Scheme 36 that the reaction leads to isomer mixtures of the desired carbamate and an amine alcohol. This side reaction can be minimized by adding the (5,10,15,20-tetraphenylporphinato)aluminium(III) acetate, Al(TPP)(O2CCH3) to the mixture. The aluminium porphyrin complex is for example found to catalyse the formation of 2-hydroxypropyl diethylcarbamate from CO2, diethylamine and 1,2-epoxypropane as shown in Scheme 37. The desired dialkylcarbamic ester is obtained at 60 °C under a pressure of 50 atm of CO2 in 47% yield.61
 | ||
| Scheme 37 Formation of dialkylcarbamic ester under aluminium porphyrin complex catalysis. | ||
The catalytic formation of the dialkylcarbamic ester is believed to proceed by the insertion of the epoxide between the aluminium–oxygen bond of the (porphinato)aluminium carbamate (A) to form an aluminium alkoxide, followed by cleavage by diethylcarbamic acid (from CO2 and diethylamine) to give the desired product and regenerate the active species (aluminium carbamato group) as seen in Scheme 38.
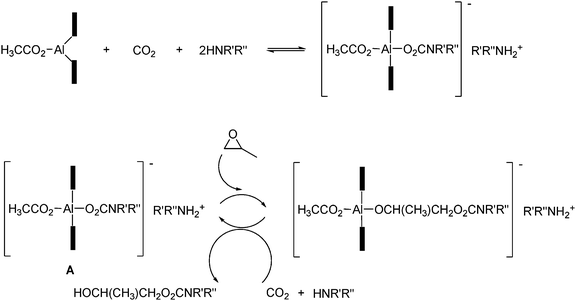 | ||
| Scheme 38 Proposed mechanism for the aluminium porphyrin complex catalyzed formation of the dialkyl carbamic ester. The black rectangle symbolizes the porphyrin.61 | ||
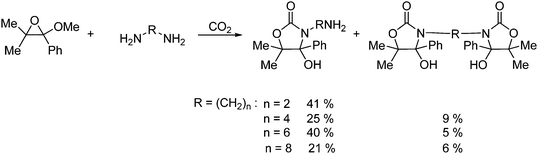
An example of the formation of a cyclic carbamate is shown in Scheme 39. A tetra substituted oxirane reacts with CO2 and an α,ω-diamine to afford cyclic carbamate bis(2-oxazolidione) derivatives. The yields for this reaction average around low to fair.62
Cyclic organic carbonates (formed from epoxide and CO2) reaction with ammonia or primary amines gives carbamates by a non-phosgene route at room temperature as shown in Scheme 40.63
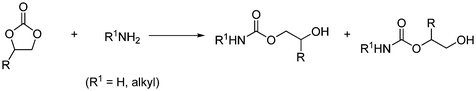 | ||
| Scheme 40 Cyclic carbonates react with ammonia or primary amines to form carbamates. | ||
Carbamate synthesis is a vast area of research and continued progress is made in the development of new synthetic routes to carbamates. Recent reviews on carbamate synthesis from CO2 are made by Chaturvedi et al.42 and Sakakura et al.7
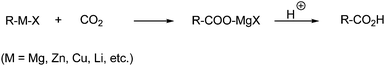 | ||
| Scheme 41 Carboxylic acids can be synthesized from reaction with, e.g., Grignard reagents and CO2. | ||
These reactions provide a convenient route to aliphatic, aromatic, olefinic and acetylenic acids.
Acrylic acids can be formed from acetylene and CO2 using catalysis. The reaction often requires a strong base, (e.g., DBU) which presumably promotes the reaction by trapping CO2 in the form of carbamate or bicarbonate.65 During the reaction a five-membered metallolactone intermediate is formed when the low-valent metal complex (catalyst in stoichiometric amounts), CO2 and the unsaturated compound reacts as shown in Scheme 42.66
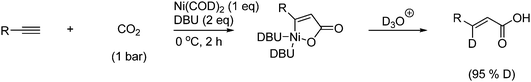 | ||
| Scheme 42 An acrylic acid is formed from acetylene with a five-membered metallolactone as intermediate. | ||
Acrylic acids can also be formed from 1,3- butadiene67 and allenes via metallacycles.68
Another recent approach to the synthesis of acrylic acids uses a palladium hydride complex (in catalytic amounts) as the active catalyst in the presence of a reducing agent as shown in Scheme 43.69
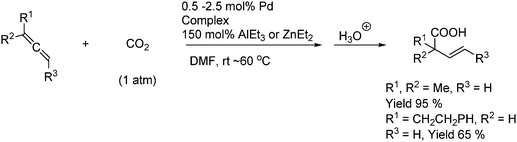 | ||
| Scheme 43 Synthesis of substituted acrylic acids from allenes under palladium hydride complex catalysis. | ||
Carboxylation with CO2 can be carried out under either basic or acidic conditions. An example of a basic carboxylation is the Kolbe–Schmidt reaction, which is described under industrial synthesis of hydroxybenzoic acid. Another example is the fixation of CO2 (at up to 50 bar pressure) into cyclopentadiene under basic conditions (DBU) to produce 1,3-dicarboxy cyclopentadiene as shown in Scheme 44.70
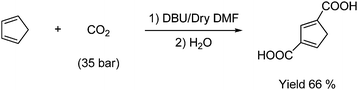 | ||
| Scheme 44 Synthesis of 1,3-dicarboxy cyclopentadiene from cyclopentadiene and CO2 under basic conditions. | ||
An example of acidic carboxylation is the synthesis of acetic acid form methane and CO2. The reaction is an example of a hydrocarbon transformation to carboxylic acid. The reaction is catalyzed by a vanadium71 or palladium72 catalyst in the presence of an oxidizing agent such as K2S2O8 as shown in Scheme 45.
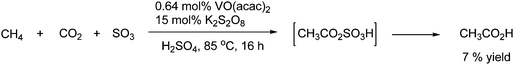 | ||
| Scheme 45 Carboxylation of methane to acetic acid. | ||
Another example of insertion of CO2 into a C–H bond is a Friedel–Crafts reaction. Aromatic carboxylic acids are synthesized in high yields by the carboxylation of aromatics with CO2 and AlCl3 (Lewis acid) under mild conditions as shown in Scheme 46.73
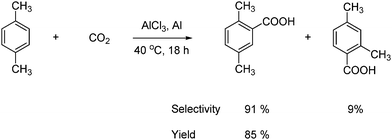 | ||
| Scheme 46 Friedel–Crafts reaction where aromatic carboxylic acids are formed from a CO2 insertion into a C–H bond. | ||
Combining various unsaturated compounds and CO2 with transition metal complexes results in the formation of esters and lactones. Unsaturated compounds such as monoolefins, dienes, allenes and acetylenes react with transition metal complexes to form metal π-complexes. Both CO2 and carbon–carbon unsaturated compounds can be activated by the same metal π-complex as exemplified in Scheme 47. An allene reacts with a palladium complex to form a metal π-complex. Oxidative addition with either CO2 or an additional allene molecule gives a five-membered palladacycle.74
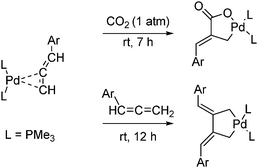 | ||
| Scheme 47 Five-membered palladacycles are formed from the oxidative addition of CO2 or an additional allene to a metal π-complex. | ||
CO2 is able to react with unsaturated compounds in the presence of transition metal complexes as catalysts to give six-membered unsaturated lactones. An example of this is shown in Scheme 48. 1,3-Butadiene reacts with CO2 in the presence of a palladium catalyst.75
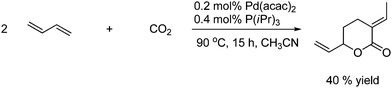 | ||
| Scheme 48 1,3 butadiene reacts with CO2 under palladium catalysis. | ||
The catalytic cycle is shown in Scheme 49. The reaction proceeds by the oxidative addition of two molecules of 1,3-butadiene to a low-valent transition metal (palladium(0)) to form a π-allyl palladium complex. Next step in the cycle is the insertion of CO2 to form a π-allyl palladium carboxylate complex. This step is followed by a reductive elimination with C–O bond formation to obtain, after isomerization, the desired lactone and regenerates the low-valent metal complex to complete the catalytic cycle.21
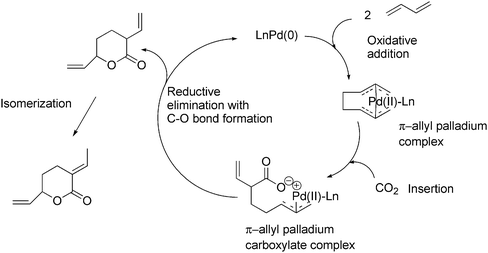 | ||
| Scheme 49 Catalytic cycle for the formation of a lactone from 1,3-butadiene. | ||
Acetylenes also react with CO2 in the presence of a low valent transition metal complex to form lactones as shown in Scheme 50.76
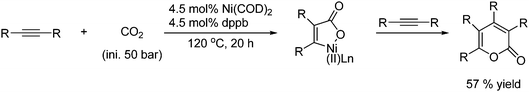 | ||
| Scheme 50 Lactones can be formed from acetylenes and CO2 with Ni(COD)2 as catalyst. | ||
The first intermediate formed in the reaction is a five-membered metallacycle, which is formed from oxidative addition of CO2 and one molecule of acetylene to a low-valent transition metal (nickel(0)). The next step is the insertion of another molecule of acetylene which leads to a ring expansion of the metallacycle to give a seven-membered intermediate. Reductive elimination with C–O bond formation gives the desired lactone and regenerates the transition metal complex as shown in Scheme 51.21
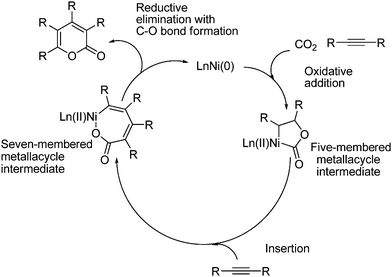
Lactones can also be formed from allenes as exemplified in Scheme 52.77
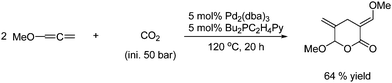
Diynes (RC≡C–(CH2)m–C≡CR) will react with CO2 in the presence of a zero-valent nickel catalyst to give either 2-pyrones or poly(2-pyrones) depending on the value of m. When the value of m is 3 or 4, the reaction proceeds by an intramolecular cycloaddition to give two cyclic 2-pyrones as shown in Scheme 53.
 | ||
| Scheme 53 Intramolecular cycloaddition to give cyclic 2-pyrones from diynes reaction with CO2 under Ni complex catalysis. | ||
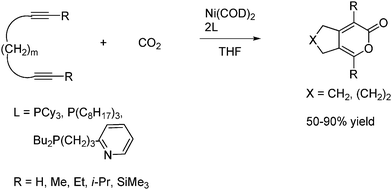
When the value of m ≤ 2 or m ≥ 6 then the reaction proceeds by an alternating copolymerization of the diynes and CO2 to give poly(2-pyrone) by an intermolecular cycloaddition, since the intramolecular cycloaddition is not feasible as shown in Scheme 54.78
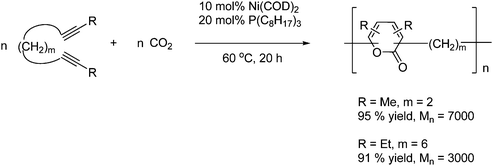 | ||
| Scheme 54 The reaction between diynes of a certain size and CO2 proceeds by an alternating copolymerization. | ||
Diynes react with CO2 and alkyl dihalides under catalysis by a copper(I) salt in the presence of K2CO3 to form poly(alkyl alkynoates) as shown in Scheme 55.79
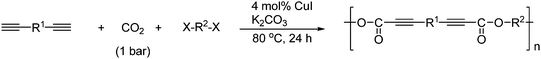 | ||
| Scheme 55 Synthesis of alkyl alkynoates from 1-alkynes, CO2 and alkyl halides mediated by copper(I) salt. | ||
The alternate copolymerization affords good yields with several aromatic diynes, CO2 and 1,4-dibromobutane.
The 2 + 2 + 2 cycloaddition between diynes and CO2 is efficiently catalyzed by nickel complexes with bulky carbene ligands under mild conditions to afford six-membered cyclic lactones as shown in Scheme 56.80
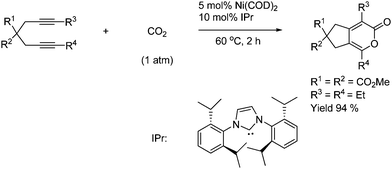 | ||
| Scheme 56 Nickel complexes with bulky carbene ligands efficiently catalyze the 2 + 2 + 2 cycloaddition. | ||
When one of the terminals on the diyne bears a bulky substitute (R = i-Pr or TMS) then a high regioselectivity is obtained as shown in Scheme 57.80
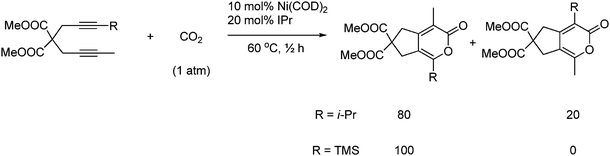 | ||
| Scheme 57 The bulky ligands provide a high degree of regioselectivity when applied to a diyne with one large terminal substitute. | ||
Recent reviews on the synthesis of carboxylic acids, esters and lactones from CO2 are made by Sakakura et al.7 and Omae.21
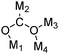
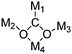
Even though CO2 is an inert molecule, it exhibits a great variety of coordination modes in its metal complexes as shown in Table 2. The nomenclature for these structures includes a simple descriptor which indicates the bonding type. ηn signifies the number of bonds between each coordinated CO2 and the metal atom or atoms, whereas μn signifies the number of metal atoms involved in bonding to each CO2 ligand.81
| Mode of bonding | Structural types of metal-CO2 complexes | Type of M [ref.] |
|---|---|---|
| η1-O | M–O–CO |
U82 |
| η1-C |
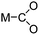
|
Rh,83 Ir84 |
| η2-C,O |
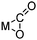
|
Ni,85 Rh,86 Fe,87 Pd88 |
| μ2-η2 |
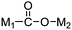
|
Pt,89 Ir/Zr,90 Ir/Os,91 Rh,92 Ru,93 Re/Ge94b |
| μ2-η3 (class I) |
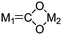
|
Re/Zr,95 Ru/Zr, Ru/Ti, Fe/Zr, Fe/Ti96 |
| μ2-η3 (class II) |
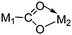
|
Re/Sn,97 Fe/Sn98 |
| μ3-η3 |

|
Os,95,99 Re100 |
| μ3-η4 |
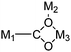
|
Co94,101 |
| μ4-η4 |
|
Ru102,103 |
| μ4-η5 |
|
Rh/Zn104 |
The CO2 molecule has three potential modes of bonding to a metal atom. CO2 can bind “end on” through an oxygen atom (η1-O), “side-on” to a C–O bond (η2-C,O) or via the central carbon atom (η1-C). CO2 can also be bound by two or more metal centers via coordination of the carbon atom to one metal and either one or both oxygen atoms of the CO2 to other metal(s). Thus, a great number of complexes with bridging CO2 ligand can be formed as shown in Table 2.3
These products are potential intermediates in the catalytic transformation of CO2, and have therefore been extensively studied.81,102,105
2.3.4.1 Reaction with transition metal hydrides. The reaction between CO2 and transition metal hydrides can be considered as the first and crucial step in the homogeneous catalytic reduction or transformation of CO2. The initial reaction between the two can proceed in either of two ways: the formation of a mono dentate formato-metal complex (1) which is in equilibrium with the bidentate formato-metal complex (2); the second is the formation of a hydroxycarbonyl-metal complex (3) as shown in Scheme 58.
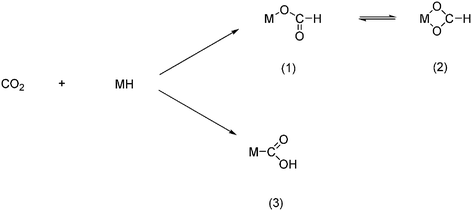 | ||
| Scheme 58 CO2 initial reaction with metal hydrides can proceed in two ways. | ||
Among the examples known to date the one that predominates is the formation of the formato complexes. The metallocarboxylic acid complex is quite unstable, and therefore a much less common product. Furthermore the known examples also show that not all transition metal hydrides react with CO2 as such but rather with carbonic acid or simple derivatives thereof, e.g., carbamic acid and HOCONR2.8,105 Recent reviews in this area can be found in the literature list.105,106
2.4 Reductive hydogenative conversion of carbon dioxide
Hydrogenation of CO2 has been widely investigated for the utilization of CO2. The reactions are carried out with both homogeneous and heterogeneous catalysts. | ||
| Scheme 59 Catalytic hydrogenative conversion of CO2 to methanol. | ||
The catalytic hydrogenation of CO2 to methanol produces water as a byproduct. A third of the hydrogen is thus converted to water, which is a considerable waste compared to the commercial production of methanol via synthesis gas. Furthermore the thermodynamics for methanol production from H2 and CO2 are not as favorable as those for production of methanol from H2 and CO. For example, at 200 °C the equilibrium yield of methanol from CO2 is slightly less than 40% while the yield from CO is greater than 80%.107 The general composition of the catalyst developed for this conversion is based on metals and their oxides, in particular copper and zinc oxide. The most widely used catalyst is Cu/ZnO/Al2O3.13,108
A crucial factor in the development of pilot scale to large scale is the availability of the raw materials namely CO2 and H2. Large amounts of CO2 can be obtained from various exhaust sources such as power plants and industrial plants, e.g., cement factories, aluminium production and fermentation plants. There are also large amounts of natural CO2 sources, e.g., CO2 accompanying natural gas and geothermal energy producing wells.
Hydrogen can either be generated by still-existing fossil fuel sources (mainly natural gas) or from splitting of water.13 The latter can be done electrochemically, thermally or photolytically. However water splitting is a very energy consuming process and it has been estimated that in water electrolyzers with a production capacity of 1000 kg of H2 per day, the cost of electricity has been estimated to represent about 80% of the cost of hydrogen produced.109 The electricity needed for this process can however come from any renewable energy source, e.g., solar, hydro, geothermal, wind, wave, tides etc.
Another way to produce methanol is via the “Carnol-process” developed at the Brookhaven National Laboratory. In this process, hydrogen is produced by thermal decomposition of methane with carbon formed as a byproduct.4,110 The generated hydrogen is then reacted with CO2 recovered from point continuous sources, such as power plants to produce methanol. The Carnol process is shown in Scheme 60.
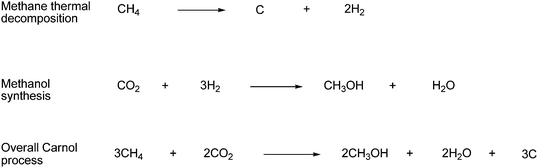 | ||
| Scheme 60 The overall Carnol process consists of two steps: methane thermal decomposition and methanol synthesis. | ||
The byproduct, solid carbon, can be handled and stored more easily than gaseous CO2. Solid carbon can be used in applications such as carbon black in the tire industry and pigments for inks and paints.13 Methanol itself can be used as a liquid energy carrier either pure or mixed with other fuels. It is found that using methanol as a fuel produces less environmentally harmful gases, e.g., hydrocarbons, SOx and NOx compared to regular gasoline. However there are some downfalls to using methanol as a fuel, it increases formaldehyde emission due to its lower heat capacity (compared to gasoline), a larger tank is needed, methanol is more corrosive than gasoline and cautionary measures have to counter this.6,111
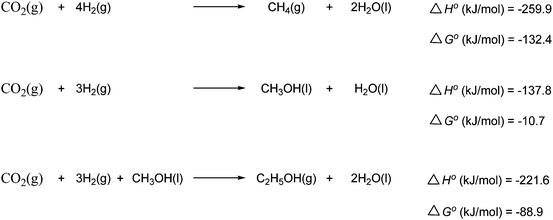
The heterogeneous catalysis of hydrogenation of CO2 to methanol and hydrocarbons is an area which has been widely investigated. However, only few reactions using homogeneous catalysis have been described.
In the first reaction (called the “Sabatier reaction”) CO2 is converted into methane by reaction with four moles of hydrogen. Heterogeneous catalysts of nickel, ruthenium and rhodium have proven to provide the best results for the Sabatier process.112
Hydrogen needed for these reactions has to be produced, which is, as mentioned above, highly energy demanding. The fact is that none of these transformations are favorable for CO2 mitigation unless the energy needed comes from renewable energy.
