Polymer wrapping technique: an effective route to prepare Pt nanoflower/carbon nanotube hybrids and application in oxygen reduction†
Wen
Yang
ab,
Yong
Wang
ab,
Jing
Li
ab and
Xiurong
Yang
*a
aState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 # Renmin Street, Changchun, Jilin 130022, China. E-mail: xryang@ciac.jl.cn; Fax: +86-431-85689278; Tel: +86-431-85262056
bGraduate School of the Chinese Academy of Sciences, Beijing, 100039, China
First published on 13th November 2009
Abstract
We reported an effective and environmentally friendly method of decorating Pt nanoflowers (Pt NFs) on the surface of carbon nanotubes (CNTs). By reduction of K2PtCl4 with L-ascorbic acid in an aqueous solution, Pt nanoparticles with a flower-like morphology were deposited onto the surface of poly(sodium 4-styrenesulfonate) wrapped CNTs (PSS-CNTs). PSS plays a dual role: dispersing stable CNTs into solution in water, and providing functional groups that bind metal ions and metal nanoparticles. As-prepared Pt NFs/PSS-CNTs showed a large electroactive specific surface area, and presented excellent electrocatalytic activity for oxygen reduction. Au and Pd nanoparticles could also be deposited on PSS-CNTs. This method could be extended to the synthesis of different kinds of metal nanoparticles on other polymer wrapped CNTs. As-prepared metal nanoparticle/CNT nanocomposites could find use as electrocatalysts in polymer electrolyte membrane fuel cells.
Broader contextPt-based materials are important electrocatalysts for proton exchange membrane fuel cell (PEMFC), both in applications and research. However, the major problem in PEMFCs is the kinetic limitations of the oxygen reduction reaction (ORR) on the cathodes. In order to solve this problem, it is necessary to maximize the catalytic activity of Pt nanoparticles by engineering their morphology, because the morphology could determine which facets are exposed on the surface of the nanoparticles. The decoration of morphology-controlled Pt nanoparticles on carbon nanotubes has been extensively studied due to carbon nanotubes’ exceptional properties. Unfortunately, at the current state of the art, to fabricate such hybrid materials is generally difficult, owing to the small size of the metal nanoparticles and the curvature of nanotubes. Theoretically, noncovalent functionalization techniques could solve this problem. In this study, we used the polymer wrapping technique as a model system of noncovalent functionalization strategies for the decoration of morphology-controlled Pt nanoflowers (Pt NFs) in one step. As-prepared nanocomposites of Pt NFs/CNTs showed a large electroactive specific surface area, and presented excellent electrocatalytic activity for oxygen reduction. |
1. Introduction
Carbon nanotubes (CNTs) have attracted an increasing amount of interest because of their outstanding electronic, mechanical, and thermal properties, as well as their potential applications in catalysis, fuel cells, solar cells, gas and vapor sensors, biosensors, and biomedical applications.1 However, the major impediment to those potential applications of CNTs is their insolubility and a strong tendency to aggregate in aqueous solution.2–6 One promising solution for the problem is noncovalent modification of CNTs with polymers.2,7–9 Since the well-known report of Smalley,2 CNTs have been successfully solubilized by wrapping them with many polymers and biopolymers, including polystyrene sulfonate (PSS),2,5 chitosan (CHIT),4 DNA,10 and even protein.1,2,7,11 This so-called polymer wrapping technique not only preserves the intrinsic CNT sp2 structure conjugation, but also maintains the electronic structure of CNTs.2,5,7,8,12,13 Thus, the polymer wrapping technique has become a promising strategy to functionalize, modify and assemble CNTs in a nondestructive way.2,7,13 This method is also particularly interesting for preparing nanocomposites of metal nanoparticles/CNTs by directly growing metal nanoparticles onto polymer-modified CNTs.14,15Strategies for the functionalization of CNTs with Pt nanoparticles are crucial for the pursuit of its applications,16–18 because the decoration of CNTs with nanoparticles may combine unique properties of two functional nanoscale materials.17,18 For example, CNT-supported Pt nanoparticles have been utilized as the main catalysts for oxygen reduction, which is considered significant in the proton exchange membrane based hydrogen fuel cell.17–21 Moreover, the hybrids of Pt nanoparticles/CNTs exhibited high catalytic ability for selective partial hydrogenation of 3-methyl-2-butenal (prenal) to 3-methyl-2-butenol (prenol).22 Furthermore, nanocomposites of Pt nanoparticles/CNTs have been constructed for electrochemical detection of hydrogen peroxide and explosive nitroaromatic compounds.9,23 Finally, nanostructures of Pt nanoparticles/CNTs have been developed for gas sensor arrays.24