Alkenes can be hydroformylated with CO2 under medium pressure to afford alcohols and aldehydes in the presence of a ruthenium catalyst as shown in Scheme 62.113
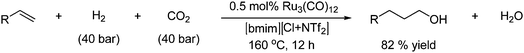 | ||
| Scheme 62 Formation of alcohols from alkenes by hydroformylation with CO2. | ||
The reaction is believed to proceed via the formation of carbon monoxide through the reverse water gas shift reaction.7 In the next reaction carbon monoxide is also believed to be the real reactant. Methanol is homologated with CO2 in the presence of hydrogen under high pressure and temperature to afford ethanol in a low yield as shown in Scheme 63.114
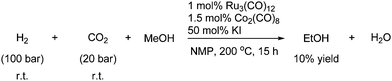 | ||
| Scheme 63 Methanol homologation with CO2 in the presence of H2. | ||
Homogeneous catalysts of, e.g., ruthenium and rhodium are used in the synthesis of a large number of compounds. A reason for this is that high turnover numbers are achieved when applied in supercritical CO2.
Hydrogenation of supercritical CO2 has gained a growing interest since CO2 can have a dual significance as both reactant and solvent, which does not produce waste. Furthermore it is possible to achieve very high H2 concentrations with supercritical CO2. Formic acid can be produced from hydrogen and supercritical CO2 under ruthenium catalysis as shown in Scheme 64. With the ruthenium catalyst, 7200 moles of formic acid are formed per 1 mole of Ru with turnover frequencies up to 1400 h−1.106,116 The presence of triethylamine and a small amount of water is a prerequisite for the formation of formic acid by the hydrogenation of CO2 in its supercritical phase.
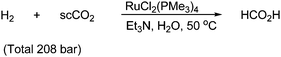 | ||
| Scheme 64 Formation of formic acid from H2 and supercritical CO2. | ||
Formic acid esters can be prepared by the hydrogenation of CO2 in alcohol solvents. For example methylformate can be synthesized very efficiently by hydrogenation of supercritical CO2 with methanol under ruthenium catalysis as shown in Scheme 65.117 Formic acid is formed initially, which then reacts with methanol to afford methylformate.
 | ||
| Scheme 65 Hydrogenation of CO2 under supercritical conditions with methanol affords methylformate. | ||
Exchanging alcohols with secondary amines affords formamide derivatives under nearly identical conditions. The hydrogenation of CO2 under supercritical conditions in the presence of dimethyl amine results in the formation of N,N-dimethylforamide as shown in Scheme 66.49c,118
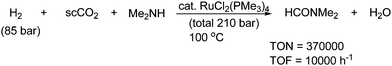 | ||
| Scheme 66 Formation of N,N-dimethylformamide by hydrogenation of supercritical CO2 with dimethyl amine. | ||
 | ||
| Scheme 67 Formation of formic acid by hydrogenation of CO2 under rhodium catalysis. | ||
3 Photochemical reduction
In artificial photosynthesis, the goal is to mimic the ability of green plants and other photosynthetic organisms in their use of CO2, which is reduced to make high energy compounds, i.e., fuels or chemicals.120 In order for the high energy compounds to be economically and environmentally attractive, the compounds must be formed from abundant, inexpensive raw materials such as water and CO2.121Transition-metal complexes have often been employed as catalysts and solar energy converters for the photochemical reduction of CO2. Transition-metal complexes have found wide application in this area, since they are able to absorb a significant portion of the solar energy spectrum, have long-lived excited states, are able to promote the activation of small molecules and are robust.122 Therefore they have been used as catalysts in the photochemical conversion of CO2 using water as the source of electrons.
The systems studied for photochemical CO2 reduction can be divided into several groups: Ru(bpy)32+ both as a photosensitizer and a catalyst;123 Ru(bpy)32+ as a photosensitizer and another metal complex as a catalyst;123a,124 ReX(CO)3(bpy) or a similar complex as a photosensitizer;124a,125 Ru(bpy)32+ and Ru(bpy)32+-type complexes as photosensitizers in microheterogeneous systems;126 metalloporphyrins both as photosensitizers and catalysts;127 and organic photosensitizers and transition-metal complexes as catalysts.122,128
Table 3 summarizes some of the systems which have been studies for photochemical CO2 reduction and their main products.129
| Sensitizer | Catalyst or relay | Donor | Product/s | Φ (mol/Einstein) | Ref. |
|---|---|---|---|---|---|
| a Pr-cyclam 6-((N-R)pyridin-4-yl)methyl-1,4,8,11-tetraazacyclotetradecane where R = p-methoxybenzyl and benzyl. b The quantum yield of product formation is defined as the formation rate divided by the light intensity. c With 15% water in DMF. d With 15% water and excess bpy in DMF. | |||||
| Ru(bpy)32+ | TEOA | HCOO− | 0.049c | 123a | |
| Ru(bpy)32+ | TEOA | HCOO− | 0.096d | 123a | |
| Ru(bpy)32+ | MV2+ | TEOA | HCOO− | 0.01 | 123b |
| Ru(bpy)32+ | Co2+/bpy | TEA | CO, H2 | 130 | |
| Ru(bpy)32+ | Co2+/Me2phen | TEA | CO, H2 | 0.012 (CO), 0.065 (H2) | 131 |
| Ru(bpy)32+ | Ru(bpy)2(CO)22+ | TEOA | HCOO− | 0.14 | 124c,d, 132 |
| Ru(bpy)32+ | Ru(bpy)2(CO)22+ | BNAH | HCOO−, CO | 0.03 (HCOO−), 0.15 (CO) | 124c,d, 132 |
| Ru(bpy)32+ | Ru(bpy)2(CO)(H)+ | TEOA | HCOO− | 0.15 | 123a |
| Ru(bpy)32+ | Ru(bpy)2(CO)(X)n+, X = Cl and Co | TEOA | HCOO− | 123a | |
| Ru(bpy)32+ | CoHMD2+ | H2A | CO, H2 | 133 | |
| Ru(bpy)32+ | Nicyclam2+ | H2A | CO, H2 | 0.001 (CO) | 124e,f |
| Ru(bpy)32+ | NiPr-cyclam2+a | H2A | CO, H2 | Ca. 0.005 (CO) | 124g |
| Ru(bpz)32+ | Ru colloid | TEOA | CH4, H2 | 10−4 (CH4)e | 126a,b |
| Ru(bpy)32+ | Bipyridinium+, Ru or Os colloid | TEOA | CH4, H2 | 10−4 (CH4)e 10−3 (H2)e | 126b |
| ReCl(bpy) (CO)3 | TEOA | CO | 0.14 | 124a, 134 | |
| ReCl(bpy) (CO)3 | TEOA | CO | 0.15 | 125a,b | |
| [ReP(OEt)3 (bpy)(CO)3]+ | TEOA | CO | 0. 38 | 125c | |
| p-Terphenyl | Cocyclam3+ | TEOA | CO, HCOO−, H2 | 0.25 (CO + HCOO−) | 135, 136 |
| p-Terphenyl | CoHMD2+ | TEOA | CO, HCOO−, H2 | 136, 137 | |
| Phenazine | Cocyclam3+ | TEOA | HCOO− | 0.07e | 138 |
| FeTPP | TEA | CO | 127a | ||
| CoTPP | TEA | HCOO−, CO | 127b | ||
The photochemical reduction is normally carried out at room temperature under 1 atm CO2, which implies that the concentration of dissolved CO2 in solution is low. The typical product for these systems is formate and carbon monoxide. DMF solutions containing Ru(bpy)32+, Ru(bpy)2(CO)Xn+ (X = CO, Cl, H, ect) and triethanolamine as a sacrificial electron donor have been used for photochemical CO2 reduction.123a,124c,d,132 These systems produce in a total quantum yield of up to 15% formate as the major product and CO as a minor product.129 Some of the best systems are able to produce a total quantum yield for the reduced products of up to 40%.122,125c The turnover number and frequency is dependant on irradiation wavelength, light intensity, irradiation time and catalyst concentration.122
A photoactive system converts sunlight into electrical energy, which is used to reduce CO2 with the help of a catalyst. The first part of a photochemical reduction is light absorption. The absorption of light is the energy input step that must trigger the photosynthetic reaction. The light is absorbed at the molecular level and the incident light energy is converted into transient stored chemical energy in the exited state. In artificial photosynthesis, ruthenium polypyridyl complexes have been used extensively to mimic plant chlorophylls, which play a major part in the light-harvesting cycle. In the ruthenium complexes the absorption of light comes from metal-to-ligand charge transfer (MLCT) transitions. The transitions involves an electron being exited from a metal based dπ orbital to a low lying π* level on the ligand as shown in Scheme 68.120,139
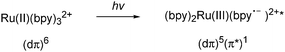 | ||
| Scheme 68 An electron is promoted from a metal-based dπ to a low lying π* level on the polypyridyl ligand. | ||
The MLCT have several desirable features. They are quite stable and their lifetime is therefore sufficiently long to undergo chemical reactions. The absorption range of these complexes can be tuned and extended by varying the transition metal or the ligand. Thereby the complexes can absorb from the near infrared to the ultraviolet region. In order for the MLCT excited state to be useful it must be reached with high efficiency following light absorption and must have a long lifetime to undergo chemical reactions.120,140 The energy in the exited state must undergo a chemical reaction, before it decays to the ground state, in order for it to be used chemically. Electron transfer chemistry provides a basis for utilizing the stored energy. The half-reactions in Scheme 69 involves net electron transfer and combined they give the overall reaction where CO2 as an example is reduced to formic acid and H2O is oxidized to O2.
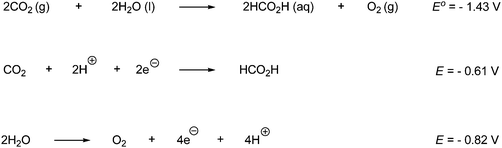 | ||
| Scheme 69 The overall reaction which produces formic acid from CO2 and H2O is an oxidation–reduction reaction and consists of two half reactions. | ||
In order to avoid recombination by back electron transfer, a directional charge-transfer character must be built into the system. In plant photosynthesis, a gradient separates the photo chemically produced oxidative and reductive equivalents by electron transfer and directs them to different places of the molecular structure. In plants there are specific catalytic sites for the reduction of CO2 and the oxidation of H2O. In artificial photosynthesis this problem have been addressed by synthesizing molecules which consist of a light absorber (chromophore) and electron transfer donors and acceptors which are chemically attached to the chromophore at spatially separated sites. In these molecules the light induced redox splitting is directional due to the existence of an intermolecular free energy gradient. The gradient arises from differences in redox potentials between the exited state couples and the quencher (ground state) couples.140,141
The stability of H2O and CO2 is evidenced by their high electrochemical overpotential for direct electron transfer. One-electron transfer to H2O and CO2 are highly unfavorable thermodynamically because they involve the formation of high-energy intermediates such as ˙OH or CO2˙⊖. The potential for the reduction of CO2 to CO2˙⊖ is for example −1.9 V versus NHE. Furthermore there is a large kinetic ‘overvoltage’ for the one-electron reduction, due to structural differences between the linear CO2 and the bent CO2˙⊖.129,140 Thus multi-electron reduction processes are thermodynamically favored over the single electron transfer transformations, as shown in Scheme 70.140 Redox reagents must therefore be designed to carry out the oxidation of water and the reduction of CO2 in synchronous multielectron steps near the thermodynamic potential of the CO2/desired reduction product.120 Thus one-electron products generated in photochemical transformations should be transferred into electron-sink entities or multi-electron charge relays, capable of inducing multi-electron oxidation or reduction processes. Catalysts should provide effective charge storage entities for such multi-electron redox transformations. A limitation of artificial photosynthetic devices is as evidenced in Scheme 70 that the reduction of CO2 could lead to five different C1-reduction products. Furthermore, the reduction of CO2 in water is expected to be accompanied by the competitive reduction of H2O to give H2 evolution.140 A further complication from performing the reduction of CO2 in aqueous media relates to the different hydration products of CO2 present in water. CO2 undergoes in water hydration to form carbonic acid that undergoes stepwise dissociation to bicarbonate HCO3− and carbonate CO32−. The predominant species in solution is dependent on pH. At pH below 4.5 CO2 is the dominant component, at pH between 7.5–8.5 bicarbonate is the major form and at pH above 11.5 is only carbonate present. The thermodynamic reduction potentials for generating certain products are strongly affected by the form of CO2 (hydrated and nonhydrated). The reduction potentials are exemplified for the formation of CO in Scheme 71.
 | ||
| Scheme 70 Potentials for the reduction of CO2 to various products and potentials for the oxidation of H2O to various products (potentials measured at pH 7 in aqueous solution versus NHE, 25 °C, 1 atmosphere gas pressure, and 1 M for the other solutes).140,141 | ||
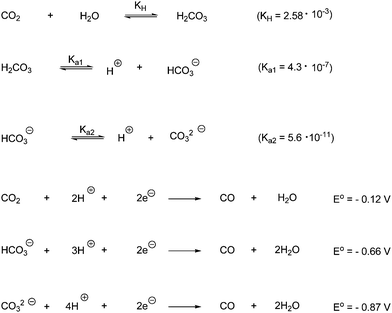 | ||
| Scheme 71 Dissociation constants for CO2 in aqueous media and reduction potentials for CO2, bicarbonate and carbonate to give CO. | ||
It is evident from Scheme 69 that tuning the pH of the aqueous solution provides a means to thermodynamically control CO2 reduction and eliminate H2 evolution.140
Thus the photochemical process could yield a mixture of products. By employing heterogeneous or homogeneous catalysts it is possible to induce selectivity into the system and control a desired route that utilizes the electron transfer products. The catalysts should furthermore activate the substrate towards the redox transformation and act as a multielectron redox relay.140
The basic requirements of the artificial photosynthetic system are summarized in Fig. 3.
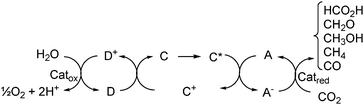 | ||
| Fig. 3 Photocatalysis of water or another electron donor and CO2-fixation in an artificial photochemical system. | ||
The system should comprise a light absorber (chromophore), which upon photoexcitation results in an electron transfer and formation of the redox products A− and D+. Subsequent catalytic reduction of CO2 and concomitant catalytic oxidation of H2O or another electron donor, (e.g., triethanolamine and triethylamine) by A− and D+ respectively recycles the system components and light energy is converted into chemical energy, provided that the process has a favorable free-energy change with ΔG° < 0. The photochemical system should be placed in a membrane, which facilitates the physical separation of the formed products.121,140
Since renewable energy is used for the CO2 transformation, this technology leads to CO2 mitigation. However while some progress has been made on each aspect of artificial photosynthesis, integration of various components in a working system has not yet been achieved. There are some unresolved issues with selectivity of the reactions and in particular limited efficiency, which has to be solved before it is possible to assess the potential of photoreduction for CO2 mitigation.6,121
4 Electrochemical and chemical reduction of carbon dioxide
4.1 Chemical reduction
As mentioned earlier, the catalytic hydrogenation of CO2 to methanol produce water as a byproduct. This means that one third of the energy-costly hydrogen is used to produce water. One way to avoid this is by reducing CO2 to CO to minimize water formation. Methanol can then be produced by adding the right amount of hydrogen to CO and thereby make synthesis gas, which in turn can be converted to methanol with the right catalyst.This can be done chemically by the reverse Boudouard reaction via the thermal reaction of CO2 with carbon or coal itself, as seen in Scheme 72.13
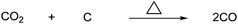 | ||
| Scheme 72 Chemical reduction of CO2 with carbon or coal to produce CO. | ||
The reaction is endothermic and is run at temperatures above 800 °C. This reaction has been investigated for the conversion of solar heat into liquid fuels. This reaction allows solar energy to be stored and transported in the form of a liquid energy carrier such as methanol. The reverse Boudouard reaction is not limited to coal; in general, all carbonaceous materials can be gasified into CO. An example of this is gasification of waste material.6
4.2 Electrochemical reduction
For the reduction of CO2 to liquid fuels or fuel precursors such as CO/H2 (synthesis gas) proton-coupled electron steps are, as mentioned earlier and exemplified in Scheme 67, generally more favorable than single electron reductions, as thermodynamically more stable compounds are produced.141 The multielectron reductions of CO2 become more favorable, in a thermodynamic sense, the more reduced the product. However the difficulty of transfer of multiple electrons to the site of the reduction is a limiting factor in the feasibility of such processes.142Electrochemical reduction using an unreactive metal or carbon electrode gives a CO2 radical anion, which may undergo dimerization to oxalate or disproportionation to CO and carbonate. By contrast, active metals, through active sites on their surface, can direct CO2 reduction to hydrogenated products at a much lower applied voltage because of the high efficiency of the heterogeneous catalysis. In these systems the metal serves a dual role, both delivering electrons and stabilizing the reduced fragments.143 The electrochemical reduction has been studied with various metal cathodes in aqueous media, and studied in some organic solvents.144 The most common reduction products are formic acid, carbon monoxide and oxalic acid, although some examples of successful 6-electron and 8-electron conversions to methanol and methane, respectively, have been described.141,144b,145 When the reaction is performed in water or methanol, hydrogen will also be formed in competition with CO2 reduction. It can, however, be advantageous to generate CO and H2 concurrently at the cathode in a H2/CO ratio close to 2:1, and thereby producing synthesis gas. The synthesis gas can then by further reaction be transformed into methanol. This reaction is however still in the research phase, and has some efficiency problems which must be overcome.146 The reaction has an advantage over commercial production of synthesis gas from natural gas or coal in that no purification step is necessary and no impurities such as sulfur are present that can deactivate the catalyst used for methanol production.13
The nature of the electrode metal for CO2 reduction in aqueous electrolytes is able to strongly effect the product composition. Metallic electrodes such as Hg, Cd, Pb, Tl, In and Sn can reduce CO2 with a high current efficiency. However these metals are poor catalysts in the sense that the primary product is formate, (i.e., there is no breaking of the carbon–oxygen bond). Pb and Hg have been shown to give oxalate in nonaqueous media.144a,147 Metals like Pt, Ni, Fe, Al, Ga and Ti will reduce CO2 to form CO, however due to the low turnover of CO the principle product for these electrodes is hydrogen.148 Electrode materials such as Au, Ag, Zn and Cu will produce CO with a high current efficiency. However Cu is able to convert CO into more-reduced species such as methane, ethane, aldehydes and alcohols in significant amounts.145,149 Cu electrodes are able to reduce CO2 into methane in bicarbonate solutions with current efficiencies as high as 65%, although the overpotential is very large (1.5 V).122,150 The overvoltage or overpotential can be considered to be the difference between the applied electrode potential and the equilibrium potential.141,151 The product composition for the electrochemical reduction of CO2 can also be affected by the electrolyte medium. Zinc electrodes in 0.1 M KHCO3 at potentials of −1.5 to −1.7 V (vs. Ag/AgCl) will give a mixture of CO and formic acid, while in 0.05 M K2SO4, the predominant product is CO, in up to 80% Faradaic efficiency, with small yields of formic acid. This electrolyte dependency can be explained by the higher rate of dissolution of the Zn electrode in the K2SO4 solution (pH 4.2) than in the KHCO3 medium (pH 6.8). The dissolved Zn2+ ions promote the formation of CO.4,152
The electrochemical reduction to hydrocarbons will give rise to bond breaking and bond formation and hence will pose some kinetic challenges. One possibility is to identify a single catalyst which can direct the complete sequence of steps for reducing CO2 to firstly CO, then to CH2O and further to hydrocarbons or alcohols. All steps take place with low kinetic barriers. A second option is to create catalyst panels where each panel catalyzes the specific step in the overall transformation of CO2 to a hydrocarbon or alcohol. An electrocatalyst is able to participate in the electron transfer reaction and increase the reaction rate and hence the current at a potential as close as possible to the equilibrium potential. The electron transfer and chemical kinetics must be fast for an efficient electrocatalyst. These factors can be optimized by chemical variations of the electrocatalyst metal centre via appropriate ligand design. The catalyzed electrochemical reduction of CO2 is illustrated in Fig. 4.141,151
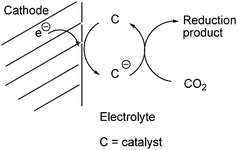 | ||
| Fig. 4 Schematic representation of the catalyzed electrochemical reduction of CO2. | ||
Direct electrochemical reduction of CO2 on most electrode surfaces requires large overvoltages which consequently lowers the conversion efficiency. Electrocatalysts can be employed to lower the overpotential, improve selectivity and increase the reaction kinetics for the CO2 reduction.141,151
Homogeneous electrocatalysts for the reduction of CO2 can be divided into three major categories that depend on the ligand type: (1) metal catalysts with macrocyclic ligands; (2) metal catalysts with bipyridine ligands; and (3) metal catalysts with phosphine ligands.141
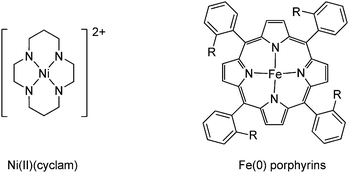 | ||
| Fig. 5 The metal complexes with macrocyclic ligands used in the mentioned examples. | ||
:1 DMF–H2O solution. The reaction shows a solvent dependency, since increased amounts of water will decrease the selectivity for CO. Under an argon atmosphere the reduction product will be solely H2. The electrocatalyst gives high current efficiencies (98%) and excellent selectivity for CO over H2 formation; however the limiting factor for this reduction is the low turnover frequency at 21.4 h−1.155 Rhodium complexes, cis-[Rh(bpy)2X2]+ (X is Cl or OTf), reduce CO2 to predominantly formate at −1.55 V vs. SCE. CO is not formed in any of the experiments; however H2 is formed presumably by the degradation of the supporting electrolyte. The electrocatalyst gives poor current efficiencies for formate (64%) and H2 (12%).156 It is unusual for homogeneous catalysts to form reduction products that require more than two electrons. However by using [Ru(tpy)(bpy)(CO)]2+ complexes (tpy = 2,2′:6′,2′′-terpyridine) as electrocatalysts CO2 can be reduced to a mixture of glycolate (HOCH2COO−), glyoxylate (OCHCOO−), formic acid, formaldehyde and methanol in a 8:2 EtOH–H2O solution at −20 °C. Although turnover numbers are not given for these highly reduced species, their formation shows that a single-site catalyst can give rise to a multielectron reduction of CO2.157
The relatively mild conditions and low overpotentials required for some of the homogeneous catalysts make them attractive for future studies; however a number of efficiency problems must be overcome before this technology is useful for fuel production.122
In this review electrochemical reductions employing semiconductors, alloy electrodes, gas diffusion electrodes4 and bioelectrochemical reductions have been omitted.159
4.3 Photocatalysis
The difference between electrochemical reduction and photocatalysis is the source of electrons. Electrons from electrochemical process are supplied by an applied current; electrons from photocatalysis are supplied by a semiconductor exposed to light.160 Semiconductors161 are used to reduce CO2 to give products like CO, HCOOH, HCHO, CH4 and CH3OH.3,160,162 Unlike metals which have continuum of electronic states, semiconductors exhibit a void energy region, or band gap, that extends from the top of the filled valence band (VB) to the bottom of the vacant conduction band (CB) when exposed to light as illustrated in Fig. 7.160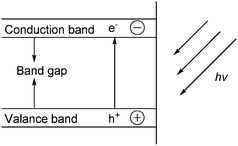 | ||
| Fig. 7 Illustration of the band-gap formation in semiconductors arising from light.160,163 | ||
The photocatalysis over semiconductors is initiated by the absorption of a photon with equal or greater energy than the band gap of the semiconductor. The excitation of an electron from the VB to the CB gives an electron vacancy or a positive charge called a hole (h+) in the VB and the electron–hole (e−/h+) pair is produced. The generation of the electron–hole pair and its reverse process are represented in Scheme 73.160,163
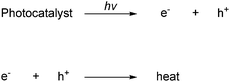 | ||
| Scheme 73 Generation of electron–hole pairs by light absorption and the recombination of the pair. (hv is the photon energy, e− represents a conduction band electron, and h+ a hole in the valance band). | ||
The separated electron–hole pair can follow one of four pathways. Migration of electrons and holes to the semiconductor surface is followed by a transfer of electrons to acceptors and the holes can combine with electrons from donor species. The electron-transfer process is more efficient if the species are absorbed on the surface.164 The last two pathways are recombination producing thermal energy either in the volume of the semiconductor particle or on the surface. The lifetime of an excited electron–hole pair is a few nanoseconds,165 but this is adequate for promoting redox reactions in the solution or gas phase in contact with the semiconductor.
When the photoreduction of CO2 is performed with water as the reductant the amount of organic products are very low. This may be ascribed to the solubility of CO2 in water, which is low at neutral pH, and the CO2 photoreduction process competing with H2 and H2O2 formation, which consumes H+ and e−. However, it is primarily because H2O is a much poorer electron donor compared to organic solvents. Therefore improving the efficiency by employing sacrificial electron donors such as triethylamine,166 triethanolamine,167 dimethylformamide166b and isopropyl alcohol169 have been extensively studied. Photocatalysis of CO2 can also be achieved in the gas phase using sacrificial electron donors such as H2S,170 H2165,171 and CH4.172 Sulfide and sulfite ions are often available as waste products from the petrochemical industry and from fossil-burning fuel stations. The oxidation of these waste products is environmentally beneficial and the development of efficient photocatalytic reduction of CO2 using sulfides or sulfites is therefore highly beneficial for CO2 mitigation and removal of waste compounds.4
A number of semiconductor materials such as TiO2,159,168,173 ZnO,174 ZrO2,175 CdS,166b168 Fe2O3,176 WO3161,176 and their various combinations have been employed as photocatalysts. Some of the studied catalysts and their products are summarized in Table 4.
| Photocatalyst | Reductant | Primary product(s) | Ref. |
|---|---|---|---|
| TiO2/zeolite | Water | CH3OH | 168, 173 |
| TiO2 | H2 | CO | 175b |
| TiO2 (P-25) | Isopropyl alcohol | CH4, HCOOH | 169 |
| TiO2 nanocrystals in SiO2 | Lithium nitrate/2-propanol | Formate, CO, NH3, urea | 173k |
| Rh/TiO2 | H2 | CO, CH4 | 177 |
| Pd/RuO2/TiO2, Pd/TiO2 | NaOH, aqueous Na2SO3 | Formate | 173c |
| MgO | H2 | CO | 178 |
| ZnO on activated carbon | — | CO, H2 | 174 |
However, it is generally accepted that TiO2 and its related materials are the most reliable materials for photocatalytic reactions, due to its low cost, high catalytic activity and high stability under irradiation with light.163,179 Furthermore, large-band gap semiconductors such as TiO2 are more suitable as photocatalysts for CO2 reduction, because they provide sufficient negative and positive redox potentials in conductance and valance bands, respectively. The large band-gap requires a high energy input and TiO2 (anatase) is only active in the ultraviolet region of the solar spectrum.160 Photocatalytic CO2 reduction using TiO2 as the catalyst has been performed in liquid168,169,173,180 and gas phase173e,o,181 systems. The efficiency of the photocatalytic system is reduced by the recombination of the photoexcited electron–hole pair. Studies have shown that doping the semiconductor with a metal improves the photocatalytic efficiency, since the metal acts as an electron trap. The metals suppress the recombination of the electron–hole pair and increase the lifetime of the separated electrons and holes.160,179 Methane production is observed for the photocatalytic reduction of CO2 when using a Cu/TiO2 suspension in water.182 Photoirradiation to CO2 saturated water with 3 wt% CuO-doped TiO2 shows the best results for methanol production. The quantum efficiency of the catalyst reaches 0.19, whereas unmodified TiO2 shows only 0.06.179,180d The metal loading must be optimized and uniformly dispersed over the semiconductor surface. An excess metal loading will result in a decrease in the photocatalytic activity, since the semiconductor surface cannot be illuminated as photons cannot be absorbed due to reflection.160
High photocatalytic efficiency and selectivity for the formation of methanol can be achieved by employing zeolite or silicate containing highly dispersed Ti-oxide species. The zeolite or silicate framework offers unique nanoscaled pores, unusual internal surface topologies, and ion-exchange capacities.173o,181a,b,c
Photocatalysis is affected by the increase of CO2 pressure since a larger amount of CO2 is dissolved in the water or organic medium, thus improving the CO2 reduction selectivity towards liquid products.160,173o
Most of the photocatalytic CO2 reduction reactions require the presence of sacrificial electron donors such as iso-propanol, tertiary amines or DMF in order to achieve substantial yields. These compounds are usually more valuable than the CO2 reduction products. In the absence of such sacrificial electron donors the yields are very low. Therefore it can be concluded that photocatalytic CO2 reduction is not a useful CO2 mitigation technology at the present stage and further research is needed.4
Photoinduced CO2 fixation systems containing enzymes are omitted, however reviews can be found in the reference list.183
5 Biological transformations of CO2
Photosynthetic organisms may be quite different, but all of them use basically the same strategy in which light is initially absorbed by antenna proteins containing many chromophores, followed by energy transfer to a specialized reaction center protein, in which the captured energy is converted into chemical energy by means of electron-transfer reactions.184Fossil fuels, by which modern day society depends upon, are formed from anaerobic decomposition of then-living plants and microorganisms. The fossil fuels: coal, oil and natural gas were formed during the carboniferous period—roughly 360 to 290 million years ago. Oil and gas were formed from the organic remains of prehistoric zooplankton and algae, which have settled down on the sea floor in large quantities under anaerobic conditions. As the sediment pile becomes deeper the organisms within it are subjected to heat and pressures which lead to formation of oil and then gas. Coal, by contrast, is typically formed in non-marine settings from the remains of land vegetation in lowland and swampy environments. Due to the anaerobic conditions the accumulated plant debris is prevented from breaking down. The plant debris initially forms a material known as peat. Then by action of the heat and pressures of geological forces, peat is eventually hardened into coal in a process called coalification.185
Biological carbon sequestration using technologies such as controlled photosynthetic reactions helps to alleviate greenhouse gas problems in a sustainable way. An example of this is the use of a photo-bioreactor system, where natural photosynthesis takes place in a controlled environment, i.e., the light flux is delivered in the right wavelength, since the uniform distribution of light will affect the CO2 uptake rates. A constant climate is kept to promote the photosynthetic process, where light, heat and CO2 is converted into useful products such as carbohydrates, hydrogen and oxygen as shown in Scheme 74.186 The type of biological strains used in the bioreactor depends on the product outcome.

It is estimated that glucose is formed at a rate of 1 g per hour per square meter of leaf surface, which means that approximately 200 Gt of glucose is produced annually by the photosynthetic process.63
The development of closed system bioreactors have been an ongoing process for the last 50 years, despite this commercial viability has yet to be achieved.5
5.1 Algae
In most plants, photosynthesis is an inefficient reaction with slow kinetics. This is especially the case for larger plant species which have to use significant amounts of energy to build their structure, uptake and transport water, and reproduce. With simpler and smaller plants, the efficiency of the photosynthesis reaction is moderately higher as they do not need to invest as much energy to build large structures. At the extreme end of this scale are single-cell algae. They are the smallest and simplest forms of plants and they lack traditional plant structures relying instead on water as their supporting structure, which allows the cells to use all their energy for reproduction. Microalgae have the ability to fix CO2 using solar energy with an efficiency 10 times greater than that of terrestrial plants.187 There have therefore been considerable efforts to apply the microalgae culture for both CO2 fixation and the production of valuable materials. Research has looked into finding and isolating suitable algal strains. Furthermore, photobioreactors have with higher fixation rates and possible scale-up have been investigated. It has been proposed to combine the wastewater treatment with fixation of CO2 from exhaust gases. Algal cultures may be combined with wastewater, which is rich in nutrients such as nitrate and phosphate, with exhaust gases from, e.g., power stations and steel mills for fixation of CO2.4 The development of suitable algal strains with not only a high tolerance for increasing CO2 concentrations but also towards temperature and toxic compounds (NOx and SOx) is therefore ongoing.188Photobioreactor systems are of high importance since the CO2 fixation rate is very low, which implies that a large area is required to perform the CO2 sequestration. The most widely used photobioreactor for commercial production of microalgae is an open pond called a raceway pond. It is found that an area of 1.5 km2 is required for the fixation of CO2 emitted from a 150 MW thermal power plant by using a raceway pond.188 Open ponds have the advantage of using free light from the sun. However, these systems are easily contaminated by other organisms, which make it difficult to maintain a monoculture in open systems.189 Therefore research have gone into designing closed systems, which give rise to an improved environmental control over important parameters such as temperature, pH and partial pressure of CO2. Closed systems gives furthermore an increased biomass concentration which makes harvesting easier and allows for an easier upholding of a monoculture.189 The productivities of various photobioreactors have been compared in Table 5.
| Photobioreactor | Microalgae stains | Culture volume/L (light path/cm) | Highest productivity/g m−2day−1 | Ref. |
|---|---|---|---|---|
| Raceway pond (open) | Chlorella sp. | 200 (20.0) | 13.2 | 190 |
| Raceway pond (open) | Chlorophyta sp. | 200 (20.0) | 8.2 | 190 |
| Helical tubular | Chlorella sp. | 30 (2.5) | 28.1 | 191 |
| Vertical flat-plate (closed) | Synechocytis aquatilis | 24 (1.5) | 31.0 | 192 |
| Vertical flat-plate (continuous) | Nannochloropsis sp. | 1000 (10.0) | 12.0 | 193 |
| Tubular (in vertical arrangements) | Chlorella sp. | 700000 (5.0) |
35.7 | 194 |
| Tubular (horizontal) | Haematococcus pluvialis | 25000 (41.0) |
13.0 | 195 |
| Tubular (inclined) | Chlorella pyrenoidosa | 50 (1.2) | 130.0 | 196 |
| Annular (closed) | Nannochloropsis sp. | 140 (3.5) | 52.5 | 197 |
Only three commercial closed photobioreactors have been constructed and are under operation. One of the systems consists of running compact glass tubes of a total length of 500000 meters and a total reactor volume of 700 m3. The systems occupies 10000 m2 and fixate 260–300 tons of CO2, which results in the annual production of 130–150 tons of dry biomass.188
Recent research shows that certain specialized algae can convert 50 to 80% of their energy into lipids. The process begins with flue gases being passed over clear tube bioreactors that are filled with water and suspended algae. The bioreactors are oriented to receive maximum exposure to the sun. As the algae mix with the flue gas, they fixate CO2. The surplus of algae is continually collected and removed from the bioreactor in order to maintain a relatively constant concentration of algae to water in the bioreactor. The harvested algae are passed through a two-stage dewatering process. The recovered water is returned to the reactors, leaving a dewatered high lipid containing algae cake. The productivity for the pilot plant is 100 g dry mass m−2 per day. After passing through the photobioreactor, the flue gas is vented to the atmosphere. Results from the pilot plant shows that CO2 in the flue gas slip stream was reduced by 82.3% ±12.5% on sunny days and 50.1% ±6.5% on cloudy days. The dewatered algae cake can be collected and converted into biofuels using commercial available processes.198
5.2 Nonphotosynthetic pathways
Nonphotosynthetic pathways for CO2 fixation are those performed by the bacteria methanogens. In these anaerobic microorganisms, the Calvin cycle for CO2 fixation does not work, and acetyl-coenzyme A (CoA) is the central intermediate in carbon metabolism.199 The methanogens grow in freshwater and marine sediments, peats, swamps and wetlands, rice paddies, landfills, sewage sludge, manure piles and the gut of animals. The bacteria grow optimally in temperatures between 20 and 95 °C. They are able to use CO and H2 or CO2 and H2 as their only source of carbon and energy. They convert CO or CO2 together with H2 into methane, and more than half of the methane released into the atmosphere is due to the actions of the methanogens.4It has been proposed to use the methanogenic bacteria for fixation of CO2 from waste gases from blast furnaces. The bacteria will then transform the waste gases into methane, which may be used as fuel for steam boilers. Experiments show that by using H2 as the reducing agent and mixtures of CO and CO2 as carbon source, methane is formed by thermophilic methanogens in a column bioreactor. When the gas recirculation rate is 18 L h−1, the daily rates of H2 consumption and CH4 production are 1380 and 300 mmol L−1.200
By employing a mixture of three bacteria cultures it is possible produce methane in a completely biocatalytic conversion from a mixture of CO, CO2 and H2. The photosynthetic bacterium Rhodospirillum rubrum transforms CO and H2O into CO2 and H2via the water–gas shift reaction resulting in a 100% conversion. Simultaneously, a mixture of two methanogens, Metanobacterium formicium and Methanosarcina barkeri converts CO2 into methane in 83% of the theoretically required by the reaction shown in Scheme 75.4
 | ||
| Scheme 75 Biocatalytic conversion of CO2 into CH4 by two methanogens. | ||
Conventional gas-phase catalytic methods for methanation of CO2 require temperatures in the range of 300 to 700 °C and pressures in the range of 3 to 20 atm. The catalysts used are furthermore sensitive to catalyst poisoning, e.g., by sulfur compounds from exhaust gases. In contrast to this is the biological conversion with the triculture system able to operate at 37 °C, and is not affected by the presence of sulfur compounds or variations in the composition of the feedstock gases.201
A possible way to obtain methane from biogas, which consists of a mixture of methane, CO2, H2S, H2 and N2 and at the same time completely removing H2S from the off-gases, is by employing the chemoautotrophic methanogen Methanobacterium thermoautotrophicum as a biocatalyst. This bacterium has a specific requirement for H2S, and is therefore able to remove it from exhaust gases. Under optimized conditions the biocatalyst gave a purified sulfur-free biogas containing about 96% methane.202
Certain bacteria cultures are able to transform methane into methanol, which may be used as a liquid energy carrier. Methanol is produced by using whole-cell cultures of Methylosinus trichosporium on a 1:1 mixture of methane and oxygen, which gives methanol by oxidation of methane, with a yield of 30% based on the methane utilized.203
Large-scale methane production by methanogens using CO2 and H2 is at present not a feasible solution for CO2 mitigation, since H2 is currently formed by steam reforming (see chapter 6) of natural gas, which is an energy demanding process.
6 Reforming with carbon dioxide and methane
Synthesis gas can be formed by reacting CO2 with methane. This process is called “dry reforming”, since it does not involve steam, as shown in Scheme 76. | ||
| Scheme 76 Dry reforming: CO2 reacts with methane to produce synthesis gas. | ||
The reaction is strongly endothermic and is carried out at temperatures around 800–1000 °C using a catalyst based on nickel (Ni/MgO, Ni/MgAl2O4, etc.).13,204 With 3 mol % Ni/uscMgO (ultrafine single-crystal magnesium oxide) as catalyst system, methane conversion at 800 °C was 96%.205 The composition of the formed synthesis gas makes it ideal as feed gas for iron ore reduction and Fischer–Tropsch synthesis of long–chain alkanes. The composition is, however not suitable for commercial production of methanol in which a H2/CO ratio 2:1 is needed. Additional H2 can be tuned via the water–gas shift reaction as shown in Scheme 77.6
 | ||
| Scheme 77 Water–gas shift reaction forms CO2 and H2. The latter can be added to the gas mixture from dry reforming, thereby getting the right composition of synthesis gas. | ||
The right composition of synthesis gas can be produced from methane directly by combining CO2 (dry reforming) and H2O (steam reforming) in a process called bireforming as illustrated in Scheme 78. Thereby large amounts of CO2 is consumed, while the water–gas shift reaction is avoided, which reduces the process cost. The reactions are performed in a temperature range between 800 and 1000 °C.6,13
 | ||
| Scheme 78 Bireforming involves a 3:2:1 ratio of CH4/H2O/CO2. | ||
The catalysts for bireforming can be those used for the separate steam and dry reforming, combining the two streams afterwards. However a process which combines the dry and steam reforming in a single step has been reported.13
In practical use, natural gas is the major source of methane. Besides methane, natural gas consists of higher hydrocarbon is various concentrations, which also can undergo bireforming to give synthesis gas as shown in Scheme 79.