Due to the potential applications of the hybrids of Pt nanoparticles/CNTs, several routes have been developed to deposit Pt nanoparticles onto the surface of CNTs. Most current techniques introduce functional groups or molecules onto the surface of CNTs through chemical or electrochemical oxidation at the defect sites of CNTs.19,22,25 Through this way, the adhesion between Pt nanoparticles and CNTs may improve.25 However, the intrinsic properties of CNTs may be disturbed. Other strategies have also been exploited, such as electroless plating,26 electrodeposition,27 spinning disc processing,11 physical adsorption techniques,28 organic molecule crossing-linking,29 noncovalent functionalization techniques9,12,13 and supramolecular self-assembly techniques.15 However, the morphology-controlled deposition of Pt nanoparticles onto the surface of CNTs is still a challenge. It is well known that the morphology (including size and shape) of Pt nanoparticles could determine which crystallographic planes are exposed on the surface of the nanoparticles. In our previous work, Pt nanocubes have been successfully decorated on CNTs via a noncovalent functionalization technique, and the resulting nanocomposites exhibited good electrocatalytic activity for oxygen reduction.13
Herein, we report an alternative strategy for the decorating of morphology-controlled Pt nanoparticles onto the surface of CNTs. Two different polymers, PSS and CHIT, were used as model polymers to facilely disperse CNTs in water. These functionalized CNTs were controllably decorated with morphology-controlled Pt nanoflowers (Pt NFs) through in situ reduction of the metal precursor by aqueous L-ascorbic acid solution at room temperature. Au and Pd nanoparticles can also be deposited onto the surface of CNTs using a similar method. The whole process of our experiment followed the concepts of green chemistry, which is more environmentally benign and economically beneficial.11,30,31 Furthermore, as-prepared Pt NF/PSS-CNT nanocomposites also exhibited high catalytic activity for the reduction of oxygen, which has a great potential application in polymer electrolyte fuel cells.
2. Experimental
2.1 Materials
K2PtCl4, 20 wt% Pt, on carbon black was purchased from Alfa Aesar (USA). KAuCl4, K2PdCl4, PSS, Mw 70000, were all purchased from Aldrich (USA). CHIT, Mw 6000, was obtained from YuHuan Ocean Biochemical. Co. Ltd (China). Multiwalled CNTs (MWCNTs) (D = 40–60 nm) were purchased from Shenzhen Nanotech Port Co. Ltd (Shenzhen, China). MWCNTs were purified by refluxing in 3 M HNO3 for 24 h before experiments. L-ascorbic acid was obtained from Shanghai Sangon Biological Engineering Technique & Services Co (Shanghai, China). Ltd. Sodium borohydride (NaBH4, 99%) and formic acid (88%) were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China).2.2 Preparation of PSS-wrapped MWCNTs
PSS wrapped MWCNTs were prepared according to the reported method.7,13 MWCNTs were dispersed in a 1 wt% aqueous solution of PSS to a concentration of 0.15 mg mL−1 by combination of strong stirring and sonication. Then, the solution was stored at 50 °C for more than 12 h. The PSS modified MWCNTs (PSS-MWCNTs) were separated and washed using a membrane filtration apparatus with a cellulose filter with a pore size of 0.45 μm. Finally, the PSS-MWCNTs were dried under vacuum at 60 °C for further assembly.The procedure of the preparation of CHIT wrapped MWCNTs is similar to PSS wrapped MWCNTs, except that CHIT solution was used.
2.3 Decoration of Pt NFs onto the surface of PSS-MWCNTs
PSS-MWCNTs (3 mg) were added to 17.9 mL distilled water in a beaker, and dispersed in water by strong sonication. 0.1 mL of 0.1 M K2PtCl4 (freshly prepared within 5 min) was added to the solution of PSS-MWCNTs. Then, 2 mL of 0.1 M L-ascorbic acid was added all at once, followed by shaking for 2 min. Finally, the beaker was kept at room temperature for 24 h. As-prepared Pt NFs/MWCNTs were separated from solution by three washing/centrifugation cycles and dried under vacuum at 60 °C.Control experiments were also carried out by introducing other kinds of reducing agents. The procedure was similar to the synthesis of Pt NFs/MWCNTs, except 2mL of aqueous 0.1M NaBH4 or 2 ml formic acid was used.