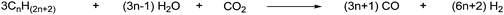 | ||
| Scheme 79 Bireforming performed with natural gas. | ||
It is advantageous to use natural gas from geothermal sources, since it often contains substantial amounts of CO2.13
In order to produce synthesis gas without either consuming or producing much heat, some plants combine the exothermic partial oxidation with the endothermic CO2 reforming to give synthesis gas with a H2/CO ratio of 1:1 in a thermal neutral reaction. The exothermic oxidation reaction generates the heat needed for the process but produces water as a byproduct as shown in Scheme 80.4,13

By employing a Ni–CaO catalyst the simultaneous catalytic partial oxidation and dry reforming can be achieved with more than 95% conversion and with more than 90% H2 selectivity.4,206
In the present commercial production of methanol from synthesis gas with traces of CO2 Cu/ZnO based catalysts are used. Hydrogen from other sources has to be added to this mixture in order to produce methanol as shown in Scheme 81.13
 | ||
| Scheme 81 Commercial synthesis of methanol from carbon monoxide and hydrogen in a ratio 1:2. | ||
The main problem which has hindered industrial applications of dry reforming is the formation of coke, which is thermodynamically favored except at very high temperatures, above 900 °C. Formation of coke quickly deactivates conventional reforming catalysts if used without the presence of steam. Carbon deposition may occur by the exothermic Boudouard reaction and by the endothermic cracking of methane as shown in Scheme 82.4,207
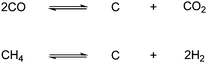 | ||
| Scheme 82 Formation of carbon from the Boudouard reaction and by cracking of methane. | ||
Coking is also a problem with steam reforming, however it is less severe since the carbon formed can be volatilized by the reaction shown in Scheme 83.4
 | ||
| Scheme 83 Carbon may be volatilized during steam reforming to produce CO and H2. | ||
The main contributor to carbon deposition during dry reforming is the Boudouard reaction; however its equilibrium is shifted to the left by increasing the temperature. Hence by performing dry reforming at high temperatures carbon deposition is minimized.208
There is no effective commercial catalyst to date exists which operates without carbon formation. In the past decade, efforts have focused on the development of catalysts which show high activity and stability for methane dry reforming with CO2 to syngas.209,210 Nickel-based catalysts209,211 and noble metal-supported catalysts (Rh, Ru, Pd, Pt, Ir)210,211,212 were found to have promising catalytic performance in terms of conversion and selectivity. The catalysts based on noble metals are reported to be less sensitive to coking compared to the nickel-based catalysts for dry reforming.210 However, considering the high cost and limited availability of noble metals, it is more practical to develop improved Ni-supported catalysts which exhibit stable operation for a long period of time.
The formation of synthesis gas by dry reforming of methane could provide a substantial use for CO2 from industrial and natural sources. This capture provides a renewable, inexhaustible carbon source and could also provide a means for the continued use of derived carbon fuels in an environmental friendly and carbon neutral way. New methods of combined partial oxidation and dry reforming considerably improve the energy economy for the production of synthesis gas.
7 Inorganic transformation of carbon dioxide
CO2 can be captured from the atmosphere using basic absorbents such as calcium hydroxide (Ca(OH)2) or potassium hydroxide (KOH), which react with CO2 to form calcium carbonate (CaCO3) and potassium carbonate (K2CO3) respectively.208 The CO2 absorption is an exothermic reaction, which liberates heat, and is readily achieved by mixing CO2 with an adequate base. The reverse step, the desorption, is however an endothermic process which requires energy to regenerate the base and recover CO2. Both calcium and sodium carbonate requires a large input of energy to recover the base, and is therefore not well suited for the capture and release of CO2. There is an ongoing effort to find suitable absorbents to remove CO2 from the atmosphere for its recycling with minimal input of energy. One of the promising absorbents is KOH, where it is shown that the electrolysis of K2CO3 in water could efficiently produce not only CO2 but also H2 with relatively modest input of energy.13,213Limestone (major components are calcium carbonate and magnesium calcium carbonate) is used industrially to produce synthetic precipitated calcium carbonate (PCC) for which the production was more than 7 Mt in 2003 worldwide. The limestone is calcinated to form lime (CaO), which is treated with water to form Ca(OH)2. The PCC is formed by carbonating Ca(OH)2 using an industrial (in few cases natural) CO2 source. About 75% of the PCC production is used in the paper industry.10,214
The annual market and fixed CO2 for various inorganic carbonates is illustrated in Fig. 8.
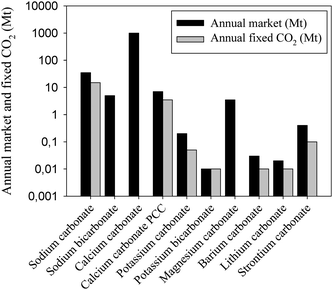 | ||
| Fig. 8 The annual market volume and fixed CO2 in megatons. Note the logarithmic scale on the y-axis. For the minerals where part of the annual market stems from mining the materials in their mineral form, the annual fixation of CO2 is smaller. | ||
SrCO3 is produced at 0.3–0.4 Mt y−1 primarily from Celestine (strontium sulfate) mineral by carbonation using coal or soda ash, which implies CO2 fixation of around 0.1 Mt y−1.10,215 SrCO3 is used in glass (monitor screens), batteries, photochemicals and catalysts.
Besides CaCO3, CO2 is also used to prepare NaHCO3 and Na2CO3. CO2 is also used to prepare carbonates of La3+, Nd3+, Sm3+, Eu3+, Gd3+, Dy3+ and Ho3+. The carbonates are formed by reaction with an aqueous solution of the corresponding oxide M2O3 and supercritical CO2. The reaction occurs to 300–320 K and at pressures around 70–250 bar, yielding above 95% of the desired product.6
The market volumes for inorganic carbonates have been summarized in Table 6.10
| Product | Year | Market/Mt y−1 | CO2 fixed/Mt y−1 | Ref. |
|---|---|---|---|---|
| Na-carbonate | 2003 | 35, ∼25% is mined | ∼15 | 216 |
| Na-bicarbonate | 2003 | 5 | — | 10 |
| Ca-carbonate | 2005 | Several 1000 | Mainly mined | 217 |
| Ca-carbonate PCC | 2003 | >7 | >3.5 | 214 |
| K-carbonate | 2005 | 0.1–0.2 | 0.03–0.05 | 10 |
| K-bicarbonate | 2005 | <0.01 | <0.01 | 10 |
| Mg-carbonate | 2005 | 3.5 | Mainly mined | 218 |
| Ba-carbonate | 2005 | 0.02–0.03 | ∼0.01 | 219 |
| Li-carbonate | 2003 | 0.01–0.02 | ∼0.01 | 220 |
| Sr-carboate | 2003 | 0.3–0.4 | ∼0.1 | 215 |
Different lithium derivatives have been investigated for the purpose of the development of a suitable adsorbent for the separation, capture and release of CO2 from flue gases. Lithium zirconate (Li2ZrO3) has been investigated as a high temperature absorbent. Li2ZrO3 reacts with CO2 to form lithium carbonate as shown in Scheme 84. The reaction is reversible in a temperature range of 450–590 °C.221
 | ||
| Scheme 84 Lithium zirconate reacts with CO2 to form lithium carbonate. | ||
Lithium silicate (Li4SiO4) also reacts with CO2 as shown in Scheme 85. It is found to have a larger CO2 adsorption capacity than that of lithium zirconate. Furthermore Li4SiO4 is found to have desirable features like rapid absorption at a wide range of temperatures and concentrations of CO2 and to be stable under the different conditions. Li4SiO4 adsorbs CO2 below 720 °C and releases CO2 above 720 °C.222
 | ||
| Scheme 85 Lithium silicate adsorbs CO2 below 720 °C to give lithium carbonate. | ||
8 Carbon dioxide capture and storage (CCS)
One of the countermeasures for global warming is CO2 capture from massive emission sources such as thermal power plants and coal fired power plants. The purpose of the capture is to produce a concentrated stream of CO2 at a high pressure that can be transported readily to a storage site.2 This process is known as carbon dioxide capture and storage (CCS). CCS technologies attract a lot of attention because they would reduce our CO2 emissions to the atmosphere while continuing to use fossil fuels. Fossil-fueled power plants are responsible for roughly 40% of the total CO2 emission, coal-fired plants being the main contributor.5 The capture of CO2 from flue gases of coal-, oil- or gas-fired power plants is a mature technology which is commercially available. Three processes are available for the capture of CO2 from large point sources: post-combustion capture, pre-combustion capture and oxygen-fired combustion. In post-combustion systems CO2 is separated from the flue gases produced by the combustion of the primary fuel in air. These systems primarily uses a liquid solvent to capture the small fraction of CO2 (typically 10–15% by volume) present in the flue gas stream in which the main constituent is nitrogen (from air). This process is usually employed for pulverized coal power plants or a natural gas combined cycle power plant. In pre-combustion systems the primary fuel is treated in a reactor with steam and air or oxygen to produce synthesis gas (CO and H2). Additional hydrogen, together with CO2, is produced by reacting CO with steam in a second reactor. The production of synthesis gas comprises the steam reforming, partial oxidation and water gas shift reaction as shown in Scheme 86.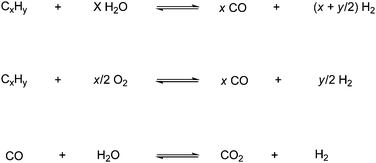 | ||
| Scheme 86 Steam reforming, partial oxidation and the water gas shift reaction takes place in the pre-combustion system process. | ||
The capture of CO2 from the resulting CO2 and hydrogen mixture can then applied at high CO2 concentrations and take place at pressures which are at least 50 times higher than in the post-combustion process. The capture is usually done by physical absorption, where the CO2 and hydrogen mixture can be separated into two streams by reduction of pressure. Oxygen-fired combustion proceeds by an approach where air separation precedes combustion. The hydrocarbon fuel is then combusted in a mixture of O2 and CO2 rather than air to produce an exhaust of CO2 and water vapor. Because oxygen is used instead of air, the nitrogen part and its combustion products are ideally eliminated from the exhaust gas stream. The net flue gas, after cooling to condense the water vapor, contains ideally 80–98% CO2 depending on the fuel used and the particular type of oxygen-fired combustion process so only simple CO2 purification is required prior to transportation and storage.2,223 A challenge of oxygen-fired combustion is improving the necessary technology for air separation needed to produce oxygen. Negative pressure and air leaks in the system are common problems for this technology, which makes it difficult to remove nitrogen from the system. The most common method is to separate oxygen from air by cryogenic separation, which is highly energy demanding, thus reducing overall plant efficiency.223
The technologies for capture from the three processes are based on either absorption into a liquid solution, adsorption onto suitable solids, cryogenic separation and permeation into membranes.224 Amine solution based CO2 absorption/desorption systems using the liquids mono-ethanolamine (MEA), diethanolamine (DEA) and methyl-diethanolamine (MDEA) are some of the most widely employed capture technologies. MEA, the least expensive of the alkanolamines, is the traditional absorbent for CO2 removal from flue gas stream as its reaction is fast even at low CO2 pressure.225 The aqueous amine based CO2 absorption can proceed by to different paths, where carbamate and bicarbonate formation are possible to occur as shown in Scheme 87.226
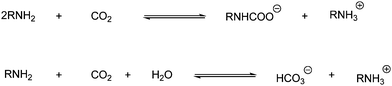 | ||
| Scheme 87 In the CO2–alkanolamine reaction, it is possible for two different paths of carbamate and bicarbonate formation to occur. | ||
When carbamate formation is the dominant reaction, two moles of amine react with one mole of CO2, whereas a one-to-one ratio is required to form bicarbonate. This indicates that the bicarbonate formation has a capacity for CO2 absorption twice as high as for the formation of carbamate.226,227
The relative amounts of formed carbamate and bicarbonate which result from carbamate instability can largely be attributed to structural types of amines.226,228 It is shown that the increase of structural bulkiness of the substituents bound to the nitrogen atom makes the general carbamate stability decrease and makes the CO2 loading capacity increase. The decrease in carbamate stability favors carbamate reversion to bicarbonate and free amine, leading to loadings approaching one mole of CO2 per mole amine.227
MEA, being a primary amine, reacts with CO2 to give a high percentage of MEA carbamate. The CO2 rich MEA solution is then sent to a stripper where it is reheated to release almost pure CO2. The CO2 recovery rate is 98% for MEA. The MEA solution is then recycled to the absorber.5,229 However there are some major drawbacks for this technology including high energy requirements for the regeneration step (regenerated at 110 °C230) and limited loadings of the amine, due to corrosion problems and amine degradation. MEA has some specific disadvantages since it has a relative low absorption capacity caused by the formation of carbamates as the main reaction product. There is therefore an ongoing research into replacing MEA with other sterically hindered alkanolamines for the purpose of increasing CO2 loading capacity.225
The adsorption process onto suitable porous solids such as zeolites and activated carbon is based on the same principle as the amine liquids.5 Polymeric membranes have more recently been introduced for CO2 separation. The flue gas passes through the polymeric membrane by a solution-diffusion mechanism. Some of the most widely employed polymers are illustrated in Fig. 9.231 Polymers used in the construction of gas separation membranes include polyacetylenes,232 polyanilines,233 poly(arylene ethers)s,234 polyarylates,235 polycarbonates,236 polyetherimides,237 poly(ethylene oxide),238 polyimides,239 poly(phenylene oxide)s,240 poly(pyrrolone)s241 and polysulfones.236a,b,242 Membrane materials with a high selectivity for CO2 over oxygen or nitrogen have been developed, however at the present stage a sufficiently high selectivity with a large flux has yet to be achieved for membranes. Membranes are more expensive than, e.g., MEA, but less space demanding.5,13,243
There is however a number of issues associated with the capture of CO2 from flue gases, which limits the applications for which polymeric membranes can be used. The high temperature of flue gases will rapidly destroy the membrane; therefore the gases need to be cooled below 100 °C prior to membrane separation. The polymeric membranes need to be chemically resistant, due to the harsh chemicals within the flue gases, or these chemicals need to be removed prior to the separation process. Furthermore, the low concentration of CO2 in the flue gases is a problem, since large quantities of gases need to be processed.5 Recent research has been directed towards the development of inorganic membranes, due to the demand in new application fields such as fuel cells, membrane-reactors and other high-temperature separations.5 In the cryogenic separation method, compressed and liquefied CO2 gas is expanded adiabatically to separate a certain element gas depending on its different evaporation point. This method is more costly than other separation methods and is not suitable for separating CO2 from gases with a low CO2 concentration. This method is applied to obtain CO2 in a high purity.244
The capture of CO2 contributes 75 percent to the overall CCS cost and CCS increases the electricity production cost by 10 to 40 percent. Hence there is a need for further development in CO2 separation and capture to reduce the overall energy cost before CCS technologies successfully can enter the energy market.2
The captured CO2 is then transported either as a liquid, gas or in its supercritical state to the storage site. The transportation is done via pipelines and/or shipping. 50 million tones of CO2 is annually transported in pipelines that extend over more than 2500 km in the western USA, carrying CO2 from natural and anthropogenic sources to enhanced oil recovery projects. Upon designing CO2 pipelines, factors like the properties of CO2, corrosion rates, and the gas mixture are important considerations toward establishing the material specifications. The water content in the CO2 gas is crucial since corrosive carbonic acid is formed. In order to inhibit hydrate formation and prevent excessive corrosion rates the water content is reduced to ppm-levels by employing either molecular sieves, glycol (MEG/TEG) or alumina desiccants.2,223 Ships can be used for long distance transport of CO2 across oceans. Ships have the advantage of introducing flexibility in the CO2 value chain, allowing collection of concentrated CO2 from various sources at volumes below the critical size for pipeline transportation. Ships are not suitable for large-scale transport of CO2, because at these pressures, the ship must be constructed as a pressure vessel, which is very costly. Ship-based transport requires liquefaction plants, intermediate storage facilities, ships, loading and unloading systems at each site, which makes ship-based transport a costly solution. Currently ships carrying up to 900–1200 tons of CO2 are in operation.2,223
8.1 Sequestration
Following the capture and transport process CO2 needs to be stored. CO2 can be disposed of in natural sites such as deep geological cavities, saline aquifers, spent oil or gas fields, coal mines or on the ocean floor, or may be chemically fixed into solid substances, e.g., inorganic carbonates.245 Large scale geological storage has already demonstrated feasibility, while other technologies like ocean and carbonate CO2 storage is still in the research phase.245 Geological storage involves injecting CO2 at depths greater than about 1 km into porous sedimentary formations using technologies derived from the oil and gas industry.246 CO2 can be stored in its supercritical state at depths below 800–1000 m, which provide the potential for efficient utilization of the space, due to the liquid-like density of supercritical CO2. Ocean storage involves injecting CO2 into the deep ocean below the depth of 3500 m taking advantage of the very slow natural interchange between the deep ocean layers and its surface layer. Below 3000 m stored CO2 would form a lake of liquid CO2 or CO2 hydrate.2 However each of the mentioned storage options has its limitations and uncertainties that will require further research before there is a guarantee of no significant losses of confined CO2.245The CCS technology is already implemented in Norway and Algeria where the technology is employed for the separation of liquefied natural gas into CO2 and methane. The captured CO2 is stored underground in the scale of one million tons per year. In Canada the technology is used to separate and capture CO2 from gasified coal. The captured CO2 is utilized for the enhanced oil recovery on the scale of one million tons per year.244 Carbon dioxide injection into geological formations for enhanced oil recovery (EOR) is a mature technology. In 2000, 84 commercial or research-level CO2-EOR projects were operational world-wide. In most CO2-EOR projects, much of the CO2 injected into the oil reservoir is only temporarily stored. This is due to the technique used to maximize oil recovery. This technique results in CO2 being released, with a small but significant amount of the injected CO2 remaining dissolved in the immobile oil.247 This point can be illustrated by the fact that 48 Mt y−1 CO2 (from natural and anthropogenic (25%) sources) are used annually in USA for EOR and it is estimated that 9 Mt y−1 CO2 is sequestered.248 However, in the Canadian field, a different technique has been employed for the EOR, which will allow for permanent CO2 storage. Over the anticipated 25-year life of the project, it is expected that the injection of some 18 million tons of CO2 will produce around 130 million barrels of enhanced oil. This has been calculated to be equivalent to approximately 14 million tons of CO2 being sequestered.247
Another way to reduce the amount of CO2 in the atmosphere is to enhance the natural sequestration of CO2. Options which could increase the amount of fixed CO2 could be an enhancement of the natural sinking process, such as forestation, ocean fertilization and mineral carbonation.5 Forestation, reforestation of arid lands and greening of deserts will increase the amount of fixed CO2. At the beginning of the forestation the amount of fixed CO2 is higher than the amount which is released by decomposition of organic matter. At a later stage when the forestation is fully developed, the net CO2 capture becomes zero, due to a balance between what is captured and what is released. The potential for CO2 sequestration in terrestrial systems is estimated to be 5–10 Gt of carbon annually.229 A second option is ocean fertilization where fertilizer is added to areas with limited nutrients to increase the production of phytoplankton. It is estimated that the current phytoplankton production has an annually uptake of 50–100 Gt of carbon, which is considerably higher than that of terrestrial vegetation.229 Part of the carbon is released back into the atmosphere by the respiration process, and the remaining part descends deeper into the ocean in the form of organic matter, either by the death of the phytoplankton or by grazing.5 There are some considerable drawbacks to this sequestration method, since ocean fertilization may interfere with the marine ecosystem, which could give rise to unforeseen consequences and possible fatal impact for the ecosystem. Furthermore, an increase in the decomposition of sinking organic matters could give raise to an increased production of stronger greenhouse gases, such as methane and nitrogen monoxide. As pointed out there some unresolved areas connected to ocean fertilization and the method is therefore still in the research phase.5
Large amounts of CO2 is fixed by a process called mineral carbonation, which is natural or artificial fixation of CO2 into rocks. Mineral carbonation results in the storage of CO2 in solid form as a stable and environmentally benign mineral carbonate. Mineral carbonates are thermodynamically very stable, and the storage of CO2 into rocks is therefore a safe and long-term solution. The natural weathering of rocks such as silicates, containing calcium or magnesium happens over geologic time scales. The silicate rocks are turned into carbonates by reaction with CO2 as shown in Scheme 88.5,249
 | ||
| Scheme 88 Formation of calcium or magnesium carbonate from natural weathering of silicate rocks. | ||
Artificial mineral carbonation seeks to accelerate the fixation of CO2 into rocks. Two methods have been studied the first is to perform a direct carbonation, which binds CO2 from its gaseous form with minerals in the solid state at suitable temperature and pressure levels. The second is an aqueous process which extracts magnesium and calcium ions from minerals into solution, followed by precipitation of either the carbonate or an intermediate product which is carbonated in a separate step. The advantage of the direct carbonation approach is its inherent simplicity. However, the direct gas-solid reactions are to slow for practical applications for the most abundant silicate rocks and are only feasible at reasonable pressures for rare, refined materials like the oxides and hydroxides of calcium and magnesium. The increased CO2 pressure will furthermore give rise to increased energy consumption.5,249,250,251,252
The solution of minerals in water and subsequent carbonation is believed to proceed by the equations in Scheme 89.253
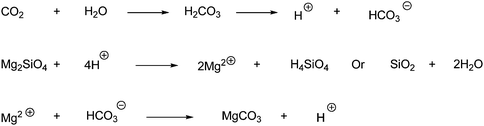 | ||
| Scheme 89 The solution of a mineral (olivine) in order to react with bicarbonate ions to give solid carbonate. | ||
In Scheme 89 magnesium carbonate is formed when CO2 is dissolved in water to give carbonic acid (H2CO3), which dissociates to H+ and HCO3−. Then, H+ ions hydrolyze the mineral, liberating Mg2+ cations and forming silicic acid or free silica and water. Finally the free Mg2+ cations react with the bicarbonate ions to form the solid carbonate.
Dissolution catalysts can be added to the aqueous solution such as strong and week acids,249,254 bases255 and chelating agents to extract SiO2 or MgO groups from the mineral.254 All three approaches have been investigated and it is found that catalyst recovery is a significant issue, which needs to be addressed.9 Hydrochloric acid dissolution of silicates is performed in a number of steps in order to precipitate magnesium hydroxide, which can then react with gaseous CO2 and thereby form the carbonate species. The steps alternate between being exothermic (from which heat recovery is not always possible) to being endothermic. It is found that the overall reaction has a negative CO2 balance and is therefore at present not viable.9 The use of a strong acid can furthermore give environmental problems and there are also problems associated with corrosion of the reaction facilities.5,249,250,251,252
Recent studies show that by employing a NaHCO3 (0.64 M)/NaCl (1 M) solution the reaction rate compared to distilled water can be dramatically increased.256 It is believed that the bicarbonate ion hydrolyzes the silicate as shown in Scheme 90, forming the carbonate, hydroxide (OH−) ions, and free silica (SiO2). The OH− ion is believed to react immediately with the additional CO2 being injected into the solution to reform the bicarbonate maintaining a relatively constant solution chemistry.253
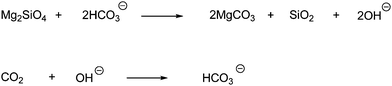 | ||
| Scheme 90 Mineral carbonation performed in a NaHCO3/NaCl solution. | ||
Four different minerals (olivine (Mg2SiO4), lizardite (Mg2Si2O5(OH)4), antigorite ((Mg, Fe(II))3Si2O5(OH)4) and wollastonite (CaSiO3)) were tested under these reaction conditions. Their reactivity, measured as the extent of the carbonation reaction after one hour under specified reaction conditions, is found. All the minerals have been subjected to a pretreatment, which is mineral specific. The activated minerals have been subjected to an ultra-fine grinding and in some cases thermal activation. The results of these experiments are shown in Table 7.256
| Mineral | Conversion after 1 h (%) | Reaction conditions |
|---|---|---|
| Olivine | 61 | 185 °C, 15 MPa |
| Olivine (activated) | 81 | 185 °C, 15 MPa |
| Lizardite | 9 | 155 °C, 11.5 MPa |
| Lizardite (activated) | 40 | 155 °C, 11.5 MPa |
| Antigorite | 62 | 155 °C, 11.5 MPa |
| Antigorite (activated) | 92 | 155 °C, 11.5 MPa |
| Wollastonite | 43 | 100 °C, 4 MPa |
| Wollastonite (activated) | 82 | 100 °C, 4 MPa |
The Table shows that the more energy used to activate the mineral the higher the conversion rate. However, the heat treatment and the grinding make the economics of the process unattractive.256 Further improvements are needed to make this process viable.
The capacity for CO2 fixation by employing artificial carbonation is vast; Mineral carbonates have been estimated to have a carbon storage capacity in the range of 100000 to 1000000 carbon equivalents.257 However, as mentioned throughout this section, there are some considerable drawbacks to this technology and mineral carbonation is therefore still in the research phase and considerable research is still needed prior to commercialization.245,251
Estimated storage capacities for various sequestration methods are summarized in Table 8.
| Sequestration option | Worldwide capacity (order of magnitude in Gt-carbon) |
|---|---|
| Mineral carbonates | 10000s–1000000s GtC |
| Ocean | 1000s GtC |
| Deep saline formations | 100s–1000s GtC |
| Depleted oil and gas reservoirs | 100s GtC |
| Coal seams | 10s–100s GtC |
| Terrestrial | 10s GtC |
| Enhanced oil recovery | 10s GtC |
| Utilization (chemical conversion) | <0.1 GtC per year |
Current estimates predict that the remaining fossil fuel resources exceed 5000 GtC,259 which implies that the world energy consumption will be based on fossil fuels for many years to come. There is, therefore, an urgent need to find sinks which can sequester some if not all of the emitted CO2. Mineral sequestration has, as shown in Table 8, the capacity to bind all the CO2 that could ever be generated and thereby limit the environmental impact that growing CO2 concentrations in the atmosphere may have. However, this option is currently too energy-intensive, and therefore still in the research phase.257 Saline aquifers imply huge storage capacities. However, because of uncertainties in storage lifetimes, seismic instability, and potential migration of CO2, long-term integrity must be established for each site, which makes this technology costly.257
The 900 Gt CO2 (245 GtC) which is currently in excess in the atmosphere can ideally be sequestrated by many of the options mentioned in Table 8. However, most sequestration methods require a concentrated stream of CO2.
EOR is a proven technology and therefore probably the easiest route to large scale sequestration. Injecting CO2 into reservoirs in which it displaces and mobilizes oil or gas will also give additional revenues that partly will offset sequestration costs. However, oil and gas sites have limited capacity and this technology is only able to temporally mitigate the growing CO2 concentrations.257
Capacity constraints leave many sequestration methods, such as biomass sequestration and CO2 utilization, insignificant for mitigation the growing CO2 concentration.257
Table 8 shows that there are sequestering options which ideally could store all of the emitted CO2 and future CO2 emissions, but these options are still and in the foreseeable future in the research phase.
9 Summary and outlook
Warming of the planet due to man-made increases in the atmospheric CO2 concentration is now an indisputable fact. There have been observations of an increase in the global average air and ocean temperature, observed widespread melting of snow and ice and an increase in the global average sea level. These changes will in time give rise to irrevocable changes in both local and global climate. The main contributor to these climate changes is the release of CO2 into the atmosphere from the combustion of fossil fuels. This leads to an increased level of CO2 in the atmosphere, which traps heat and prevents it from being radiated back to space. This effect is named the greenhouse effect. The consequence of the increase in average global temperature is, among others, increasing desert formation and a rise in sea level from melting of the glaciers, the Antarctic ice caps and expansion of sea water. Options for reducing the current annual emissions from fossil fuel consumption, which is about 24 Gt CO2 are a reduction of the amount of CO2 produced, usage of the emitted CO2 and capture and storage of the emitted CO2. Utilization of CO2 in industrial applications is not expected to mitigate the increased CO2 concentration with current available technologies. At present the typical lifetime of the CO2 currently used in chemical applications is only days to months. The stored carbon is then degraded to CO2 again and emitted into the atmosphere. With such short lifetimes it is difficult to contribute significantly to the mitigation of the CO2 problem by the industrial utilization of CO2. However there are some promising processes which in time potentially could utilize some of the emitted CO2. A possible large scale use of CO2 could potentially be in the polymer (polycarbonates and polyurethanes) synthesis area. In particular plastics and laminates used in the construction industry, where the lifetime of the materials can be decades.4 Another promising area is the production of liquid carbonaceous transportation fuels. Methanol can be formed from CO2 and H2 under catalysis and subsequently catalytically dehydrated to a hydrocarbon fuel such as gasoline. The production of synthetic liquid fuels is attractive due to their high energy density and ease of use in an already well-established infrastructure (cars, petrol stations, ships, cargo transport, mass transit systems, planes, etc.). The energy requirement for the production depends of the method used for capture of CO2 from large scale emitters and the method used for production of H2. However, the production of liquid carbon-based fuels from CO2 only reduces CO2 emissions if the energy used for the conversion is not based on fossil energy. The hydrogen needed for the reductive conversion of CO2 could be produced from water hydrolysis, using hydropower, nuclear energy, solar energy or wind energy. As long as some power generation using fossil fuels remains, CO2 for this conversion will be available.9 Alternatively, it might be possible to develop a recycling system, where CO2 is being captured from the atmosphere by biological or chemical means. This cycle would rely on the availability of cheap and abundant non-fossil energy and the same applies for the H2 economy.9 Even though the lifetime of methanol may only be days, this proposed closed cycle would ideally be CO2 emission neutral.Another area is the utilization of the physical aspects of CO2, which is done in enhanced oil recovery. As fossil fuel resources become scarce it is projected that enhanced oil recovery and enhanced coal bed methane recovery using supercritical CO2 will provide additional revenues for the oil and gas industry. This in turn will increase the demand for CO2, which could be provided from large stationary CO2 emitters. The storage time for the injected CO2 is expected to be 10000 years or more.9
However, in the near future improvements in the energy efficiency, a significant growth in renewable energies and further developments in emission free methods for separating and capturing CO2 from flue gases will help to stabilize the concentration of CO2 in the atmosphere.
10. Conclusions
This review has covered a number of CO2 transformations, which are all still in the research phase, but are potential technologies for mitigating the still-increasing atmospheric CO2 concentration. The utilization of CO2 as a raw material could well be a technology contributing to the reduction of the atmospheric CO2 loading in conjunction with the CCS technology. An example of this could be development of ways to recycle CO2via its chemical reduction with hydrogen to produce a liquid energy carrier such as methanol. The recycling would preferably be done from the atmosphere to deal with small and dispersed emitters, which contribute to more than half of the CO2 emission induced by humans. Recycling of CO2 from anthropogenic sources provides a renewable, inexhaustible carbon source and could allow the continued use of derived carbon fuels in an environmentally friendly, carbon neutral way. In principle when the recycling of CO2 becomes a feasible technology we would no longer rely on the diminishing and nonrenewable fossil fuels for our energy needs. However there are still many years of research ahead before the utilization of CO2 as a raw material becomes a CO2 mitigating technology, therefore efforts must be directed towards fast and low energy pathways for extraction and the utilization of CO2.| A | acceptor |
| acac | acetylacetonato |
| Ac | acetyl |
| Ar | aryl |
| bmim | 1-butyl-3-methylimidazolium |
| BMImCl | 1-n-butyl-3-methyl imidazolium chloride |
| BNAH | 1-benzyl-1,4-dihydronicotinamide |
| BOC | tert-butyloxycarbonyl |
| bpy | 2,2′-bipyridine |
| Bu | butyl |
| C | carbon |
| CB | conduction band |
| CCS | carbon dioxide storage and capture |
| CF | fluorocarbon |
| CFC | chlorofluorocarbons |
| CoA | acetyl-coenzyme A |
| COD | 1,5-cyclooctadiene |
| COT | cyclooctatetraene |
| cyclam | 1,4,8,11-tetraazacyclotetradecane |
| D | donor |
| DBAD | ditbutylazodicarboxylate |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-ene |
| DCC | dicyclohexyl carbodiimide |
| dcpb | 1,4-bis(dicyclohexylphosphino)butane |
| DEA | diethanolamine |
| DEC | diethyl carbonate |
| DMC | dimethyl carbonate |
| DMF | dimethylformamide |
| dppb | 1,4-bis(diphenylphosphino)butane |
| EOR | enhanced oil recovery |
| Et | ethyl |
| hfacac | hexafluoroacetylacetonate |
| Gt | gigatons |
| H2A | ascorbic acid |
| HMPA | hexamethylenephosphoric triamide |
| IPCC | Intergovernmental Panel on Climate Change |
| IPr | 1,3-bis-(2,6-diisopropylphenyl)-imidazole-2-ylidene |
| i-Pr | iso-propyl |
| IR | infrared radiation |
| L | ligand |
| M | metal |
| MDEA | methyldiethanolamine |
| Me | methyl |
| MEA | monoethanolamine |
| MEG | monoethylene glycol |
| Me2phen | 2,9-dimethyl-1,10-phenanthroline |
| MLCT | metal ligand charge transfer |
| M n | number average molecular weight |
| Mt | megatons |
| MV2+ | methylviologen |
| n | integer |
| NHE | normal hydrogen electrode |
| NMP | 1-methyl-2-pyrolidione |
| NMR | nuclear magnetic resonance |
| OTf | trifluoromethanesulfonato |
| PCC | precipitated calcium carbonate |
| PCy3 | tricyclohexylphosphine |
| Ph | phenyl |
| Py | pyridine |
| PVC | polyvinyl chloride |
| rt | room temperature |
| sc | supercritical |
| SCE | standard calomel electrode |
| TEA | triethylamine |
| TEG | triethylene glycol |
| TEOA | triethanolamine |
| Tf | trifluoromethanesulfonyl |
| TMS | trimethylsilyl |
| TOF | turnover frequency |
| TON | turnover number |
| TPP | 5,10,15,20-tetraphenylporphinato |
| tpy | 2,2′:6′,2′′-terpyridine |
| Ts | p-tolunenesulfonyl |
| usc | ultrafine single-crystal |
| VB | valence band |
| X | halogen |
12. Acknowledgements
This work was supported by The Danish Research Council for Technology and Production Sciences (FTP 274-05-0356).13. References
- B. Schrader, Eur. Environ., 2002, 12, 173 Search PubMed.
- H. Balat and C. Öz, Energy Explor. Exploit., 2007, 25, 357 Search PubMed.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975 RSC.
- M. M. Halmann and M. Steinberg, Greenhouse Gas Carbon Dioxide Mitigation: Science and Technology, Lewis Publishers, Boca Raton, Florida. 1999 Search PubMed.
- H. Yang, Z. Xu, M. Fan, R. Gupta, R. B. Slimane, A. E. Bland and I. Wright, J. Environ. Sci., 2008, 20, 14 Search PubMed.
- X. Xiaoding and J. A. Moulijn, Energy Fuels, 1996, 10, 305 CrossRef.
- T. Sakakura, J. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS.
- R. P. A. Sneeden, in Comprehensive Organometallic Chemistry, ed. G. Wilkinson, F. A. G. Stone, and A. W. Abel, Pergamon Press, New York, 1982, vol. 8, ch. 50.4, pp. 225 Search PubMed.
- Intergovernmental Panel on climate change Special report on Carbon Dioxide Capture and Storage, ed. B. Metz, O. Davidson, H. C. De Coninck, M. Loos and L. A. Meyer, Cambridge University Press, Cambridge, UK and New York, NY, USA, 2005. See: http://www.ipcc.ch/pdf/special-reports/srccs/srccs_wholereport.pdf Search PubMed.
- R. Zeverhoven, S. Eloneva and S. Teir, Catal. Today, 2006, 115, 73 CrossRef CAS.
- (a) Y. Ono, Catal. Today, 1997, 35, 15 CrossRef CAS; (b) M. A. Pachero and C. L. Marshall, Energy Fuels, 1997, 11, 2 CrossRef CAS.
- J. H. Messen and H. Petersen, 3rd ed., Ullmann's Encyclopedia of Industrial Chemistry, Vol. 37, Wiley-VCH, 2003, p. 683 Search PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487 CrossRef CAS.
- (a) A. Vatani, M. Mehrpooya and F. Gharagheizi, Int. J. Mol. Sci., 2007, 8, 407 Search PubMed; (b) M. Aresta, E. Quaranta and I. Tommasi, Energy Convers. Manage., 1992, 33, 495 CrossRef CAS.
- Y. Sun, in Proceedings of the Seventh International Conference on Carbon Dioxide Utilization, Seoul, Korea, October 2003, Stud. Surf. Sci. Catal., vol. 153, Elsevier, Amsterdam, 2004, p. 9 Search PubMed.
- J.-C. Choi, L.-N. He and T. Sakakura, Green Chem., 2002, 4, 230 RSC.
- T. Sakakura, Y. Saito, M. Okano, J.-C. Choi and T. Sako, J. Org. Chem., 1998, 63, 7095 CrossRef CAS.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC.
- (a) T. Sakakura, J.-C. Choi, Y. Saito, T. Masuda, T. Sako and T. Oriyama, J. Org. Chem., 1999, 64, 4506 CrossRef CAS; (b) J.-C. Choi, T. Sakakura and T. Sako, J. Am. Chem. Soc., 1999, 121, 3793 CrossRef CAS.
- J.-C. Choi, K. Kohno, Y. Ohshima, H. Yasuda and T. Sakakura, Catal. Commun., 2008, 9, 1630 CrossRef CAS.
- I. Omae, Catal. Today, 2006, 115, 33 CrossRef CAS.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951 CrossRef CAS.
- (a) Organic and Bio-organic Chemistry of Carbon Dioxide, ed. S. Inoue and Y. Yamazaki, Kodansha, Tokyo, 1982 Search PubMed; (b) Carbon Dioxide as a Source of Carbon, NATO ASI Ser., ed. M. Aresta and G. Forti, Reidel, Dordrect, The Netherlands, 1987 Search PubMed.
- L.-N. He, H. Yasuda and T. Sakakura, Green Chem., 2003, 5, 92 RSC.
- (a) M. Aresta, A. Dibenedetto and I. Tommasi, Appl. Organomet. Chem., 2000, 14, 799 CrossRef CAS; (b) M. Aresta and A. Dibenedetto, J. Mol. Catal. A: Chem., 2002, 182–183, 399 CrossRef CAS; (c) N. Eghbali and C. J. Li, Green Chem., 2007, 9, 213 RSC.
- (a) Y. Du, D. L. Kong, H. Y. Wang, F. Cai, H. S. Tian, J. Q. Wang and L. N. He, J. Mol. Catal. A: Chem., 2005, 241, 233 CrossRef CAS; (b) S. Y. Huang, S. G. Liu, J. P. Li, N. Zhao, W. Wei and Y. H. Sun, Catal. Lett., 2007, 118, 290 CrossRef CAS; (c) S. Y. Huang, J. Ma, J. P. Li, N. Zhao, W. Wei and Y. H. Sun, Catal. Commun., 2008, 9, 276 CrossRef CAS.
- M. Aresta, A. Dibenedetto, C. Dileo, I. Tommasi and E. Amodio, J. Supercrit. Fluids, 2003, 25, 177 CrossRef CAS.
- (a) K. Uemura, T. Kawaguchi, H. Takayama, A. Nakamura and Y. Inoue, J. Mol. Catal. A: Chem., 1999, 139, 1 CrossRef CAS; (b) W. Yamada, Y. Sugawara, H. M. Cheng, T. Ikeno and T. Yamada, Eur. J. Org. Chem., 2007, 2604 CrossRef CAS; (c) Y. Kayaki, M. Yamamoto and T. Ikariya, J. Org. Chem., 2007, 72, 647 CrossRef CAS; (d) H. F. Jiang, J. W. Zhao and A. H. Wang, Synthesis, 2008, 763 CrossRef CAS.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618 CrossRef CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388 CrossRef CAS.
- (a) S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287 CrossRef CAS; (b) S. Inoue, Chemtech, 1976, Sept., 588 Search PubMed; (c) S. Inoue, M. Kobayashi, H. Koinuma and T. Tsuruta, Makromol. Chem., 1972, 155, 61 CrossRef CAS; (d) M. Kobayashi, Y.-L. Tang, T. Tsuruta and S. Inoue, Makromol. Chem., 1973, 169, 69 CrossRef CAS; (e) S. Inoue, J. Macromol. Sci., Part A: Pure Appl. Chem., 1979, 13, 651 Search PubMed.
- S. Inoue, H. Koinuma and T. Tsuruta, Makromol. Chem., 1969, 130, 210 CrossRef CAS.
- W. Kuran and T. Listos, Macromol. Chem. Phys., 1994, 195, 977 CrossRef CAS.