2.4 Decoration of Au and Pd nanoparticles onto the surface of PSS-MWCNTs
The preparation of Au nanoparticles/PSS-MWCNTs was as follows: 0.1 mL of 0.1 M KAuCl4 was added to 17.9 ml of a solution of PSS-MWCNTs with sonication. Then, 1 mL of 0.1 M L-ascorbic acid was added with strong shaking. Finally, 1 mL of 0.6 M NaOH was added, and Au nanoparticles were deposited on the surface of PSS-MWCNTs. The solution was kept at room temperature for 24 h.The procedure for the synthesis of Pd nanoparticles/PSS-MWCNTs was similar to the preparation of Pt NFs/PSS-MWCNTs, except 0.1 M K2PdCl4 was used.
The procedure for the synthesis of Pt or Au nanoparticle/CHIT wrapped MWCNTs was analogous to the above mentioned synthesis of nanoparticle/PSS-MWCNTs, except CHIT wrapped MWCNTs was used.
2.5 Preparation of Pt NF/CNT composite modified electrode
A glassy carbon electrode (GCE, 3 mm in diameter) was polished with 1.0 and 0.3 μm alumina slurry sequentially and then washed ultrasonically in water for a minute. Then the cleaned GCE was dried with a nitrogen stream.10 μl of a 2 mg mL−1 as-prepared Pt NF/CNT composite suspension was dropped onto the GCE. After evaporation of the water at 4 °C in a refrigerator, 10 μl of a diluted Nafion solution (0.05 wt% in ethanol) was put on top of the catalyst, and then dried at 4 °C in a refrigerator.
2.6. Instrumentation
TEM measurements were performed on a JEOL 2010 transmission electron microscope operating at an accelerating voltage of 200 kV. Samples for XPS were prepared by placing a Pt NF/CNT composite solution onto a clean silicon wafer. XPS measurements were conducted with an ESCALAB MK II spectrometer (VG Co., UK) with Mg Kα radiation (hν = 1253.6 eV) as the X-ray source and a pass energy of 20 eV. The pressure inside the analyzer was maintained at 6.5 × 10−7 Pa. The Pt loadings of Pt NF/CNT composites were measured using inductively coupled plasma atomic optical emission spectrometry (ICP-OES). A sample of a known weight was soaked in aqua regia overnight,8 and the concentration of Pt ions was characterized by ICP-OES. The ICP-OES measurement was performed on a Thermo Scientific ICAP 6000 ICP-OES (USA). Electrochemical experiments were performed with a CHI 660B electrochemical workstation in a conventional three-electrode cell using a GCE (3 mm diameter) as the working electrode, Pt foil as the auxiliary electrode, and Ag/AgCl as a reference electrode.3. Results and discussion
We used PSS as a model polymer to facilely disperse CNTs into solution by direct mixing.6,7,13 It is proposed that the PSS wraps around CNTs with the aromatic base interacting with the SWNT wall and the charged backbone staying at the exterior of the micelles.6,7,13 After reduction by L-ascorbic acid, Pt and other different kinds of metal nanoparticles were facilely deposited onto the surface of PSS wrapped CNTs.11,31The formation of Pt nanoparticles on the surface of PSS-CNTs was confirmed by TEM observation. Fig. 1A shows many Pt nanoparticles with flower nanostructures that were successfully deposited onto the surfaces of PSS-CNTs (the initial concentration of K2PtCl4 was 0.5 mM). The enlarged TEM images reported in Fig. 1B reveal that the Pt NFs were composed of many interconnected blocks forming flower-shaped Pt nanoparticles. Higher-magnification images of as-prepared Pt NFs (Fig. S1†) indicated that these flower nanostructures are single structures rather than the result of aggregated small nanoparticles. The dimension of those as-formed Pt NFs mainly ranged from 20.2 to 37.3 nm, and the average size was 33.8 nm (Fig. S2A†). Selected-area electron diffraction (SAED) patterns (as shown in Fig. S3†) for the Pt NFs reveal several bright concentric rings, attributed to {111}, {200}, {220}, and {311} Pt face-centered cubic (fcc) crystal diffractions.13 This confirms that the Pt NFs grown on the surface of PSS-CNTs by this method are crystallized in a phase similar to bulk Pt.13 It is well known that the formation of Pt NFs on the surface of PSS-CNTs is dependent on the concentration and ratio of reagents. The concentrations of L-ascorbic acid (10 mM) and PSS-CNTs (0.15 mg mL−1) were held constant while the concentration of K2PtCl4 was varied. TEM images of the resulting products are shown in Fig. 1C, and D for the K2PtCl4 concentration of 1, and 1.5 mM, respectively. As expected, the dimensions of Pt NFs increased greatly with increasing initial concentration of K2PtCl4. When the concentration of K2PtCl4 was 1 mM, the dimensions of Pt NFs mainly ranged from 56.6 to 79.9 nm, and the average size was 69.9 nm (Fig. S2B†). When the concentration of K2PtCl4 was 1.5 mM, the dimension of as-formed Pt NFs mainly ranged from 49.7 to 86.7 nm, and the average size was 73.4 nm (Fig. S2C†). The concentration of Pt ions was found by ICP-OES. The Pt loadings of Pt NF/CNT composites for the sample in Fig. 1A, C, and D were 39.3 wt%, 54.4 wt%, and 68.1 wt%, respectively.