- (a) D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911 CrossRef CAS; (b) D. Walther, K. Wermann, M. Lutsche, W. Gunther, H. Gorls and E. Anders, J. Org. Chem., 2006, 71, 1399 CrossRef CAS; (c) D. J. Darensbourg, S. J. Lewis, J. L. Rodgers and J. C. Yarbrough, Inorg. Chem., 2003, 42, 581 CrossRef CAS; (d) D. J. Darensbourg and M. W. Holtcamp, Macromolecules, 1995, 28, 7577 CrossRef CAS; (e) Y. L. Xiao, Z. Wang and K. L. Ding, Chem.–Eur. J., 2005, 11, 3668 CrossRef CAS; (f) M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 8738 CrossRef CAS; (g) W. J. van Meerendonk, R. Duchateau, C. E. Koning and G. J. M. Gruter, Macromolecules, 2005, 38, 7306 CrossRef CAS; (h) R. Eberhardt, M. Allmendinger, G. A. Luinstra and B. Rieger, Organometallics, 2003, 22, 211 CrossRef CAS; (i) S. D. Allen, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 14284 CrossRef CAS; (j) M. Kroger, C. Folli, O. Walter and M. Doring, Adv. Synth. Catal., 2005, 347, 1325 CrossRef; (k) B. Y. Lee, H. Y. Kwon, S. Y. Lee, S. J. Na, S. I. Han, H. S. Yun, H. Lee and Y. W. Park, J. Am. Chem. Soc., 2005, 127, 3031 CrossRef CAS; (l) D. J. Darensbourg, J. R. Wildeson, J. C. Yarbrough and J. H. Reibenspies, J. Am. Chem. Soc., 2000, 122, 12487 CrossRef CAS.
- (a) R. L. Paddock and S. T. Nguyen, Macromolecules, 2005, 38, 6251 CrossRef CAS; (b) Z. Q. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484 CrossRef CAS; (c) C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869 CrossRef CAS; (d) C. T. Cohen, C. M. Thomas, K. L. Peretti, E. B. Lobkovsky and G. W. Coates, Dalton Trans., 2006, 237 RSC; (e) X. B. Lu and Y. Wang, Angew. Chem., Int. Ed., 2004, 43, 3574 CrossRef CAS; (f) X. B. Lu, L. Shi, Y. M. Wang, R. Zhang, Y. J. Zhang, X. J. Peng, Z. C. Zhang and B. Li, J. Am. Chem. Soc., 2006, 128, 1664 CrossRef CAS.
- (a) D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers and A. L. Phelps, Inorg. Chem., 2004, 43, 1831 CAS; (b) D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers, C. C. Fang, D. R. Billodeaux and J. H. Reibenspies, Inorg. Chem., 2004, 43, 6024 CrossRef CAS; (c) D. J. Darensbourg and R. M. Mackiewicz, J. Am. Chem. Soc., 2005, 127, 14026 CrossRef CAS; (d) D. J. Darensbourg and J. C. Yarbrough, J. Am. Chem. Soc., 2002, 124, 6335 CrossRef CAS; (e) R. Eberhardt, M. Allmendinger and B. Rieger, Macromol. Rapid Commun., 2003, 24, 194 CrossRef CAS; (f) D. J. Darensbourg, R. M. Mackiewicz and D. R. Billodeaux, Organometallics, 2005, 24, 144 CrossRef CAS; (g) D. J. Darensbourg and A. L. Phelps, Inorg. Chem., 2005, 44, 4622 CrossRef CAS; (h) S. Mang, A. I. Cooper, M. E. Colclough, N. Chauhan and A. B. Holmes, Macromolecules, 2000, 33, 303 CrossRef CAS.
- (a) D. V. Vitanova, F. Hampel and K. C. Hultzsch, J. Organomet. Chem., 2005, 690, 5182 CrossRef CAS; (b) D. M. Cui, M. Nishiura and Z. M. Hou, Macromolecules, 2005, 38, 4089 CrossRef CAS.
- H. Sugimoto, H. Ohshima and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3549 CrossRef CAS.
- S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497 RSC.
- (a) S. Fukuoka, Chem. Ind., 1997, 6, 757; (b) S. Fukuoka, Eur. Chem. News, 1999, 15–21, 44 Search PubMed.
- A. Baba, H. Kashiwagi and H. Matsuda, Organometallics, 1987, 6, 137 CrossRef CAS.
- D. Chaturvedi and S. Ray, Curr. Org. Chem., 2007, 11, 987 CrossRef CAS.
- M. Yoshida, N. Hara and S. Okuyama, Chem. Commun., 2000, 151 RSC.
- (a) Y. Hori, Y. Nagano, J. Nakao, T. Fukuhara and H. Taniguschi, Chem. Expr., 1986, 224 Search PubMed; (b) W. D. McGhee, Y. Pan and D. P. Riley, Chem. Commun., 1996, 699 RSC; (c) W. D. McGhee, D. P. Riley, K. Christ, Y. Pan and B. Parnas, J. Org. Chem., 1995, 60, 2820 CrossRef CAS.
- M. Aresta and E. Quaranta, Tetrahedron, 1992, 48, 1515 CrossRef CAS.
- K. J. Butcher, Synlett, 1994, 825 CrossRef CAS.
- M. Alba, J.-C. Choi and T. Sakakura, Chem. Commun., 2001, 2238 RSC.
- F. Shi, Y. Q. Deng, T. L. SiMa, J. J. Peng, Y. L. Gu and B. T. Qiao, Angew. Chem., Int. Ed., 2003, 42, 3257 CrossRef.
- (a) W. D. McGhee, D. P. Riley and M. K. M. Christ, Organometallics, 1993, 12, 1429 CrossRef CAS; (b) M. Aresta and E. Quaranta, Chem. Tech., 1997, 32 CAS; (c) P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259 CrossRef CAS.
- V. L. K. Valli and H. Alper, J. Org. Chem., 1995, 60, 257 CrossRef CAS.
- (a) T. E. Waldman and W. D. McGhee, J. Chem. Soc., Chem. Commun., 1994, 957 RSC; (b) D. Saylik, M. J. Horvath, P. S. Elmes, W. R. Jackson, C. G. Lovel and K. Moody, J. Org. Chem., 1999, 64, 3940 CrossRef CAS.
- (a) H. Kawanami, H. Matsumoto and Y. Ikushima, Chem. Lett., 2005, 34, 60 CrossRef CAS; (b) A. W. Miller and S. T. Nguyen, Org. Lett., 2004, 6, 2301 CrossRef CAS; (c) A. Sudo, Y. Morioka, F. Sanda and T. Endo, Tetrahedron Lett., 2004, 45, 1363 CrossRef CAS; (d) A. Sudo, Y. Morioka, E. Koizumi, F. Sanda and T. Endo, Tetrahedron Lett., 2003, 44, 7889 CrossRef CAS; (e) M. Shi, J. K. Jiang, Y. M. Shen, Y. S. Feng and G. X. Lei, J. Org. Chem., 2000, 65, 3443 CrossRef CAS.
- P. Tascedda and E. Duñach, Chem. Commun., 2000, 449 RSC.
- K. I. Tominaga and Y. Sasaki, Synlett, 2002, 307 CrossRef CAS.
- (a) O. Mitsunobu, Synthesis, 1981, 1 CrossRef CAS; (b) D. L. Huges, Org. Prep. Proced. Int., 1996, 28, 127 CrossRef CAS.
- C. J. Dinsmor and S. P. Mercer, Org. Lett., 2004, 6, 2885 CrossRef CAS.
- (a) Y. Sasaki and P. H. Dixneuf, J. Chem. Soc., Chem. Commun., 1986, 790 RSC; (b) R. Mahé and P. H. Dixneuf, Tetrahedron Lett., 1986, 27, 6333 CrossRef CAS.
- T. Mitsudo, Y. Hori, Y. Yamakawa and Y. Watanabe, Tetrahedron Lett., 1987, 28, 4417 CrossRef CAS.
- (a) O. Ihata, Y. Kayaki and T. Ikariya, Chem. Commun., 2005, 2268 RSC; (b) B. Ochiai, S. Inoue and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6613 CrossRef CAS; (c) O. Ihata, Y. Kayaki and T. Ikariya, Macromolecules, 2005, 38, 6429 CrossRef CAS; (d) O. Ihata, Y. Kayaki and T. Ikariya, Angew. Chem., Int. Ed., 2004, 43, 717 CrossRef CAS.
- D. B. Dell'Amico, F. Calderazzo, L. Labella, F. Marchetti and G. Pampaloni, Chem. Rev., 2003, 103, 3857 CrossRef.
- F. Kojima, T. Aida and S. Inoue, J. Am. Chem. Soc., 1986, 108, 391 CrossRef CAS.
- N. Saito, K. Hatakeda, S. Ito, T. Asano and T. Toda, Bull. Chem. Soc. Jpn., 1986, 59, 1629 CrossRef CAS.
- A. Behr, Carbon Dioxide Activated by Metal Complexes, VCH, Weinheim, Germany, 1988 Search PubMed.
- (a) N. P. Mankad, T. G. Gray, D. S. Laitar and J. P. Sadighi, Organometallics, 2004, 23, 1191 CrossRef CAS; (b) K. Chiba, H. Tagaya, S. Miura and M. Karasu, Chem. Lett., 1992, 923 CAS; (c) H. Abe and S. H. Inoue, J. Chem. Soc., Chem. Commun., 1994, 1197 RSC; (d) R. P. Quirk, J. Yin, L. J. Fetters and R. V. Kastrup, Macromolecules, 1992, 25, 2262 CrossRef CAS.
- (a) E. R. Perez, R. H. A. Santos, M. T. P. Gambardellea, L. G. M. de Macedo, U. P. Rodrigues, J. C. Launay and D. W. Franco, J. Org. Chem., 2004, 69, 8005 CrossRef CAS; (b) D. J. Heldebrant, P. G. Jessop, C. A. Thomas, C. A. Eckert and C. L. Liotta, J. Org. Chem., 2005, 70, 5335 CrossRef CAS.
- S. Saito, S. Nakagawa, T. Koizumi, K. Hirayama and Y. Yamamoto, J. Org. Chem., 1999, 64, 3975 CrossRef CAS.
- M. Takimoto and M. Mori, J. Am. Chem. Soc., 2001, 123, 2895 CrossRef CAS.
- M. Takimoto, M. Kawamura and M. Mori, Org. Lett., 2003, 5, 2599 CrossRef CAS.
- J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2008, 130, 15254 CrossRef CAS.
- E. Haruki, T. Hara and H. Inoue, Chem. Expr., 1990, 5, 493 Search PubMed.
- M. Zerella, S. Mukhopadhyay and A. T. Bell, Org. Lett., 2003, 5, 3193 CrossRef CAS.
- (a) Y. Fujiwara, Y. Taniguchi, K. Takaki, M. Kurioka, T. Jintoku and T. Kitamura, Stud. Surf. Sci. Catal., 1997, 107, 275 CAS; (b) Y. Taniguchi, T. Hayashida, H. Shibasaki, D. Piao, T. Kitamura, T. Yamaji and Y. Fujiwara, Org. Lett., 1999, 1, 557 CrossRef CAS; (c) Y. Fujiwara, K. Takaki and Y. Taniguchi, Synlett, 1996, 591 CrossRef CAS.
- G. A. Olah, B. Törok, J. P. Joschek, I. Bucsi, P. M. Esteves, G. Rasul and G. K. Surya Prakash, J. Am. Chem. Soc., 2002, 124, 11379 CrossRef CAS.
- K. Shiraishi, J.-C. Choi and T. Sakakura, 50th Symposium on Organometallic Chemistry, Osaka, Japan, September 28–30, 2003, Kinki Chemical Society, Abstract B304 Search PubMed.
- (a) A. Behr and M. Heite, Chem. Ing. Tech., 2000, 72, 58 CrossRef CAS; (b) A. Behr and V. A. Brehme, J. Mol. Catal. A: Chem., 2002, 187, 69 CrossRef CAS; (c) A. Behr and K. D. Juszak, J. Organomet. Chem., 1983, 255, 263 CrossRef CAS.
- (a) Y. Inoue, Y. Itoh, H. Kazama and H. Hasimoto, Bull. Chem. Soc. Jpn., 1980, 53, 3329 CAS; (b) H. Hoberg, D. Schefer, G. Burkhart, C. Kruger and M. J. Romao, J. Organomet. Chem., 1984, 266, 203 CrossRef CAS.
- T. Tsuda, T. Yamamoto and T. Saegusa, J. Organomet. Chem., 1992, 429, C46 CrossRef CAS.
- (a) T. Tsuda, H. Yasukawa and K. Komori, Macromolecules, 1995, 28, 1356 CrossRef CAS; (b) T. Tsuda, H. Yasukawa, H. Hokazono and Y. Kitaike, Macromolecules, 1995, 28, 1312 CrossRef CAS; (c) T. Tsuda and H. Hokazono, Macromolecules, 1994, 27, 1289 CrossRef CAS; (d) T. Tsuda, O. Ooi and K. Maruta, Macromolecules, 1993, 26, 4840 CrossRef CAS; (e) T. Tsuda, Y. Kitaike and O. Ooi, Macromolecules, 1993, 26, 4956 CrossRef CAS; (f) T. Tsuda, K. Maruta and Y. Kitaike, J. Am. Chem. Soc., 1992, 114, 1498 CrossRef CAS; (g) T. Tsuda and K. Maruta, Macromolecules, 1992, 25, 6102 CrossRef CAS; (h) T. Tsuda, S. Morikawa, R. Sumiya and T. Saegusa, J. Org. Chem., 1988, 53, 3140 CrossRef CAS; (i) T. Tsuda, S. Morikawa, N. Hasegawa and T. Saegusa, J. Org. Chem., 1990, 55, 2978 CrossRef CAS.
- S. Oi, Y. Fukue, K. Nemoto and Y. Inoue, Macromolecules, 1996, 29, 2694 CrossRef CAS.
- (a) J. Louie, J. E. Gibby, M. V. Farnworth and T. N. Tekavec, J. Am. Chem. Soc., 2002, 124, 15188 CrossRef CAS; (b) T. N. Tekavec, A. M. Arif and J. Louie, Tetrahedron, 2004, 60, 7431 CrossRef CAS.
- D. H. Gibson, Chem. Rev., 1996, 96, 2063 CrossRef CAS.
- K. Meyer and I. Castro Rodriguez, J. Am. Chem. Soc., 2005, 127, 11242 CrossRef CAS.
- J. C. Calabrese, T. Herskovitz and J. B. Kinney, J. Am. Chem. Soc., 1983, 105, 5914 CrossRef CAS.
- T. Herskovitz, J. Am. Chem. Soc., 1977, 99, 2391 CrossRef.
- (a) M. Aresta and C. F. Nobile, J. Chem. Soc., Dalton Trans., 1977, 708 RSC; (b) M. G. Mason and J. A. Ibers, J. Am. Chem. Soc., 1982, 104, 5153 CrossRef CAS; (c) A. Doehring, P. W. Jolly, C. Krueger and M. Romao, Z. Naturforsch., B, 1985, 40, 484.
- M. Aresta and C. F. Nobile, Inorg. Chim. Acta, 1977, 24, L49 CrossRef CAS.
- (a) H. H. Karsch, Chem. Ber., 1977, 110, 2213 CAS; (b) S. Komiya, M. Akita, N. Kasuga, V. Hirano and A. Fukuoka, J. Chem. Soc., Chem. Commun., 1994, 1115 RSC.
- M. Sakamoto, L. Shimizu and A. Yamamoto, Organometallics, 1994, 13, 407 CrossRef CAS.
- (a) S. M. Tetrick, F. S. Thom and A. R. Cutler, J. Am. Chem. Soc., 1997, 119, 6193 CrossRef CAS; (b) S. M. Tetrick, C. Xu, J. R. Pinkes and A. R. Cutler, Organometallics, 1998, 17, 1861 CrossRef CAS.
- T. A. Hanna, A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 3292 CrossRef CAS.
- J. D. Audett, T. J. Collins, B. D. Santarsiero and G. H. Spies, J. Am. Chem. Soc., 1982, 104, 7352 CrossRef CAS.
- C. P. Kubiak, C. Woodcock and R. Eisenberg, Inorg. Chem., 1982, 21, 2119 CrossRef CAS.
- (a) J. S. Field, R. J. Haines, J. Sundermeyer and S. F. Woollam, J. Chem. Soc., Chem. Commun., 1990, 985 RSC; (b) J. S. Field, R. J. Haines, J. Sundermeyer and S. F. Woollam, J. Chem. Soc., Dalton Trans., 1993, 2735 RSC.
- (a) C. Floriani and G. Fachinetti, J. Chem. Soc., Chem. Commun., 1974, 615 RSC; (b) G. Fachinetti, C. Floriani and P. F. Zanazzi, J. Am. Chem. Soc., 1978, 100, 7405 CrossRef CAS; (c) S. Gambarotta, F. Arena, C. Floriani and P. F. Zanazzi, J. Am. Chem. Soc., 1982, 104, 5082 CrossRef CAS.
- C. T. Tso and A. R. Cutler, J. Am. Chem. Soc., 1986, 108, 6069 CrossRef CAS.
- (a) J. C. Vites, B. D. Steffey, M. E. Giuseppetti-Dery and A. R. Cutler, Organometallics, 1991, 10, 2827 CrossRef CAS; (b) J. R. Pinkes, B. D. Steffey, J. C. Vites and A. R. Cutler, Organometallics, 1994, 13, 21 CrossRef CAS.
- D. R. Senn, J. A. Gladysz, K. Emerson and R. D. Larsen, Inorg. Chem., 1987, 26, 2737 CrossRef CAS.
- D. H. Gibson, M. Ye, B. A. Sleadd, J. M. Mehta, O. P. Mbadike, J. F. Richardson and M. S. Mashuta, Organometallics, 1995, 14, 1242 CrossRef CAS.
- (a) C. R. Eady, J. J. Guy, B. F. G. Johnson, J. Lewis, M. C. Malatesta and G. M. Sheldrick, J. Chem. Soc., Chem. Commun., 1976, 602 RSC; (b) G. R. John, B. F. G. Johnson, J. Lewis and K. C. Wong, J. Organomet. Chem., 1979, 169, C23 CrossRef CAS.
- B. K. Balbach, F. Helus, F. Oberdorfer and M. L. Ziegler, Angew. Chem., 1981, 93, 479 CrossRef CAS.
- S. Sakaki and Y. Musashi, Inorg. Chem., 1995, 34, 1914 CrossRef CAS.
- D. H. Gibson, Coord. Chem. Rev., 1999, 185–186, 335 CrossRef CAS.
- D. H. Gibson, Compr. Coord. Chem. II, 2004, 1, 595 Search PubMed.
- E. G. Lundquist, J. C. Huffman, K. Folting, B. E. Mann and K. G. Caulton, Inorg. Chem., 1990, 29, 128 CrossRef CAS.
- X. Yin and J. R. Moss, Coord. Chem. Rev., 1999, 181, 27 CrossRef CAS.
- P. G. Jessop, F. Joó and C. C. Tai, Coord. Chem. Rev., 2004, 248, 2425 CrossRef CAS.
- H. Arakawa, in Advances in Chemical Conversions for Mitigating Carbon Dioxide, Stud. Surf. Sci. Catal., ed. T. Inui, M. Anpo, K. Izui, S. Yanagida and T. Yamaguchi, Elsevier Science, New York, 1998, 114, p. 19 Search PubMed.
- (a) M. Saito and K. Murata, Catal. Surv. Asia, 2004, 8, 285 CrossRef CAS; (b) M. Saito, Catal. Surv. Jpn., 1998, 2, 175 CrossRef CAS; (c) M. Saito, M. Takeuchi, T. Fujitani, J. Toyir, S. Luo, J. Wu, H. Mabuse, K. Ushikoshi, K. Mori and T. Watanabe, Appl. Organomet. Chem., 2000, 14, 763 CrossRef CAS; (d) X. An, J. Li, Y. Zuo, Q. Zhang, D. Wang and J. Wang, Catal. Lett., 2007, 118, 264 CrossRef CAS.
- J. Ivy, Summary of Electrolytic Hydrogen Production, Milestone Completion Report, NREL/MP-560-35948, NREL, Golden, CO, April 2004 Search PubMed.
- M. Steinberg, Int. J. Hydrogen Energy, 1999, 24, 771 CrossRef CAS.
- (a) L. Pettersson and K. Sjöström, Combust. Sci. Technol., 1991, 80, 265 CrossRef CAS; (b) C. B. von der Decken, H. Dötsch, B. Höhlein, H. Fedders, E. Riensche, P. Bröckerhoff, F. Pischinger, U. Hilger, H. Barnert, H. Niesen and M. Walbeck, Energy Alcohols, Production and Application of a Synthetic Liquid Energy Carrier, Institut für Reaktorbauelemente, Kernforschungsanlage, Jülich, Germany, 1987 (ISSN 0343–7639) Search PubMed; (c) D. Sperling, New Transportation Fuels: A Strategic Approach to Technological Change, University of California Press, Berkeley, CA, 1989 Search PubMed; (d) C. L. Gray Jr and J. A. Alson, Sci. Am., 1989, Nov, 108 Search PubMed.
- A. Behr, Chem. Eng. Technol., 1987, 10, 16 CrossRef.
- K. Tominaga, Catal. Today, 2006, 115, 70 CrossRef CAS.
- (a) K. Tominaga, Y. Sasaki, M. Saito, K. Hagihara and T. Watanabe, J. Mol. Catal., 1994, 89, 51 CrossRef CAS; (b) K. Tominaga, Y. Sasaki, T. Watanabe and M. Saito, Stud. Surf. Sci. Catal., 1998, 114, 495 CAS.
- A. Baiker, Appl. Organomet. Chem., 2000, 14, 751 CrossRef CAS.
- (a) P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231 CrossRef CAS; (b) P. G. Jessop, J. Supercrit. Fluids, 2006, 38, 211 CrossRef CAS.
- (a) P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 344 CrossRef CAS; (b) P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Chem. Soc., Chem. Commun., 1995, 707 RSC.
- P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1994, 116, 8851 CrossRef CAS.
- W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207 CrossRef CAS.
- T. J. Meyer, Acc. Chem. Res., 1989, 22, 163 CrossRef CAS.
- V. Balzani, A. Credi and M. Venturi, Chem. Sus. Chem., 2008, 1, 26 CrossRef CAS.
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953 CrossRef CAS.
- (a) J.-M. Lehn and R. Ziessel, J. Organomet. Chem., 1990, 382, 157 CrossRef CAS; (b) N. Kitamura and S. Tazuke, Chem. Lett., 1983, 1109 CrossRef CAS.
- (a) J. Hawecker, J. M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990 CrossRef CAS; (b) R. Ziessel, In Catalysis by Metal Complexes: Photosensitization and Photocatalysis Using Inorganic and Organometallic Compounds, ed. K. Kalyanasundaram and M. Grätzel, Kluwer Academic, Dordrecht, The Netherlands, 1993, p 217 Search PubMed; (c) H. Ishida, K. Tanaka and T. Tanaka, Chem. Lett., 1988, 339 CrossRef CAS; (d) H. Ishida, T. Terada, K. Tanaka and T. Tanaka, Inorg. Chem., 1990, 29, 905 CrossRef CAS; (e) J. L. Grant, K. Goswami, L. O. Spreer, J. W. Otvos and M. Calvin, J. Chem. Soc., Dalton Trans., 1987, 2105 RSC; (f) C. A. Craig, L. O. Spreer, J. W. Otvos and M. Calvin, J. Phys. Chem., 1990, 94, 7957 CrossRef CAS; (g) E. Kimura, S. Wada, M. Shionoya and Y. Okazaki, Inorg. Chem., 1994, 33, 770 CrossRef CAS; (h) K. Mochizuki, S. Manaka, I. Takeda and T. Kondo, Inorg. Chem., 1996, 35, 5132 CrossRef CAS.
- (a) C. Kutal, M. A. Weber, G. Ferraudi and D. Geiger, Organometallics, 1985, 4, 2161 CrossRef CAS; (b) C. Kutal, A. J. Corbin and G. Ferraudi, Organometallics, 1987, 6, 553 CrossRef CAS; (c) H. Hori, F. P. A. Johnson, K. Koike, O. Ishitani and T. Ibusuki, J. Photochem. Photobiol., A, 1996, 96, 171 CrossRef CAS.
- (a) R. Maidan and I. Willner, J. Am. Chem. Soc., 1986, 108, 8100 CrossRef CAS; (b) I. Willner, R. Maidan, D. Mandler, H. Dürr, G. Dörr and K. Zengerle, J. Am. Chem. Soc., 1987, 109, 6080 CrossRef CAS.
- (a) J. Grodkowski, D. Behar, P. Neta and P. Hambright, J. Phys. Chem. A, 1997, 101, 248 CrossRef CAS; (b) D. Behar, T. Dhanasekaran, P. Neta, C. M. Hosten, D. Ejeh, P. Hambright and E. Fujita, J. Phys. Chem. A, 1998, 102, 2870 CrossRef CAS; (c) T. Dhanasc Kaman, J. Gradowski, P. Neta, P. Hambright and E. Fujita, J. Phys. Chem., 1999, 103, 7742 Search PubMed.
- H. Gunardson, Industrial Gases in Petrochemical Processing, Marcel Dekker, New York, 1989 Search PubMed.
- E. Fujita, Coord. Chem. Rev., 1999, 185–186, 373 CrossRef CAS.
- J.-M. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 701 CAS.
- R. Ziessel, J. Hawecker and J.-M. Lehn, Helv. Chim. Acta, 1986, 69, 1065 CAS.
- H. Ishida, K. Tanaka and T. Tanaka, Organometallics, 1987, 6, 181 CrossRef CAS.
- A. T. A. Tinnemans, T. P. M. Koster, D. H. M. W. Thewissen and A. Mackor, Recl. Trav. Chim. Pays Bas, 1984, 103, 288 CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536 RSC.
- S. Matsuoka, K. Yamamoto, C. Pac and S. Yanagida, Chem. Lett., 1990, 2099.
- S. Matsuoka, K. Yamamoto, T. Ogata, M. Kusaba, N. Nakashima, E. Fujita and S. Yanagida, J. Am. Chem. Soc., 1993, 115, 601 CrossRef CAS.
- T. Ogata, S. Yanagida, B. S. Brunschwig and E. Fujita, J. Am. Chem. Soc., 1995, 117, 6708 CrossRef CAS.
- T. Ogata, Y. Yamamoto, Y. Wada, K. Murakoshi, M. Kusaba, N. Nakashima, A. Ishida, S. Takamuku and S. Yanagida, J. Phys. Chem., 1995, 99, 11916 CrossRef CAS.
- (a) G. A. Crosby, Acc. Chem. Res., 1975, 8, 231 CrossRef CAS; (b) J. Ferguson, F. Herren, E. R. Krausz and J. Vrbanich, Coord. Chem. Rev., 1985, 64, 21 CrossRef CAS; (c) T. J. Meyer, Pure Appl. Chem., 1986, 58, 1193 CrossRef CAS; (d) V. Balzani, F. Bolleta, M. T. Gandolfi and M. Maestri, Top. Curr. Chem., 1978, 75, 1 CAS; (e) K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159 CrossRef; (f) A. Jursi, F. Barigelletti, S. Campagna, V. Balzani, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- I. Willner, and B. Willner, in Artificial Photosynthetic Model Systems Using Light Induced Electron Transfer Reactions in Catalytic and Biocatalytic Assemblies, in Top. Curr. Chem., Photoinduced Electron Transfer III, ed. J. Mattay, Springer-Verlag, Berlin Heidelberg, 1991, vol. 159, pp. 153–218 Search PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89 RSC.
- F. R. Keene, in Electrochemical and Electrocatalytic Reactions of Carbon Dioxide, ed. B. P. Sullivan, K. Krist and H. E. Guard, Elsevier, Amsterdam, Holland, 1993, 1 Search PubMed.
- C. Creutz and E. Fujita, in Carbon Dioxide as a Feedstock, Carbon Management: Implications for R&D in Chemical Science and Technology, A Workshop Report to the Chemical Sciences Roundtable, National Academy Press 2001, pp. 83 Search PubMed.
- (a) M. Jitaru, D. A. Lowy, M. Toma and L. Oniciu, J. Appl. Electrochem., 1997, 27, 875 CrossRef CAS; (b) M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1 CrossRef CAS; (c) S. Kaneco, K. Iiba, S. K. Suzuki, K. Ohta and T. Mizuno, J. Phys. Chem. B, 1999, 103, 7456 CrossRef CAS; (d) S. Kaneco, R. Iwao, K. Iiba, S. I. Itoh, K. Ohata and T. Mizuno, Environ. Eng. Sci., 1999, 16, 131 CrossRef CAS; (e) S. Kaneco, H. Katsumata, T. Suzuki and K. Ohta, Chem. Eng. J., 2006, 116, 227 CrossRef CAS.
- Y. Hori, in Handbook of Fuel Cells, ed. W. Vielstich, A. Lamm and H. A. Gasteiger, Wiley, West Sussex, England, 2003, 2, pp. 720–733 Search PubMed.
- G. A. Olah and G. K. S. Prakash, Electrolysis of Carbon Dioxide in Aqueous Media to Carbon Monoxide and Hydrogen for Production of Methanol, US Provisional Pat. Appl. 60/949,723, 2007 Search PubMed.
- B. R. Eggins, C. Ennis, R. McConnell and M. Spence, J. Appl. Electrochem., 1997, 27, 706 CrossRef CAS.
- (a) Y. Hori and A. Murata, Electrochim. Acta, 1990, 35, 1777 CrossRef CAS; (b) S. Taguchi and A. Aramata, Electrochim. Acta, 1994, 39, 2533 CrossRef CAS.
- (a) Y. Hori, K. Kikuchi and S. Suzuki, Chem. Lett., 1985, 1695 CAS; (b) Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833 CrossRef CAS; (c) M. Azuma, M. Hashimoto, M. Hiramoto, M. Watanbe and T. Sakata, J. Electrochem. Soc., 1990, 137, 1772 CAS.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 897 CAS.
- J.-M. Savéant, Chem. Rev., 2008, 108, 2348 CrossRef CAS.
- A. Hattori, S. Ikeda, M. Maeda, H. Einaga and K. Ito, Electrochemistry, 2000, 68, 257.
- (a) M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315 RSC; (b) M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461 CrossRef CAS; (c) J. P. Collin, A. Jouaiti and J. P. Sauvage, Inorg. Chem., 1988, 27, 1986 CrossRef CAS.
- I. Bhugun, D. Lexa and J. M. Saveant, J. Am. Chem. Soc., 1996, 118, 1769 CrossRef CAS.
- J. Hawecker, J. M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328 RSC.
- C. M. Bolinger, N. Story, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1988, 27, 4582 CrossRef CAS.
- (a) H. Nagao, T. Mizukawa and K. Tanaka, Chem. Lett., 1993, 955 CAS; (b) H. Nagao, T. Mizukawa and K. Tanaka, Inorg. Chem., 1994, 33, 3415 CrossRef CAS; (c) K. Toyohara, K. Tsuge and K. Tanaka, Organometallics, 1995, 14, 5099 CrossRef CAS; (d) D. Ooyama, T. Tomon, K. Tsuge and K. Tanaka, J. Organomet. Chem., 2001, 619, 299 CrossRef CAS; (e) K. Toyohara, H. Nagao, T. Mizukawa and K. Tanaka, Inorg. Chem., 1995, 34, 5399 CrossRef CAS.
- (a) D. L. Dubois, Comments Inorg. Chem., 1997, 19, 307 CAS; (b) D. L. Dubois, A. Miedaner and R. C. Haltiwanger, J. Am. Chem. Soc., 1991, 113, 8753 CrossRef CAS; (c) J. W. Raebiger, J. W. Turner, B. C. Noll, C. J. Curtis, A. Miedaner, B. Cox and D. L. Dubois, Organometallics, 2006, 25, 3345 CrossRef CAS.
- K. Sugimura, S. Kuwababa and H. Yoneyama, J. Am. Chem. Soc., 1989, 111, 2361 CrossRef CAS.
- P. Usubharatana, D. McMartin, A. Veawab and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2006, 45, 2558 CrossRef CAS.
- T. Inoue, A. Fujisima, S. Konishi and K. Honda, Nature, 1979, 277, 637.
- A. Mackor, A. H. A. Tinnemans and T. P. M. Koster, in Carbon dioxide as a source of carbon: Chemical and biochemical uses, Nato ASI Ser., Ser. C, ed. M. Aresta, and G. Forti, D. Reidel, Publishing Company, Dordrecht, Holland, 1987 Search PubMed.
- K. Kabra, R. Chaudhary and R. L. Sawhney, Ind. Eng. Chem. Res., 2004, 43, 7683 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735 CrossRef CAS.
- J. Bussi, M. Ohanian, M. Vazquez and D. A. Dalchiele, J. Environ. Eng., 2002, 128, 733 CrossRef CAS.
- (a) M. Kanemoto, K.-I. Ishihara, Y. Wada, T. Sakata, H. Mori and S. Yanagida, Chem. Lett., 1992, 835 CAS; (b) H. Fujiwara, H. Hosokawa, K. Murakoshi, Y. Wada, S. Yanagida, T. Okada and H. Kobayashi, J. Phys. Chem. B, 1997, 101, 8270 CrossRef CAS; (c) M. Kanemoto, M. Nomura, Y. Wada, T. Akano and S. Yanagida, Chem. Lett., 1993, 1687 CAS.
- P. Johne and H. Kisch, J. Photochem. Photobiol., A, 1997, 111, 223 CrossRef CAS.
- B.-J. Liu, T. Torimoto and H. Yoneyama, J. Photochem. Photobiol., A, 1998, 115, 227 CrossRef CAS.
- S. Kaneco, Y. Shimizu, K. Ohta and T. Mizuno, J. Photochem. Photobiol., A, 1998, 115, 223 CrossRef CAS.
- S. M. Aliwi and K. F. Aljubori, Sol. Energy Mater., 1989, 18, 223 CrossRef CAS.
- Y. Kohno, T. Tanaka, T. Funabiki and S. Yoshida, Phys. Chem. Chem. Phys., 2000, 2, 2635 RSC.
- Y. Kohno, T. Tanaka, T. Funabiki and S. Yoshida, Phys. Chem. Chem. Phys., 2000, 2, 5302 RSC.
- (a) M. Anpo, H. Yamashita, Y. Ichihashi, Y. Fujii and M. Honda, J. Phys. Chem. B, 1997, 101, 2632 CrossRef CAS; (b) M. Subrahmanyam, S. Kaneco and N. Alonso-Vante, Appl. Catal., B, 1999, 23, 169 CrossRef; (c) T. Xie, D. Wang, L. Zhu, T. Li and Y. Xu, Mater. Chem. Phys., 2001, 70, 103 CrossRef CAS; (d) X.-H. Xia, Z.-J. Jia, Y. Yu, Y. Liang, Z. Wang and L.-L. Ma, Carbon, 2007, 45, 717 CrossRef CAS; (e) S. S. Tan, L. Zou and E. Hu, Catal. Today, 2006, 115, 269 CrossRef CAS; (f) S. S. Tan, L. Zou and E. Hu, Sci. Technol. Adv. Mater., 2007, 8, 89 CrossRef; (g) N. Sasirekha, S. J. S. Basha and K. Shanthi, Appl. Catal., B, 2006, 62, 169 CrossRef CAS; (h) J. C. S. Wu, H.-M. Lin and C.-L. Lai, Appl. Catal., A, 2005, 296, 194 CrossRef CAS; (i) I.-H. Tseng, W.-C. Chang and J. C. S. Wu, Appl. Catal., B, 2002, 37, 37 CrossRef CAS; (j) B.-J. Liu, T. Torimoto, H. Matsumoto and H. Yoneyama, J. Photochem. Photobiol., A, 1997, 108, 187 CrossRef CAS; (k) K. Adachi, K. Ohta and M. Mizuno, Sol. Energy, 1994, 53, 187 CrossRef CAS; (l) M. Anpo, H. Yamashita, Y. Ichinashi and S. Ehara, J. Electroanal. Chem., 1995, 396, 21 CrossRef CAS; (m) H. Yamashita, A. Shiga, S. Kawasaki, Y. Ichihashi, S. Ehara and M. Anpo, Energy Convers. Manage., 1995, 36, 617 CrossRef CAS; (n) T. Mizuno, K. Adachi, K. Ohta and A. Saji, J. Photochem. Photobiol., A, 1996, 98, 87 CrossRef CAS; (o) H. Yamashita, Y. Fujii, Y. Ichinashi, S. G. Zhang, K. Ikeue, D. R. Park, K. Koyano, T. Tatsumi and M. Anpo, Catal. Today, 1998, 45, 221 CrossRef CAS.
- N. Gokon, N. Hasegawa, H. Kaneko, H. Aoki, Y. Tamaura and M. Kitamura, Sol. Energy Mater. Sol. Cells, 2003, 80, 335 CrossRef CAS.
- (a) K. Sayama and H. Arakawa, J. Phys. Chem., 1993, 97, 531 CrossRef CAS; (b) Y. Kohno, T. Tanaka, T. Funabiki and S. Yoshida, Chem. Commun., 1997, 841 RSC.
- H. Yoneyama, Y. Yamashita and H. Tamura, Nature, 1979, 282, 817 CAS.
- Y. Kohno, H. Hayashi, S. Takenaka, T. Tanaka, T. Funabiki and S. Yoshida, J. Photochem. Photobiol., A, 1999, 126, 117 CrossRef CAS.
- Y. Kohno, H. Ishikawa, T. Tanaka, T. Funabiki and S. Yoshida, Phys. Chem. Chem. Phys., 2001, 3, 1108 RSC.
- Y. Shiraishi and T. Hirai, J. Photochem. Photobiol., C, 2008, 9, 157 CrossRef CAS.
- (a) G. R. Dey, A. D. Belapurkar and K. Kisshore, J. Photochem. Photobiol., A, 2004, 163, 503 CrossRef CAS; (b) A. H. Yahaya, M. A. Gondal and A. Hameed, Chem. Phys. Lett., 2004, 400, 206 CrossRef CAS; (c) F. Saladin, L. Forss and I. Kamber, J. Chem. Soc., Chem. Commun., 1995, 533 RSC; (d) Slamet, H. W. Nasution, E. Purnama, S. Kosela and J. Gunlazuardi, Catal. Commun., 2005, 6, 313 CrossRef.
- (a) K. Ikeue, H. Yamashita and M. Anpo, Chem. Lett., 1999, 1135 CrossRef CAS; (b) K. Ikeue, H. Yamashita, M. Anpo and T. Takewaki, J. Phys. Chem. B, 2001, 105, 8350 CrossRef CAS; (c) H. Yamashita, N. Nishiguchi, N. Kamada, M. Anpo, H. Hatano, K. Kikui, Y. Teraoka, S. Kagawa, S. Ehara, L. Palmisano, A. Sclafani, M. Schiavello and M. A. Fox, Res. Chem. Intermed., 1994, 20, 815 CrossRef CAS; (d) H. Yamashita, K. Ikeue, T. Takewaki and M. Anpo, Top. Catal., 2002, 18, 95 CrossRef CAS; (e) K. Ikeue, S. Nozaki, M. Ogawa and M. Anpo, Catal. Today, 2002, 74, 241 CrossRef CAS; (f) Y. Shioya, K. Ikeue, M. Ogawa and M. Anpo, Appl. Catal., A, 2003, 254, 251 CrossRef CAS.
- (a) K. Hirano, K. Inoue and T. Yatsu, J. Photochem. Photobiol., A, 1992, 64, 255 CrossRef CAS; (b) I.-H. Tseng, J. C. S. Wu and H.-Y. Chou, J. Catal., 2004, 221, 432 CrossRef CAS.
- (a) D. Mandler and I. Willlner, J. Am. Chem. Soc., 1984, 106, 5352 CrossRef CAS; (b) I. Willlner, D. Mandler and A. Riklin, J. Chem. Soc., Chem. Commun., 1986, 1022 RSC; (c) D. Walther, M. Ruben and S. Rau, Coord. Chem. Rev., 1999, 182, 67 CrossRef.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Blackwell Science, Oxford, 2002 Search PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed.
- C. Steward and M.-A. Hessami, Energy Convers. Manage., 2005, 46, 403 CrossRef CAS.
- N. Usui and M. Ikenouchi, Energy Convers. Manage., 1997, 38, S487 CrossRef CAS.
- J.-S. Lee and J.-P. Lee, Biotechnol. Bioprocess Eng., 2003, 8, 354 Search PubMed.
- I. S. Suh and C.-G. Lee, Biotechnol. Bioprocess Eng., 2003, 8, 313 Search PubMed.
- R. Hase, H. Okigawa, C. Sasao, M. Morita and Y. Watanabe, J. Biosci. Bioeng., 2000, 89, 157 CrossRef CAS.
- M. Morita, Y. Watanabe and H. Saiki, Proceedings of 4th International Conference on Greenhouse Gas Control Technologies, 1998, August 30-September 2, Interlaken, Switzerland Search PubMed.
- K. Zhang, S. Miyachi and N. Kurano, Biotechnol. Lett., 2001, 23, 21 CrossRef CAS.
- C. W. Zhang, O. Zmora, R. Kopel and A. Richimond, Aquaculture, 2001, 195, 35 CrossRef.
- O. Pulz, Proceedings of Symposium on Microalgae and seaweed Products in Plant/Soil-Systems, 2001, June 20–22, Mosonmagyarovar, Hungary Search PubMed.