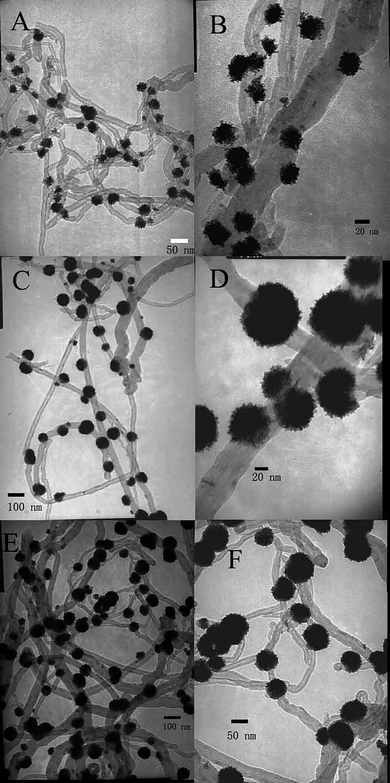 | ||
| Fig. 1 TEM images of the products of Pt NF/PSS-CNT composites obtained with different of amounts of K2PtCl4: (A) 0.5, (C) 1, (E) 1.5 mM, respectively, and their corresponding higher-magnification images (B), (D) and (F). | ||
XPS characterizations confirmed the decoration of Pt NFs onto the surface of CNTs. Fig. 2 displays a representative XPS spectrum of CNTs coated with Pt NFs. The C(1s) peak (Fig. 2A) at 284.7 eV was assigned to a π–π interaction with the extensively delocalized undamaged structure of CNTs.13,32 Distinct Pt(4f7/2) and Pt(4f5/2) peaks at 71.1 eV and 74.4 eV, respectively, are clearly visualized (Fig. 2B), which are in good agreement with those of pure bulk platinum.13,32 Based on these results, we believe that the Pt NFs are successfully deposited on CNTs.
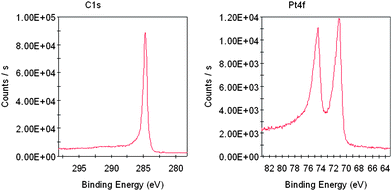 | ||
| Fig. 2 XPS spectra of the C1s and Pt4f of the as-prepared Pt NF/PSS-CNT composites deposited on a clean silicon wafer. | ||
The decoration of Pt nanoparticles on the surface of PSS-CNTs most likely takes place through ion exchange at the sulfonic acid functional groups of PSS attached to CNTs. PSS has a high density of negatively charged sulfonated groups.6,13,20,33 As a cation-exchange polyelectrolyte, PSS has been used in ultrafiltration for the removal of heavy metal ions from aqueous solution, for the separation of metal ion species, and also as a binding layer for the measurement of Cu2+ and Cd2+.34
A mechanism consisting of nanoparticle seeding followed by fast autocatalytic surface growth could be discussed for the formation of Pt NFs on the surface of PSS-CNTs.35,36 Recently, Wang and co-workers reported the synthesis of highly branched Ag nanostructures of silver nanostructures from the reduction of silver nitrate (AgNO3) by L-ascorbic acid.37 In this example, they suggested that such structures formed due to higher growth rates occurring at areas with high curvature.37 Moreover, this type of overgrowth has previously been used to explain the formation of dendritic and other branched particles.38,39
In our system, PSS is not as effective as other polymers with a hydrophobic backbone in stabilizing smaller nanoparticles.40,41 It could not prevent the aggregation of discrete Pt particles facilitated by L-ascorbic acid (L-AA) in the early stages of a reaction.37,42,43 As a result, the necessary platinum nanonuclei with high curvature would likely form on the surface of PSS-CNTs.37,42,43 Once nucleation of Pt(0) particles with high curvature on the surface of PSS-CNTs has started, the reduction of platinum ions was accelerated through an autocatalytic reduction process.35,36 Thus, supersaturation was achieved in a very short time because of the high precursor concentration and the effect of the autocatalytic ability of Pt, which led to formation of Pt NFs.35,36 This mode of formation might explain the growth mechanism of Pt NFs on the surface of PSS-CNTs. In this light, it should be noted that the use of NaBH4 gave few Pt nanoparticles on the surface of PSS-CNT under similar conditions (Fig. S4A†). This is because NaBH4 is a well-known strong reducing agent, and it can reduce K2PtCl4 in a short time. Therefore, Pt nanoparticles nucleated in solution instead of on the surface of PSS-CNTs. When NaBH4 was replaced by another milder reducing agent, i.e. formic acid,36,44 Pt nanoparticles were also deposited on the surface of PSS-CNTs; however, the morphology of Pt nanoparticles was mainly spherical (Fig. S4B†).36,45
The key idea of modifying PSS-CNTs for supporting Pt nanoparticles may be extended to other metals by using L-ascorbic acid as the reductant.37,46,47 When KAuCl4 was chosen as the metal precursor, nothing happened, because L-ascorbic acid is a mild reducing reagent and it could not reduce KAuCl4 to Au(0) at room temperature in the absence of gold seed particles.46,47 However, when we introduced NaOH, ascorbic acid was deprotonated to ascorbate, which has been found to be an effective reductant for the formation of gold nanocrystals on the surface of PSS-CNTs.46,47 Au nanoparticles with an irregular shape were formed on the surface of PSS-CNTs (as shown in Fig. 3A).16 The XPS spectrum of the as-prepared Au nanoparticle/PSS-CNT composites shows the Au 4f7/2 and 4f5/2 doublet with binding energies of 84.0 and 87.6eV, respectively (Fig. S5A†). These are typical values for Au0, indicating that Au NPs are readily deposited onto the surface of PSS-CNTs.16 Pd nanoparticles can also be deposited onto the surface of PSS-CNTs (as shown in Fig. 3B). When the metal precursor was supplied by K2PdCl4, instead of K2PtCl4, the products obtained showed different morphologies (see Fig. 3B), with the sample exhibiting irregular nanostructures with a nonuniform size range. However, XPS results of the Pd nanoparticle/PSS-CNT nanocomposites showed four binding energy peaks in the Pd 3d region at 335.7, 337.9, 340.9, and 342.6 eV, which indicated that the Pd was in the Pd(0)/Pd(II) mixed-valence state (Fig. S4B†). Further analysis of the XPS data gave the Pd 3d binding energies of Pd(0) and Pd(II), respectively, as follows: (1) Pd(0), 335.7 (Pd 3d5/2) eV; (2) Pd(II), 337.9 (Pd 3d5/2) eV; Pd(0), 340.9 (Pd 3d3/2) eV; Pd(II), 342.6 (Pd 3d3/2) eV. The percentages of Pd(0) and Pd(II), calculated from the relative areas of these peaks, were 82.6% and 17.4%, respectively. Therefore, Pd(0) was the main component in the nanocomposites of Pd nanoparticles/PSS-CNTs.48
 | ||
| Fig. 3 TEM images of 1D Au/PSS-CNTs (A), and Pd/PSS-CNTs (B). | ||
We could also deposit the mentioned nanoparticles on other polymer wrapped CNTs by using the same methods. CHIT is a cationic biomacropolymer with a primary amine.4 Cationic CHIT was coated onto MWNTs by virtue of electrostatic interactions, because purified CNTs were slightly coated by a small quantity of carboxylic groups.16 The fabrication of Pt and other nanoparticles on the surface CHIT-CNTs is feasible because of the abundance of amines on the surface of CHIT-CNTs.3,4 Pt NFs were also decorated onto the surfaces of the CHIT-CNTs (as shown in Fig. 4A). However, the structure of Pt NFs was much more irregular, and they were composed of many small Pt nanoparticles. It is very well known that amino groups form covalent and electrostatic interactions with metal nanoparticles.49 Considering the strong capping and stabilizing effects of CHIT on Pt nanoparticles, it is unsurprising that its presence could prevent the aggregation of discrete Pt nanoparticles in the early stages of a reaction.49 As a result, the necessary seed with high curvature structures did not form, and the formation of Pt NFs was inhibited.37,50Fig. 4B shows TEM images of 1D CHIT-CNT/Au NP nanocomposites prepared by a similar method. Au NPs with flower nanostructures are also decorated onto the surface of CNTs.
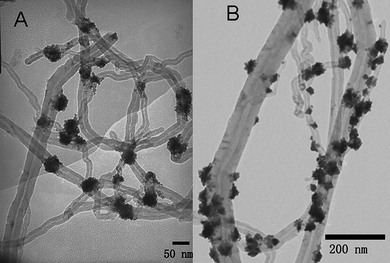 | ||
| Fig. 4 TEM images of 1D Pt/CHIT-CNTs (A), and Au/CHIT-CNTs (B). | ||
We studied the electrochemical properties of the Pt NF/PSS-CNT composites using cyclic voltammetry. Electrochemical measurements were conducted with a GCE modified with the Pt NF/CNT composites with a metal loading of 39.1 wt%. Fig. 5 shows the cyclic voltammogram (CV) of the Pt NF/CNT composites in 0.5 M H2SO4 saturated with ultrahigh purity nitrogen at a scan rate of 20 mV s−1 from −0.2 V to 1.5 V. It can be seen that the electrode with 39.1 wt% catalyst produced the highest specific current in the hydrogen region (−0.2 V–0.2 V).13,19–21 This result is believed to be due to the catalyst dispersion of 39.1 wt% being the highest. The platinum oxide reduced at about 0.46 V was also observed. The electrochemical properties of the Pt NF/PSS-CNT composites were consistent with those of a bare platinum electrode, indicating the as-prepared composites consisting of Pt NFs and Pt NFs had an active surface.13,19–21 To compare our catalyst (39.1 wt% Pt loading as a model) with commercial fuel catalysts, the cyclic voltammogram of a commercial catalyst under identical conditions was obtained. The commercial catalyst is 20.0 wt% Pt (3 nm in diameter) on carbon black (Alfa Aesar).13 The cyclic voltammogram of the Alfa Aesar catalyst is displayed in Fig. 5. The electroactive specific surface area of the Pt NF/PSS-CNT electrode determined from the hydrogen adsorption peak was 76.1 cm2 mg−1, while that for the Alfa Aesar electrode was 64.2 cm2 mg−1.13,20
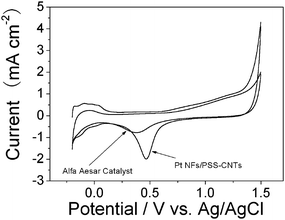 | ||
| Fig. 5 Cyclic voltammogram of the Pt NF/CNT and commercial catalysts (Alfa Aesar). Measurements were performed in 0.5 M H2SO4 saturated by nitrogen. Scan rate: 20 mV s−1. | ||
The electroreduction of oxygen to water has been extensively studied, because of its considerable importance for polymer electrolyte fuel cells, batteries, and many other electrode applications. The electrocatalysis of the Pt NF/PSS-CNT composites toward oxygen reduction were investigated. Fig. 6 shows the typical cyclic voltammograms of oxygen reduction at the GCE modified with Pt NF/PSS-CNT composites with a metal loading of 39.1 wt% in 0.5 M H2SO4 in the presence and absence of oxygen. Curve a corresponds to the CV of the modified electrode in N2-saturated 0.5 M H2SO4. In the presence of oxygen, the Pt NF/CNT composites exhibited high electrocatalytic activity towards oxygen, and the reduction peak was present at about 0.43 V in 0.5 M H2SO4 saturated by air (line b). Hence, the as-prepared Pt NC/CNT composites show a high electrocatalytic activity for oxygen reduction.13,21
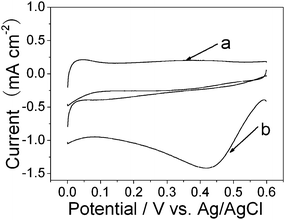 | ||
| Fig. 6 Typical cyclic voltammograms of oxygen reduction at the Pt NF/PSS-CNT composite modified GCE in N2-saturated 0.5 M H2SO4 (a) and oxygen-saturated 0.5 M H2SO4 (b). Scan rate: 100mV s−1. | ||
4. Conclusion
In summary, we have demonstrated that the polymer wrapping technique could not only readily disperse CNTs into solution, but also provide functional groups for the synthesis of morphology-controlled and electrochemically active Pt NF/CNT heterogeneous nanostructures. These flower-shaped Pt nanostructures are easily deposited on the surface of PSS-CNT or CHIT-CNTs through the reduction of K2PtCl4 with L-ascorbic acid. As-prepared nanocomposites of Pt NFs/PSS-CNTs showed high electroactive specific surface areas, and presented excellent electrocatalytic activity for oxygen reduction. The method is simple and could also be extended to the synthesis of Au or Pd/CNT nanocomposites. The polymer wrapping technique may act as a general technique for the fabrication of other kinds of nanoparticles decorated on the surface of nanoparticles. In addition, as-prepared nanocomposites have considerable potential in catalysis, fuel cells and the fabrication of biosensors.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 90713022), the National Key Basic Research Development Project of China (No. 2007CB714500) and the Project of Chinese Academy of Sciences (No. KJCX2-YW-H09) and (No. KJCX2-YW-H11). We thank Zhonghua Zhang for his advice on Pt electrochemical area measurements. We appreciate Dr Fan Yang and Dr Wei Hui for helpful discussion. Dr Zoe Schnepp is thanked for her help with English.Notes and references
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS.
- M. J. O'Connell, P. Boul, L. M. Ericson, C. Huffman, Y. H. Wang, E. Haroz, C. Kuper, J. Tour, K. D. Ausman and R. E. Smalley, Chem. Phys. Lett., 2001, 342, 265 CrossRef CAS.
- L. Qian and X. Yang, Talanta, 2006, 68, 721 CrossRef CAS.
- M. G. Zhang, A. Smith and W. Gorshi, Anal. Chem., 2004, 76, 5045 CrossRef CAS.
- K. Gong, P. Yu, L. Su, S. Xiong and L. Mao, J. Phys. Chem. C, 2007, 111, 1882 CrossRef CAS.
- A. Kongkanand, K. Vinodgopal, S. Kuwabata and P. V. Kamat, J. Phys. Chem. B, 2006, 110, 16185 CrossRef CAS.
- M. A. Correa-Duarte, N. Sobal, L. M. Liz-Marzan and M. Giersig, Adv. Mater., 2004, 16, 2179–2184 CrossRef CAS.
- M. Grzelczak, M. A. Correa-Duarte, V. Salgueirino-Maceira, M. Giersig, R. Diaz and L. M. Liz-Marzan, Adv. Mater., 2006, 18, 415 CrossRef CAS.
- S. Hrapovic, Y. L. Liu, K. B. Male and J. H. T. Luong, Anal. Chem., 2004, 76, 1083 CrossRef CAS.
- G. N. Ostojic, J. R. Ireland and M. C. Hersam, Langmuir, 2008, 24, 9784 CrossRef CAS.
- S. K. Iyer and C. L. Raston, J. Mater. Chem., 2007, 17, 4872 RSC.
- M. Grzelczak, M. A. Correa-Duarte, V. Salgueiriño-Maceira, B. Rodríguez-González, J. Rivas and L. M. Liz-Marzán, Angew. Chem., Int. Ed., 2007, 46, 7026 CrossRef CAS.
- W. Yang, X. L. Wang, F. Yang, C. Yang and X. R. Yang, Adv. Mater., 2008, 20, 2579 CrossRef CAS.
- D. Wang, Z. C. Li and L. Chen, J. Am. Chem. Soc., 2006, 128, 15078 CrossRef CAS.
- X. Hu, T. Wang, X. Qu and S. J. Dong, J. Phys. Chem. B, 2006, 110, 853 CrossRef CAS.
- V. Georgakilas, D. Gournis, V. Tzitzios, L. Pasquato, D. M. Guldi and M. Prato, J. Mater. Chem., 2007, 17, 2679 RSC.
- C. J. Zhong, J. Luo, Peter N. Njoke, D. Mott, B. Wanjala, R. Loukrakpam, S. Lim, L. Wang, B. Fang and Z. Xu, Energy Environ. Sci., 2008, 1, 454 RSC.
- A. Manthiram, A. V. Murugan, A. Sarkar and T. Muraliganth, Energy Environ. Sci., 2008, 1, 621 RSC.
- Y. C. Xing, J. Phys. Chem. B, 2004, 108, 19255 CrossRef CAS.
- M. M. Waje, X. Wang, W. Z. Li and Y. S. Yan, Nanotechnology, 2005, 16, S395 CrossRef.
- Y. Lin, X. Cui, C. Yen and C. M. Wai, J. Phys. Chem. B, 2005, 109, 14410 CrossRef CAS.
- V. Lordi, N. Yao and J. Wei, Chem. Mater., 2001, 13, 733 CrossRef CAS.
- S. Hrapovic, E. Majid, Y. Liu, K. Male and J. H. T. Luong, Anal. Chem., 2006, 78, 5504 CrossRef CAS.
- A. Star, V. Joshi, S. Skarupo, D. Thomas and J. P. Gabriel, J. Phys. Chem. B, 2006, 110, 21014 CrossRef CAS.
- R. Q. Yu, L. W. Chen, Q. P. Liu, J. Y. Lin, K. L. Tan, S. C. Ng, H. S. O. Chan, G. Q. Xu and T. S. A. Hor, Chem. Mater., 1998, 10, 718 CrossRef CAS.
- L. M. Ang, T. S. A. Hor, G. Q. Xu, C. H. Tung, S. P. Zhao and J. L. S. Wang, Chem. Mater., 1999, 11, 2115 CrossRef CAS.
- C. Wang, M. Waje, X. Wang, J. M. Tang, R. C. Haddon and Y. S. Yan, Nano Lett., 2004, 4, 345 CrossRef CAS.
- J. M. Planeix, N. Coustel, B. Coq, V. Brotons, P. S. Kumbhar, R. Dutartre, P. Geneste, P. Bernier and P. M. Ajayan, J. Am. Chem. Soc., 1994, 116, 7935 CrossRef CAS.
- Y. Y. Mu, H. P. Liang, J. S. Hu, L. Jiang and L. J. Wan, J. Phys. Chem. B, 2005, 109, 22212 CrossRef CAS.
- W. Yang, Y. Ma, J. Tang and X. Yang, Colloids Surf., A, 2007, 302, 628 CrossRef CAS.
- S. Guo, L. Wang, S. J. Dong and E. Wang, J. Phys. Chem. C, 2008, 112, 13510 CrossRef CAS.
- D. Q. Yang, B. Hennequin and E. Sacher, Chem. Mater., 2006, 18, 5033 CrossRef CAS.
- M. A. Correa-Duarte, M. Grzelczak, V. Salgueirino-Maceira, M. Giersig, L. M. Liz-Marzan, M. Farle, K. Sierazdki and R. Diaz, J. Phys. Chem. B, 2005, 109, 19060 CrossRef CAS.
- W. Li, P. R. Teasdale, S. Zhang, R. John and H. Zhao, Anal. Chem., 2003, 75, 2578 CrossRef CAS.
- Y. Song, Y. Yang, C. J. Medforth, E. Pereira, A. K. Singh, H. Xu, Y. Jiang, C. J. Brinker, F. V. Swol and J. A. Shelnutt, J. Am. Chem. Soc., 2004, 126, 635 CrossRef CAS.
- L. Wang, S. Guo, J. Zhai and S. J. Dong, J. Phys. Chem. C, 2008, 112, 13372 CrossRef CAS.
- Y. L. Wang, P. H. C. Camargo, S. E. Skrabalak, H. C. Gu and Y. N. Xia, Langmuir, 2008, 24, 12042 CrossRef CAS.
- A. Chernov, Sov. Phys. Crystallogr., 1972, 16, 734 Search PubMed.
- B. Wiley, Y. Sun and Y. Xia, Acc. Chem. Res., 2007, 40, 1067 CrossRef CAS.
- I. Pardinas-Blanco, C. E. Hoppe, Y. Pineiro-Redondo, M. A. Lopez-Quintela and J. Rivas, Langmuir, 2008, 24, 983 CrossRef CAS.
- J. F. Banfield, S. A. Welch, H. Zhang, T. T. Ebert and R. L. Penn, Science, 2000, 289, 751 CrossRef CAS.
- X. Lou, C. Yuan and L. A. Archer, Chem. Mater., 2006, 18, 3921 CrossRef CAS.
- A. B. R. Mayer and J. E. Mark, Eur. Polym. J., 1998, 34, 103 CrossRef CAS.
- S. S. Kumar, C. S. Kumar, J. Mathiyarasu and K. L. Phani, Langmuir, 2007, 23, 3401 CrossRef CAS.
- S. Sun, D. Yang, G. Zhang, E. Sacher and J. P. Dodelet, Chem. Mater., 2007, 19, 6376 CrossRef CAS.
- H. Wu, M. Liu and M. H. Huang, J. Phys. Chem. B, 2006, 110, 19291 CrossRef CAS.
- L. Lu, A. Kobayashi, K. Tawa and Y. Ozaki, Chem. Mater., 2006, 18, 4894 CrossRef CAS.
- C. D. Wagner, A. V. Naumkin, A. Kraut-Vass, J. W. Allison, C. J. Powell and J. R. Rumble, U.S. National Institute of Standards and Technology (NIST) XPS database, http://srdata.nist.gov/xps/ and references therein.
- B. L. V. Prasad, S. I. Stoeva, C. M. Sorensen and K. J. Klabunde, Chem. Mater., 2003, 15, 935 CrossRef CAS.
- S. Wang and H. Xin, J. Phys. Chem. B, 2000, 104, 5681 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Higher-magnification image of as-prepared Pt NFs; histogram of the Pt NF size distribution on PSS-CNTs for A: Fig. 1A , B: Fig. 1C, and C: Fig. 1D; selected-area electron diffraction of Pt NFs; TEM images of the products obtained from different reducing agents: NaBH4 (A), and formic acid (B); XPS data of Au 4f (A), and Pd 3d (B). See DOI: 10.1039/b916423e |
| This journal is © The Royal Society of Chemistry 2010 |