- M. Olaizola, Biomol. Eng., 2003, 20, 459 CrossRef CAS.
- Y. K. Lee and C. S. Low, Biotechnol. Bioeng., 1991, 38, 995 CrossRef CAS.
- G. C. Zittelli, R. Liliana and M. Tredici, J. Appl. Phycol., 2003, 15, 107 Search PubMed.
- G. Vunjak-Novakovic, Y. Kim, X. Wu, I. Berzin and J. C. Merchuk, Ind. Eng. Chem. Res., 2005, 16, 44 http://www.greenfuelonline.com/ .
- (a) C. R. Woese, L. J. Magrum and G. E. Fox, J. Mol. Evol., 1978, 11, 245 CrossRef CAS; (b) G. Fuchs and E. Stupperich, Physiol. Veg., 1983, 21, 845 Search PubMed; (c) G. Fuchs, Carbon dioxide reduction by anaerobic bacteria, NATO ASI Ser. C, Carbon Dioxide Source Carbon: Biochemical and Chemical Uses, 1987, 206, 263 Search PubMed; (d) G. Fuchs, Colloq. Biol. Chem., 1990, 41, 13 Search PubMed.
- E. C. Bugante, Y. Shimonura, T. Tanaka, M. Taniguchi and S. Oi, J. Ferment. Bioeng., 1989, 67, 419 CrossRef CAS.
- K. T. Klasson, J. P. Cowger, C. W. Ko, J. L. Vega, E. C. Clausen and J. L. Gladdy, Appl. Biochem. Biotechnol., 1990, 24–25, 317 CrossRef CAS.
- K. A. Strevett, R. F. Vieth and D. Grasso, Chem. Eng. J., 1995, 58, 71 CAS.
- R. N. Patel, C. T. Hou and A. I. Laskin, Microbiological Oxidation, Eur. Patent Appl. EP 88,602 (cl. Cl12N9/02), September 14, 1983, Chem. Abstr. 100, P33289n Search PubMed.
- (a) M. C. J. Bradford and M. A. Vannice, Catal. Rev. Sci. Eng., 1999, 41, 1 CrossRef CAS; (b) H. Holm-Larsen, Stud. Surf. Sci. Catal., 2001, 136, 441 CAS; (c) J. R. H. Ross, Catal. Today, 2005, 100, 151 CrossRef CAS.
- O. Takayasu, C. Soman, Y. Takegahara and I. Matsuura, Stud. Surf. Sci. Catal., 1994, 88, 281 CAS.
- V. R. Chaudhary, A. M. Rayput and B. Prabhakar, Catal. Lett., 1995, 32, 391 CrossRef.
- A. M. De Groote and G. F. Froment, Can. J. Chem. Eng., 1996, 74, 735 CrossRef CAS.
- M. C. J. Bradford and M. A. Vannice, Appl. Catal., A, 1996, 142, 73 CrossRef CAS.
- (a) D. Dissanayake, M. P. Rosynek, K. C. C. Kharas and J. H. Lunsford, J. Catal., 1991, 132, 117 CrossRef CAS; (b) V. R. Choudhary, S. D. Samsare and A. S. Mammam, Appl. Catal., A, 1992, 90, L1 CrossRef CAS; (c) A. M. Gadalla and B. Bower, Chem. Eng. Sci., 1988, 42, 3049 CrossRef; (d) A. M. Gadalla and M. E. Sommer, Chem. Eng. Sci., 1989, 44, 2815; (e) G. J. Kim, D. S. Cho, K. H. Kim and J. H. Kim, Catal. Lett., 1994, 28, 41 CrossRef CAS; (f) H. M. Swaan, V. C. H. Kroll, G. A. Martin and C. Mirodatos, Catal. Today, 1994, 21, 571 CrossRef CAS; (g) J. S. Chang, S. E. Park and K. W. Lee, Stud. Surf. Sci. Catal., 1994, 84, 1587 CAS; (h) Z. Zhang and X. E. Verykios, J. Chem. Soc., Chem. Commun., 1995, 71 RSC.
- (a) A. T. Ashcroft, A. K. Cheethan, M. L. H. Green and P. D. F. Vernom, Science, 1991, 352, 225 CAS; (b) J. R. Rostrup-Nielsen and J. H. B. Hansen, J. Catal., 1993, 144, 38 CrossRef CAS; (c) D. Qin and J. Lapszewicz, Catal. Today, 1994, 21, 551 CrossRef CAS; (d) J. T. Richardson and Paripatyadar, Appl. Catal., 1990, 61, 293 Search PubMed.
- P. M. Torniainen, X. Chu and L. D. Schmidt, J. Catal., 1994, 146, 1 CrossRef CAS.
- (a) A. T. Ashcroft, A. K. Cheethan, J. S. Foord, M. L. H. Green, C. P. Grey, A. J. Murrell and P. D. F. Vernom, Nature, 1990, 344, 319 CrossRef CAS; (b) R. H. Jones, A. T. Ashcroft, D. Waller, A. K. Cheetham and J. M. Thomas, Catal. Lett., 1991, 8, 169 CrossRef CAS; (c) J. K. Hochmuth, Appl. Catal., B, 1992, 1, 89 CrossRef CAS; (d) D. F. Vernon, M. L. H. Green, A. K. Cheetham and A. T. Ashcroft, Catal. Lett., 1990, 6, 181 CrossRef CAS; (e) M. G. Poirier, J. Trudel and D. Guay, Catal. Lett., 1993, 21, 99 CrossRef CAS.
- (a) S. S. Schuler and M. Constantinescu, Int. J. Hydrogen Energy, 1995, 20, 653 CrossRef; (b) J. F. Martin and W. L. Kubic, Green Freedom – A Concept for Producing Carbon-Neutral Synthetic Fuels and Chemicals, LA-UR-07–7897, Los Alamos National Laboratory, Los Alamos, NM, 2007 Search PubMed.
- S. Teir, S. Eloneva and R. Zevenhoven, Energy Convers. Manage., 2005, 46, 2954 CrossRef CAS.
- US Geological Survey Minerals Yearbook 2003, US Geological Survey, Reston, VA, USA, 2003, p. 73.1 Search PubMed.
- US Geological Survey, Mineral Commodity Summaries, US Geological Survey, Reston, VA, USA, January 2005, p. 152 Search PubMed.
- US Geological Survey, Mineral Commodity Summaries, US Geological Survey, Reston, VA, USA, January 2005, p. 156 Search PubMed.
- (a) US Geological Survey, Mineral Commodity Summaries, US Geological Survey, Reston, VA, USA, January 2005, p. 100 Search PubMed; (b) US Geological Survey Minerals Yearbook 2003, US Geological Survey, Reston, VA, USA, 2003, p. 47.1 Search PubMed.
- US Geological Survey Minerals Yearbook 2003, US Geological Survey, Reston, VA, USA, 2003, p. 9.1 Search PubMed.
- US Geological Survey Minerals Yearbook 2003, US Geological Survey, Reston, VA, USA, 2003, p. 45.1 Search PubMed.
- D. J. Fauth, E. A. Frommell, J. S. Hoffman, R. P. Reasbeck and H. W. Pennline, Fuel Process. Technol., 2005, 86, 1503 CrossRef CAS.
- (a) K. Essaki, K. Nakagawa, M. Kato and H. Uemoto, J. Chem. Eng. Jpn., 2004, 37, 772 CrossRef CAS; (b) M. Kato, K. Nakagawa, K. Essaki, Y. Maezawa, S. Takeda, R. Kogo and Y. Hagiwara, Int. J. Appl. Ceram. Technol., 2005, 2, 467 CrossRef CAS.
- R. Steeneveldt, B. Berger and T. A. Torp, Chem. Eng. Res. Des., 2006, 84, 739 CrossRef CAS.
- A. Kohl and R. Nielsen, Gas Purification, 5th ed., Gulf Publishing Co., Houston, 1997 Search PubMed.
- F. Barzagli, F. Mani and M. Peruzzini, Energy Environ. Sci., 2009, 2, 322 RSC.
- S. J. Yoon and H. Lee, Chem. Lett., 2003, 32, 344 CrossRef CAS.
- J.-Y. Park, S. J. Yoon and H. Lee, Environ. Sci. Technol., 2003, 37, 1670 CrossRef CAS.
- G. Sartori and D. W. Savage, Ind. Eng. Chem. Fundam., 1983, 22, 239 CrossRef CAS.
- A. Yamasaki, J. Chem. Eng. Jpn., 2003, 36, 361 CrossRef CAS.
- R. A. Khatri, S. S. C. Chuang, Y. Soong and M. Gray, Energy Fuels, 2006, 20, 1514 CrossRef CAS.
- C. E. Powell and G. G. Qiao, J. Membr. Sci., 2006, 279, 1 CrossRef CAS.
- S. A. Stern, J. Membr. Sci., 1994, 94, 1 CrossRef CAS.
- (a) R. Anderson, B. R. Mattes, R. B. Kaner and H. Reiss, Science, 1991, 252, 1412 CrossRef CAS; (b) R. B. Kaner, R. Anderson, B. R. Mattes and H. Reiss, Membranes having selective permeability, US Patent 5,096,586 1992 Search PubMed; (c) L. Rebattet, M. Escoubes, E. Genies and M. Pineri, J. Appl. Polym. Sci., 1995, 57, 1595 CrossRef CAS; (d) G. Illing, K. Hellgardt, R. J. Wakeman and A. Jungbauer, J. Membr. Sci., 2001, 184, 69 CrossRef CAS; (e) J. A. Conklin, T. M. Su, S.-C. Huang and R. B. Kaner in Gas and Liquid Separation Application of Polyaniline, Handbook of Conducting Polymers, 2nd Edition, Marcel Dekker, New York, Basel, Hongkong, 1998, pp. 945 Search PubMed; (f) S. Kuwabata and R. Charles, J. Membr. Sci., 1994, 91, 1 CrossRef CAS; (g) M.-J. Chang, Y.-H. Liao, A. S. Myerson and T. K. Kwei, J. Appl. Polym. Sci., 1996, 62, 1427 CrossRef CAS.
- Z.-K. Xu, C. Dannenberg, J. Springer, S. Banerjee and G. Maier, J. Membr. Sci., 2002, 205, 23 CrossRef CAS.
- (a) M. R. Pixton and D. R. Paul, Macromolecules, 1995, 28, 8277 CrossRef CAS; (b) M. R. Pixton and D. R. Paul, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 1135 CrossRef CAS; (c) M. R. Pixton and D. R. Paul, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 1353 CrossRef CAS; (d) M. R. Pixtona and D. R. Paul, Polymer, 1995, 36, 2745 CrossRef.
- (a) M. Aguilar-Vega and D. R. Paul, J. Polym. Sci., Part B: Polym. Phys., 1993, 31, 1599 CrossRef CAS; (b) J. S. McHattie, W. J. Koros and D. R. Paul, J. Polym. Sci., Part B: Polym. Phys., 1991, 29, 731 CrossRef CAS; (c) T. A. Barbari, W. J. Koros and D. R. Paul, J. Membr. Sci., 1989, 42, 69 CrossRef CAS; (d) M. W. Hellums, W. J. Koros, G. R. Husk and D. R. Paul, J. Membr. Sci., 1989, 46, 93 CrossRef CAS; (e) N. Muruganandam and D. R. Paul, J. Membr. Sci., 1987, 34, 185 CrossRef CAS.
- (a) Y. Li, M. Ding and J. Xu, J. Appl. Polym. Sci., 1997, 63, 1 CrossRef CAS; (b) T. A. Barbari, W. J. Koros and D. R. Paul, J. Polym. Sci., Part B: Polym. Phys., 1988, 26, 709 CrossRef CAS.
- (a) H. Lin and B. D. Freeman, J. Membr. Sci., 2004, 239, 105 CrossRef CAS; (b) M. Yoshino, H. Kita, K.-I. Okamoto, M. Tabuchi and T. Sakai, Trans. Mater. Res. Soc. Jpn., 2002, 27, 419 Search PubMed.
- (a) P. S. Tin, T.-S. Chung, Y. Liu, R. Wang, S. L. Liu and K. P. Pramoda, J. Membr. Sci., 2003, 225, 77 CrossRef CAS; (b) Y. Liu, M. L. Chng, T.-S. Chung and R. Wang, J. Membr. Sci., 2003, 214, 83 CrossRef CAS; (c) C. Hibshman, C. J. Cornelius and E. Marand, J. Membr. Sci., 2003, 211, 25 CrossRef CAS; (d) Z.-K. Xu, L. Xiao, J.-L. Wang and J. Springer, J. Membr. Sci., 2002, 202, 27 CrossRef CAS; (e) S. L. Liu, R. Wang, T.-S. Chung, M. L. Chng, Y. Liu and R. H. Vora, J. Membr. Sci., 2002, 202, 165 CrossRef CAS; (f) R. Wang, S. S. Chan, Y. Liu and T.-S. Chung, J. Membr. Sci., 2002, 199, 191 CrossRef CAS; (g) F. Piroux, E. Espuche, R. Mercier and M. Pineri, Desalination, 2002, 145, 371 CrossRef CAS; (h) Y. Liu, R. Wang and T.-S. Chung, J. Membr. Sci., 2001, 189, 231 CrossRef CAS; (i) W.-H. Lin, R. H. Vora and T.-S. Chung, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 2703 CAS; (j) M. Al-Masei, D. Fritsch and H. R. Kricheldorf, Macromolecules, 2000, 33, 7127 CrossRef CAS; (k) M. Al-Masei, H. R. Kricheldorf and D. Fritsch, Macromolecules, 1999, 32, 7853 CrossRef CAS; (l) B.-W. Chun, C. Ishizu, H. Itatani, K. Haraya and Y. Shindo, J. Polym. Sci., Part B: Polym. Phys., 1994, 32, 1009 CrossRef CAS; (m) S. A. Stern, Y. Liu and W. A. Feld, J. Polym. Sci., Part B: Polym. Phys., 1993, 31, 939 CrossRef CAS; (n) K. Tanaka, H. Kita, M. Okano and K.-I. Okamoto, Polymer, 1992, 33, 585 CrossRef CAS; (o) K. Tanaka, M. Okano, H. Toshino, H. Kita and K.-I. Okamoto, J. Polym. Sci., Part B: Polym. Phys., 1992, 30, 907 CrossRef CAS; (p) H. Yamamoto, Y. Mi, S. A. Stern and A. K. St. Clair, J. Polym. Sci., Part B: Polym. Phys., 1990, 28, 2291 CrossRef CAS; (q) T.-H. Kim, W. J. Koros and G. R. Husk, J. Membr. Sci., 1989, 46, 43 CrossRef CAS; (r) S. A. Stern, Y. Mi, H. Yamamoto and A. K. St. Clair, J. Polym. Sci., Part B: Polym. Phys., 1989, 27, 1887 CAS.
- (a) F. Hamad, G. Chowdhury and T. Matsuura, Desalination, 2002, 145, 365 CrossRef CAS; (b) F. Hamad, K. C. Khulbe and T. Matsuura, Desalination, 2002, 148, 369 CrossRef CAS; (c) M. Aguilar-Vega and D. R. Paul, J. Polym. Sci., Part B: Polym. Phys., 1993, 31, 1577 CrossRef CAS.
- (a) C. M. Zimmerman and W. J. Koros, Macromolecules, 1999, 32, 3341 CrossRef CAS; (b) C. M. Zimmerman and W. J. Koros, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 1235 CAS; (c) C. M. Zimmerman and W. J. Koros, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 1251 CrossRef CAS; (d) C. M. Zimmerman and W. J. Koros, Polymer, 1999, 40, 5655 CrossRef CAS; (e) D. R. B. Walker and W. J. Koros, J. Membr. Sci., 1991, 55, 99 CrossRef CAS.
- (a) Y. Dai, M. D. Guiver, G. P. Robertson, Y. S. Kang, K. J. Lee and J. Y. Jho, Macromolecules, 2004, 37, 1403 CrossRef CAS; (b) Y. Dai, M. D. Guiver, G. P. Robertson, Y. S. Kang and K. J. Lee, Macromolecules, 2003, 36, 6807 CrossRef CAS; (c) Y. Dai, M. D. Guiver, G. P. Robertson, F. Bilodeau, Y. S. Kang, K. J. Lee, J. Y. Jho and J. Won, Polymer, 2002, 43, 5369 CrossRef CAS; (d) I.-W. Kim, K. J. Lee, J. Y. Jho, H. C. Park, J. Won, Y. S. Kang, M. D. Guiver, G. P. Robertson and Y. Dai, Macromolecules, 2001, 34, 2908 CrossRef CAS; (e) C. Bonfanti, L. Lanzini, R. Sisto and C. Valentini, J. Appl. Polym. Sci., 1997, 64, 1987 CrossRef CAS; (f) M. R. Pixton and D. R. Paul, Polymer, 1995, 36, 3165 CrossRef CAS; (g) C. L. Aitken, W. J. Koros and D. R. Paul, Macromolecules, 1992, 25, 3651 CrossRef CAS; (h) C. L. Aitken, W. J. Koros and D. R. Paul, Macromolecules, 1992, 25, 3424 CrossRef CAS; (i) C. L. Aitken and D. R. Paul, J. Polym. Sci., Part B: Polym. Phys., 1993, 31, 1061 CrossRef; (j) J. S. McHattie, W. J. Koros and D. R. Paul, Polymer, 1992, 33, 1701 CrossRef CAS; (k) J. S. McHattie, W. J. Koros and D. R. Paul, Polymer, 1991, 32, 840 CrossRef CAS; (l) J. S. McHattie, W. J. Koros and D. R. Paul, Polymer, 1991, 32, 2618 CrossRef CAS.
- A. Kohl and R. Nielsen, Gas Purification, 5th ed., Gulf Publishing Co., Houston, 1997 Search PubMed.
- S. Murai and Y. Fujioka, IEEJ Trans., 2008, 3, 37 Search PubMed.
- Risø Energy Report 6, Future options for energy technologies, ed. H. Larsenand L. S. Petersen, ISBN 978-87-550-3611-6, 2007 Search PubMed.
- M. Ha-Duong and D. W. Keith, Clean Technol. Environ. Policy, 2003, 5, 181 Search PubMed.
- H. J. Herzog and D. Golomb, “Carbon Capture and Storage from Fossil Fuel Use,” in Encyclopedia of Energy, ed. C. J. Cleveland, Elsevier Science Inc., New York, pp 277–287, 2004 Search PubMed.
- National Energy Technology Laboratory, Carbon Sequestration Atlas, 2007 Search PubMed.
- M. M. Maroto-Valer, D. J. Fauth, M. E. Kuchta, Y. Zhang and J. M. Andersen, Fuel Process. Technol., 2005, 86, 1487 CrossRef CAS.
- (a) N. Liu, G. M. Bond, B. J. McPherson and J. Stinger, Fuel Process. Technol., 2005, 86, 1615 CrossRef CAS; (b) J. K. Stolaroff, G. V. Lowry and D. W. Keith, Energy Convers. Manage., 2005, 46, 687 CrossRef CAS.
- H. M. Haywood, J. M. Eyre and H. Scholes, Environ. Geol., 2001, 41, 11 CrossRef CAS.
- M. L. Druckenmiller and M. M. Maroto-Valer, Fuel Process. Technol., 2005, 86, 1599 CrossRef CAS.
- W. K. O'Connor, D. C. Dahlin, D. N. Nielsen, S. J. Gerdeman, G. E. Rush, L. R. Penner, R. P. Walters and P. C. Turner, Proceedings of the 27th Int. Conf. on Coal utilization and Fuel Systems, Clearwater, FL, ( March 2002), p. 819 Search PubMed.
- A.-H. Park, A. R. Jadhav and L.-S. Fan, Can. J. Chem. Eng., 2003, 81, 885 CAS.
- J. G. Blencoe, L. M. Anovitz, D. A. Palmer and J. S. Beard, Carbonation of metal silicates for long-term CO2 sequestration, U.S. patent application. 2003 Search PubMed.
- W. K. O'Connor, D. C. Dahlin, G. E. Rush, S. J. Gedermann, L. R. Penner and D. N. Nilsen, Aqueous mineral carbonation, Final Report, DOE/ARC-TR-04–002, US DOE, Albany Research Center ( March 15, 2005) Search PubMed.
- K. S. Lackner, Science, 2003, 300, 1677 CrossRef CAS.
- H. J. Herzog, Am. Chem. Soc. Div. Fuel Chem. Prepr., 2001, 46, 53 Search PubMed.
- H.-H. Rogner, Annu. Rev. Energy Environ., 1997, 22, 217 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |