Using “radioparagenesis” to design robust nuclear waste forms
C.
Jiang
a,
B. P.
Uberuaga
a,
K. E.
Sickafus
a,
F. M.
Nortier
b,
J. J.
Kitten
b,
N. A.
Marks
c and
C. R.
Stanek
*a
aMaterials Science and Technology Division, Los Alamos National Laboratory, Los Alamos, NM 87545, USA. E-mail: stanek@lanl.gov; Tel: +1 505 664 0361
bChemistry Division, Los Alamos National Laboratory, Los Alamos, NM 87545, USA
cNanochemistry Research Institute, Curtin University of Technology, Perth, Australia 6845
First published on 27th November 2009
Abstract
Recently, using first principles theoretical methods, we have predicted that unconventional compounds and crystal structures may form via the chemical transmutation that occurs during radioactive decay (e.g. rock salt 137BaCl formation from the β- decay of 137CsCl) (C. Jiang, C.R. Stanek, N. A. Marks, K. E. Sickafus and B. P. Uberuaga, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 132110). We refer to this phenomenon as “radioparagenesis”. In this paper, we explore the concept of applying radioparagenesis to the evaluation of long time scale crystalline nuclear waste form performance as well as to the design of chemically robust waste forms. For waste form evaluation, we specifically discuss: (1) the variation in the properties of radioparagenetic phases that govern waste form performance, as compared to conventional materials, (2) the volumetric change associated with radioparagenetic phase formation (using Cs1−xBaxCl as an example), which may lead to waste form cracking and (3) thermodynamic and mechanical stability of radioparagenetic phases. For waste form identification, we introduce the possibility of “backward design”, where the chemical evolution of the waste form is explicitly considered with the goal of achieving a defined end state. The decay of 90SrO2 to 90ZrO2 is provided as a demonstrative example. We use density functional theory to examine the performance and design of waste forms. Finally, we propose accelerated experiments to rapidly verify the formation of radioparagenetic phases, which has not occurred to date.
Broader contextAlthough public support for the expansion of nuclear power is increasing, significant growth is liable to be hindered or even halted by the seemingly intractable nuclear waste problem. A particularly difficult component of the waste problem is that any solution must be highly predictable at time scales not conducive to direct experimental verification. However, we have recently discovered a phenomenon that may permit improved predictability of long-term waste form performance. Specifically, from first principles theoretical methods, we have found that unconventional compounds and crystal structures may form via the chemical transmutation that occurs during radioactive decay (e.g. rock salt 137BaCl formation from the β- decay of 137CsCl) (C. Jiang, C.R. Stanek, N. A. Marks, K. E. Sickafus and B. P. Uberuaga, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 132110). We refer to this phenomenon as “radioparagenesis.” For crystalline nuclear waste forms, understanding this phenomenon and its consequences may allow us to examine stability far beyond what is accessible by experiments. More importantly, we may also be able to use insight gained from radioparagenesis to revise how waste forms are designed. |
1. Introduction
A combination of increasing global energy demands and the simultaneous impetus to reduce CO2 emissions has generated a renewed interest in nuclear energy. Additionally, public opinion regarding the safety of nuclear power plants has improved dramatically. Despite this revisitation of nuclear energy, there remain several outstanding issues that may prevent a so-called “nuclear renaissance”,1 including: construction cost of new plants, proliferation of nuclear material, and nuclear waste disposal. Of these, nuclear waste is the issue that possesses the highest potential to benefit from technological advances, especially in the realm of modeling and simulation. Specifically, the time and length scales that must be addressed to evaluate nuclear waste stability are decidedly extreme, and not conducive to direct experimental verification. Therefore, any solution to the waste problem must rely heavily upon computational modeling and simulation to substitute for prohibitively long experiments. In this paper, we discuss how the new concept of radioparagenesis may allow for improved predictability of long-term waste form performance, which supports our goal of designing exceptionally robust waste forms.It is well known that reprocessing spent nuclear fuel will result in a significant reduction in the volumetric load on a geologic repository. For example, Wigeland et al.2 have predicted a load increase of 225 times on a repository by removing burnable actinides (Pu, Am and Cm) and the short-lived fission products (Cs and Sr). In addition to importantly minimizing the volume in (and therefore the number of) geologic repositories, reprocessing spent nuclear fuel presents an opportunity to design customized waste forms for specific fission products. By using spent nuclear fuel as a waste form (the solution proposed in the license application for Yucca Mountain), fission products with very different chemistries are collectively encapsulated in the same waste form. As a simple example, 99Tc has a half-life of 2.13 × 105 years, while 137Cs has a half-life of only 30.07 years. Clearly, an optimized solution to the waste problem does not entail a singular disposal strategy for fission products so chemically and physically disparate. Rather, customized waste forms for specific fission products can be designed to account for the differences of fission product chemistry and physics.
For over thirty years, crystalline ceramic nuclear waste forms have been considered for the encapsulation of specific fission products.3–5 Faced with the necessity to predict the long-term behavior of ceramic waste forms, researchers have focused on candidate compositions that exhibit: (1) geological analogues, thus suggesting long-term stability, i.e. Synroc,6 (2) resistance to radiation damage (either using accelerated isotopes7 or by ion irradiation8–10) and (3) leaching resistance.11 However, these studies have not yet adequately accounted for the chemical and physical changes that may occur to the waste form during fission product transmutation, i.e. the conversion of one element or isotope into another, in this case during radioactive decay. Indeed, transmutation effects are critical to understanding the performance of a waste form. The candidate waste form selection criteria discussed above may adequately describe the performance of a waste form very early in its life, but lack the predictability required to evaluate the performance of the waste form far in to the future. The goal of this paper is to demonstrate how chemical transmutation may significantly and non-intuitively modify the structure and property relations that govern waste form performance.
The potential importance of chemical transmutation for waste form stability has been predicted previously,12,13 but the detail required to understand the effects has been limited. For example, Fortner et al.14 conducted a study of “radiogenetic transmutation effects” of 137Cs in 20 year old crystalline pollucite (CsAlSi2O6) samples. This transmission electron microscopy (TEM) study did not detect any appreciable change to the pollucite crystal structure due to 137Ba formation. However, as acknowledged by these authors, the mechanistics of Ba accommodation are not known and the samples studied may not be entirely representative of aged waste forms, since only 1.5% of the total Cs had been transmuted to Ba.
To better understand the effects of chemical transmutation, we introduce the phenomenon of “radioparagenesis”, which we define as: the formation of unconventional crystal structures (e.g. rock salt BaCl15) for compounds formed during the chemical transmutation that occurs during radioactive decay. “Paragenesis” is a term used in geology to describe the co-occurrence of minerals, which provides important insight to the sequence of and conditions for mineral formation. The conditions that govern conventional mineral formation are, of course, pressure, temperature and environment, whereas radioparagenetic phases are formed unconventionally, via the decay of an unstable isotope, in a certain crystallographic environment, to a chemically distinct element. The nuclides produced are “radiogenic”, that is, those produced by radioactive decay.16
Our initial example15 of 137CsCl → 137BaCl clearly demonstrates that radioparagenetic phases defy intuition and conventional bonding arguments (see Fig. 1). (We direct the reader to ref. 15 for additional detail of Cs1−xBaxCl). For waste forms, it is crucial to understand how the formation of these phases will impact stability. However, once we understand how the structure evolves over time, we can backward design candidate waste forms. That is, we can design a waste form from the end of their life to the beginning, rather than vice versa, which is the standard current approach. Our ultimate goal is to develop the ability to design a waste form with predictable and acceptable performance for the duration of service, and perhaps including a crystal structure that actually becomes more stable over time.
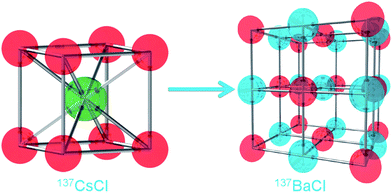 | ||
Fig. 1 Depiction of chemical transmutation of 137Cs to 137Ba via β- decay in CsCl (spacegroup: Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, no. 221), resulting in the radioparagenetic formation of rock salt BaCl (spacegroup: Fmm, no. 225). Red atoms denote Cl, green Cs and blue Ba. m, no. 221), resulting in the radioparagenetic formation of rock salt BaCl (spacegroup: Fmm, no. 225). Red atoms denote Cl, green Cs and blue Ba. | ||
Computer simulations, such as those used to discover radioparagenesis, are necessary for the evaluation and design of nuclear waste forms. Quite simply, the experiments required for direct verification are impossible. Therefore, clever accelerated aging experiments must be used to validate models, and therefore improve our confidence in them. In this paper, we introduce the concept of radioparagenesis and describe how it applies to waste form performance. Specifically, we discuss simulation results that aid the understanding of long-term waste form stability and contribute to waste form design. We also propose accelerated experiments to validate the predictions made.
2. Improved waste forms via radioparagenesis
In order to concisely demonstrate our concept of radioparagenetic phase formation effects on waste form stability, we limit our focus to the relatively short-lived fission products 90Sr and 137Cs. It is well known that these fission products represent a significant volume fraction of nuclear waste not usable as fuel, which warrants special consideration of these isotopes. However, we are particularly interested in the substantial chemical change that occurs during β− decay: 137Cs decays via β− to 137Ba, with a half-life of 30.07 years, and 90Sr β− decays to 90Y, with a half-life of 28.78 years, which then subsequently decays, again by β−, to 90Zr, with a half-life of 2.67 days. The chemical differences between the parent radioactive isotopes (137Cs and 90Sr), and the final stable isotopes (137Ba and 90Zr) are significant. For example, the charge state and size of the starting and final cations are different enough to expect variation in the structure-property relations that govern waste form stability. We have also limited our initial focus to 90Sr and 137Cs, since β− decay does not result in the radiation damage and subsequent defect creation typical of, for example, α decay. Thus, by only considering β− decay, we can focus on the effects of chemical transmutation and temporarily neglect defect structures that may well lead to even more complex radioparagenetic phases. Finally, the relative short-livedness of 137Cs and 90Sr may warrant a waste solution separate from vitrification, i.e. our approach may have the most applicability in the near term for short-lived isotopes. Although we have chosen to focus our consideration to a subset of fission products, we expect that the general concepts will be applicable to all fission products encapsulated in a crystalline waste form.The results shown in this paper are theoretical in nature. Specifically, we have employed density functional theory (DFT) to simulate the structure of example ceramic waste forms (model systems rather than representative candidates) as a function of fission product decay (and, therefore, time). Specifically, we have used the all-electron projector augmented wave method,17 within the local density approximation (PAW-LDA), as implemented in the Vienna ab initio Simulation Package (VASP).18 Large plane-wave cutoff energies (e.g. 350 eV for Cs1−xBaxCl) and dense Monkhorst-Pack k-point meshes for Brillouin zone sampling were used to ensure high accuracy for both total energy calculations. To simulate waste form structural stability far in to the future, we vary the composition of the crystalline waste form. For intermediate compositions, when both fission products and radiogenic nuclides are present, we rely on the special quasi-random structure (SQS) approach19 to generate physically meaningful solid solutions. To determine the lowest energy structure for a particular composition (and therefore, a particular time, e.g. at one half-life of 137Cs, the composition = 137Cs0.5137Ba0.5Cl), we use these theoretical methods to survey a range of possible structures. In addition to thermodynamic stability, we also use these methods to evaluate mechanical stability via consideration of elastic constants and phonon dispersion curves.15
DFT is the only viable approach to consider the effects of chemical evolution on crystalline solids. Although in some cases classical pair potentials can be used to adequately describe the starting waste form compound, e.g. CsCl, these types of models will not capture radioparagenetic phase formation where the bonding nature deviates from purely ionic in a way that is difficult to predict a priori. That is, the pair-potentials used for these models cannot be derived for compounds that have not been observed before. In addition to the inability to perform various types of simulation, experiments on actual waste products of interest are far too long to be feasible.
2.1 Implications of radioparagenetic phase formation on waste form stability
Current models of long-term waste form performance are based upon the material properties of conventionally synthesized prototypes, i.e. the starting waste form composition. However, as we have shown,15 new phases may form unconventionally via radioparagenesis during the decay of fission products. These radioparagenetic phases may exhibit dramatically different properties later in life than the original material. It then follows that a waste form demonstrating acceptable properties at t = 0 may exhibit significantly different properties due to chemical transmutation; that is, the new phase may behave very differently than what is expected from the starting phase. From a qualification and licensing perspective, it is imperative that the performance of the waste form be not only acceptable, but also predictable, thus motivating consideration of the effects of radioparagenesis on material properties.An example of material property variation is the bonding nature of rock salt 137BaCl (predicted to form from the decay of B2 structured 137CsCl).15 Electronic structure calculations revealed that BaCl exhibits a complex mixture of metallic, covalent and ionic bonding.15 One can reasonably expect, for example, the diffusivity and corrosion resistance of a material with this complex bonding nature to be markedly different from that of an ionic crystal, such as CsCl. Due to the long (in experimental terms) half-life of 137Cs, it may prove difficult to synthesize BaCl in a timely manner. Thus, experimental verification of material property variation must rely on a combination of theoretical studies (it is possible to simulate important aspects of diffusion and corrosion in radioparagenetic phases) and accelerated aging experiments (which are discussed later in this paper).
Another example of how chemical transmutation might affect waste form stability is the variation in size between the beginning and final cations. As pointed out by Gray,11 the reduction in cation radius between Cs+ and Ba2+ is 20%, while Zr4+ is 30% smaller than Sr2+. Since large volumetric changes may lead to cracking, and therefore increased surface area that will likely enhance fission product leaching, understanding how the crystal lattice responds to these changes in cation size is important. Therefore, phase transformations that result in the least amount of volumetric change are likely preferred. Fig. 2 shows DFT predictions of how the CsCl lattice changes with Ba formation. Recall that, as 137Cs decays to 137Ba in the B2 CsCl structure (red curve in Fig. 2), the rock salt structure (blue curve in Fig. 2) becomes energetically favored when about 18% of the 137Cs has decayed.15 When x is small in Cs1−xBaxCl, the volume difference between B2 and rock salt is largest, and the volumetric difference decreases with increasing x. For example, when x = 0.2 (a composition where the B1 rock salt phase begins to be preferred over B2 CsCl), Fig. 2 shows that the volume difference between the B1 and B2 phases is ∼20%, while when x = 0.8, the volume change is only ∼15%. Therefore, to avoid any deleterious effects of drastic volume change on waste form stability and performance (e.g. cracking), it is desirable for the B2 to rock salt phase transformation to occur at large x. Although our results predict that the thermodynamic preference is for this transformation to occur when x is relatively small, it may be kinetically limited. Nevertheless, a general waste form design principle should be to minimize volumetric changes that may negatively impact waste form stability. For this reason, CsCl is clearly not an optimized host for 137Cs (but is useful in demonstrating the phenomena that govern long-term performance).
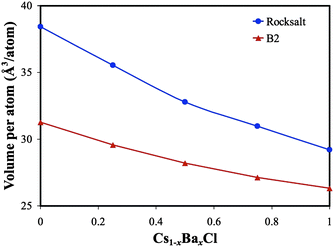 | ||
| Fig. 2 The volume decrease of CsCl as a function of Ba concentration. The structural variation in both the CsCl B2 structure (red curve) and the rock salt structure (blue curve) are shown. | ||
Finally, in addition to material property variation and cation size effects, we have also been able to predict the thermodynamic metastability of radioparagenetic phases. We did this by comparing lattice energies (calculated by the first principles approach described earlier) of possible phases for specific compositions corresponding to varying amounts of daughter product formation. For example, in the case of Cs1−xBaxCl, we considered three crystallographic structures: B1 rock salt, B2 CsCl and B3 zincblende, for x = 0, 0.25, 0.5, 0.75 and 1. We also considered the possibility of phase decomposition to Ba metal plus BaCl2.15 We found that at x ∼ 0.175, the B1 rock salt structure became favored over the original B2 CsCl structure, and this preference increased to 0.2 eV/atom by x = 1. Also, the driving force for phase decomposition was calculated to be only 0.03 eV/atom, suggesting that the formation of the BaCl phase is possible.15 In the next section, we describe the results of similar calculations for Sr1−xZrxO2. However, it may be that a thermodynamically metastable radioparagenetic phase becomes mechanically unstable at a critical concentration of daughter product. Mechanical stability can be evaluated by calculating elastic constants (the strain energy must be positive against any homogeneous elastic deformation) and phonon dispersion curves (to ensure there are no imaginary frequencies). Although BaCl satisfied these requirements for mechanical stability (see ref. 15), we expect that other radioparagenetic phases may spontaneously transform to lower energy phases due to mechanical instability. Again, complete understanding of phase evolution is required for reliable prediction of long time scale performance.
2.2 Design of waste forms using radioparagenetic insights
Once we understand the mechanistics of radioparagenetic phase formation, we may use the information gained to not only describe non-intuitive material property variation, but also to “backward” design waste form compounds. The current approach to design waste forms begins with a very well-known material, which evolves in a less well-known (or even unknown) manner. This approach relies upon sufficient waste form performance at t = 0 to withstand any changes over time due to radiation or chemical transmutation. As described in the previous section, in many cases, the unexpected formation of radioparagenetic phases may result in unstable phases, or new phases with properties significantly different than likely anticipated. However, we may be able to take advantage of insights gained from radioparagenesis and look far into the future to design waste form compositions from the future to the present. Furthermore, with complete understanding, we may manipulate the effects of radioparagenesis and design a waste form that takes advantage of non-intuitive phase formation.As a hypothetical example, on account of its well-known stability, ZrO2 may be the desired end-of-life phase of a waste form dedicated to the encapsulation of 90Sr. To investigate the possibility of using the desired end state of the waste form to define the starting compound, we performed DFT calculations for the Sr1−xZrxO2 system, analogous to those we performed for Cs1−xBaxCl. The results for Sr1−xZrxO2 are shown in Fig. 3. For the five crystal structures, we consider: CaC2, spacegroup #139 (which we calculate to be the ground state of SrO2, and therefore is the reference that the other four structures are shown with respect to), PbO2 spacegroup #136, and the three polymorphs of ZrO2, monoclinic spacegroup #13, tetragonal spacegroup #137 and cubic spacegroup #225. Thus, Fig. 3 shows that almost immediately (i.e. with only a small amount of Zr formation), the PbO2 structure is preferred over the CaC2 structure. However, the inset of Fig. 3 shows that near complete transmutation of Sr to Zr, the monoclinic ZrO2 structure becomes preferred (at x ∼ 0.9). We have not performed an exhaustive SQS study for the intermediate compositions, as consideration of the end members clearly demonstrates the concept of defining the end state to determine the beginning state. That is, for waste forms, Fig. 3 shows that if one chooses the SrO2 phase as the initial 90Sr waste form, the thermodynamic preference is to transform to monoclinic ZrO2. Although SrO is the most common strontium oxide phase, SrO2 is readily synthesized at high temperature and pressure,20 and therefore is a viable starting point. Thus, as 90Sr decays to 90Zr, the material actually becomes more thermodynamically stable, not less. Although an oversimplification, this example demonstrates how an initial waste form composition can be selected by defining the desired end state. Of course, for this approach to reach its full potential, it should be extended to understand the phase stability of a candidate waste form during its entire lifespan.
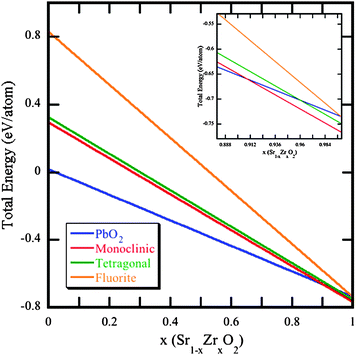 | ||
| Fig. 3 The thermodynamic stability of four crystallographic structures for the Sr1−xZrxO2 system as a function of x, where x = 0 corresponds to pure 90SrO2. The data is presented with respect to the CaC2 structure (space group #139), which is the lowest energy at x = 0. At x = 1, the typical polymorphs of ZrO2 are predicted, and the PbO2 structure (space group #136) has relaxed to the fluorite structure. | ||
Further study in this area will likely lead to optimized waste form compositions with predictable and acceptable performance during the entire lifetime of the waste form. Although we have limited our initial consideration of radioparagenetic effects to binary compounds, these materials are unlikely to be seriously considered as waste forms. Rather, complex oxides that have already been shown to be resistant to radiation induced amorphization may exhibit the chemical and structural flexibility required to also withstand chemical transmutation effects. For example, already in 1982, Vance et al.21 began the search for structures and classes of materials that could accommodate both the parent and daughter nuclides on the same lattice site. In their work, Vance et al.21 identified ABO3 perovskites as a class of materials worth further consideration,21 but coherently described the difficulty of finding an optimized compound that could account for the size and valence changes that occur during transmutation. Now, with improved understanding of the effects of chemical transmutation from our work on radioparagenesis, we are in a position to more knowledgably re-examine waste form candidates. However, to evaluate waste form performance while accounting for radioparagenetic phase formation, we must not rely solely on conventional synthesis approaches, but rather shift our dependence to first principles simulation combined with experiments that incorporate short-lived isotopes in candidate systems. Such experiments are described in the next section.
2.3 Experimental validation of radioparagenetic phase formation
At this point, the concept of radioparagenesis is only a hypothesis, and experimental verification is lacking. The reason radioparagenesis has not yet been observed experimentally is that the time scales of relevant fission product decay are sufficiently long to prohibit observation. Simply, the study of crystalline waste forms has not been carried out for over a long enough time to observe radioparagenesis. For instance, observation of radioparagenetic phase formation for 90Sr or 137Cs may take well over 100 years and crystalline waste forms have been studied for only 30 years. In order to remove this time barrier, we have developed an experimental plan that should identify radioparagenetic phases and validate our theoretical results.To rapidly invoke structural changes due to chemical transmutation, we have identified isotopes that quickly decay to chemically distinct daughter products. For example, 109Cd, 88Zr and 67Cu can be readily synthesized via nuclear reactions that occur during proton irradiation of In, Nb and Zn targets, respectively. A chemical recovery process can yield microgram quantities of 109Cd, 88Zr and 67Cu chloride (X-ray diffraction is required to characterize the structure of the separated chloride). While these isotopes are highly radioactive, emitting high energy gamma rays, the small amounts necessary to observe radioparagenesis can be handled in glove box environments. The decay chains of 109Cd (i.e. 461 day half-life, electron capture (EC) to 109Ag), 88Zr (i.e. 83.4 day half-life, EC to 88Y, 106.65 day half-life, which in turn decays via EC to stable 88Sr) and 67Cu (i.e. 2.58 day half-life, β− to 67Zn) allow us to directly and rapidly observe the effects of chemical evolution, i.e. daughter products are sufficiently different in size and valence. More important than explicit consideration of a relevant fission product is the demonstration of the effect of radioparagenesis on crystalline stability. Coincidentally, although 88Zr is not a relevant fission product, it exhibits the “inverse” of 90Sr decay (i.e. β− to 90Y and β− to 90Zr), and is therefore prototypic of the changes expected in actual nuclear waste. For 109Cd, 88Zr and 67Cu chloride, we can compare first principles predictions of decay to X-ray diffraction results (perhaps requiring synchrotron sources due to the small sample size). Diffraction patterns should be obtained throughout the decay process. Eventually, stable 109Ag, 88Sr and 67Zn chloride samples can be characterized for local atomic structure, leaching and thermodynamic stability to determine the variation of structure and material properties with composition.
Similar simulations and experiments can be performed for short-lived radionuclide reactor products. For example, 45Ca, 89Sr and 177Lu are readily available from facilities such as the Missouri University Research Reactor (MURR). Each of these isotopes decay via β−, so the daughter products (45Sc, 89Y and 177Hf, respectively) are, again, chemically distinct. Furthermore, the half-lives of these isotopes (162 days, 50 days and 160 days, respectively) are conducive to experimental consideration. Other examples of accelerated analogues exist, but those mentioned here are the most likely to allow observation of radioparagenetic phase formation.
Previously, similar accelerated experiments have been proposed using neutron irradiation to generate isotopes through neutron capture.12 Gray synthesized 134Cs from natural 133Cs in the high flux isotope reactor (HFIR) at Oak Ridge National Laboratory. Natural Cs is comprised entirely of the 133 isotope, which has a fairly large thermal neutron cross section, thus leading to the formation of 134Cs during neutron irradiation. 134Cs decays via β− to 134Ba with a 2.04 year half-life, resulting in a much shorter experiment than if the 137 isotope was considered. Although this appears an attractive experiment due to the identical decay products and short half-life, Gray was only able to convert 12% of 133Cs to 134Cs. This is because 134Cs also has a large neutron capture cross section that results in the formation of long-lived 135Cs. Therefore, although Gray observed no appreciable change in crystalline waste form structure (e.g. pollucite), the fraction of transmuted atoms was much lower than expected for actual waste.
Other examples of short-lived, neutron activated examples exist. For example, 139La is 99% abundant and has a thermal neutron capture cross section of 9 barns. With a fair amount of irradiation time, 140La could be produced, which quickly (1.678 day half life) β− decays to 140Ce. An alternative option is 175Lu, which has a 16 barn thermal neutron cross-section, producing 176Lu, which β− decays very quickly to 176Hf. In addition to the difficulties associated with synthesizing sufficient quantities of material and subsequent handling of these highly radioactive samples, a further complication may obscure the effects of chemical transmutation. That is, neutron irradiation (predominantly fast neutrons) will generate point defects that are not indicative of a waste form. For example, point defects may allow pathways for the materials to avoid metastable radioparagenetic phases by phase decomposition (most of the radioparagenetic phases are thermodynamically metastable). Gray realized this issue during his experiment and annealed the crystalline samples after irradiation in order to hopefully remove the damage.12 Furthermore, reactor produced isotopes are typically not as pure as those produced by an accelerator.
The two isotope isolation approaches described above that require chemical separation are more indicative of actual waste, since waste products will be incorporated in a waste form, not generated in the form itself. Gray12 also noted that short-lived isotopes could be used to simulate waste, but suggested that the experiments were prohibitively expensive. On the contrary, recent advances in isotope production, e.g. for the nuclear medicine community, make these types of experiments feasible. Regardless, any experiment that produces an ample quantity of a radioisotope that decays quickly enough to cleanly observe (i.e., without defects) radioparagenesis is sufficient.
3. Conclusions
We have introduced the new concept of radioparagenesis, which we define as: the formation of unconventional compounds and crystal structures due to the chemical transmutation that occurs during radioactive decay. This concept may permit the removal of a barrier preventing the expansion of nuclear energy by significantly improving the predictability of long-term waste form stability. That is, by understanding radioparagenesis, we acquire knowledge of how the material properties that govern waste form performance vary during fission product decay. However, this approach only allows us to comment on the relative performance of a waste form if radioparagenetic phases were assumed not to form. Much more powerful is the design of new waste form materials from the realization that radioparagenetic phases do form. Therefore, we have extended our consideration to apply radioparagenetic insight to design robust waste forms. The innovativeness of this approach is that the initial waste form can be determined based upon a desired end state, which accounts for chemical transmutation of the encapsulated fission product. Finally, the concept of radioparagenesis remains to be validated. To directly verify the phenomenon for an actual “short-lived” fission product (such as 90Sr or 137Cs) would likely require more than 100 years. Therefore, we have proposed an accelerated experiment that will allow rapid confirmation of the concept.The examples provided in this paper demonstrate how the concept of radioparagenesis may be applied to improve waste form design and performance. Further work is required to extend these concepts beyond theoretical considerations to experimental demonstrations and optimized waste solutions.
References
- W. J. Nuttal, Nuclear Renaissance: Technologies and Policies for the Future of Nuclear Power, Institute of Physics Publishing, 2005 Search PubMed.
- R. A. Wigeland, T. H. Bauer, T. H. Fanning and E. E. Morris, Nucl. Technol., 2006, 154, 96.
- G. J. McCarthy, Nucl. Technol., 1977, 32, 92 CAS.
- G. R. Lumpkin, Elements, 2006, 2, 365 CrossRef CAS.
- W. Lutze and R. C. Ewing, Radioactive Waste Forms for the Future, North-Holland, New York, 1988 Search PubMed.
- A. E. Ringwood, S. E. Kesson, N. G. Ware, W. Hibberson and A. Major, Nature, 1979, 278, 219 CAS.
- I. Farnan, H. Cho and W. J. Weber, Nature, 2007, 445, 190 CrossRef CAS.
- K. E. Sickafus, L. Minervini, R. W. Grimes, J. A. Valdez, M. Ishimaru, F. Li, K. J. McClellan and T. Hartmann, Science, 2000, 289, 748 CrossRef CAS.
- K. E. Sickafus, R. W. Grimes, J. A. Valdez, A. R. Cleave, M. Tang, M. Ishimaru, S. M. Corish, C. R. Stanek and B. P. Uberuaga, Nat. Mater., 2007, 6, 217 CrossRef CAS.
- J. Lian, J. Chen, L. M. Wang, R. C. Ewing, J. M. Farmer, L. A. Boatner and K. B. Helean, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 134107 CrossRef.
- E. R. Vance, Ceram. Trans., 2006, 176, 153 CAS.
- W. A. Gray, Nature, 1982, 296, 547 CrossRef CAS.
- W. J. Weber, R. C. Ewing, C. R. A. Catlow, T. Diaz de la Rubia, L. W. Hobbs, C. Kinoshita, Hj. Matzke, A. T. Motta, M. Nastasi, E. K. H. Salje, E. R. Vance and S. J. Zinkle, J. Mater. Res., 1998, 13, 1434 CAS.
- J. Fortner, S. Aase and D. Reed, Mat. Res. Soc. Symp. Proc., 2002, 713, 527 CAS.
- C. Jiang, C. R. Stanek, N. A. Marks, K. E. Sickafus and B. P. Uberuaga, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 132110 CrossRef.
- A. P. Dickin, Radiogenic Isotope Geology, Cambridge University Press, Cambridge, 2005 Search PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169 CrossRef CAS.
- C. Jiang, C. Wolverton, J. Sofo, L. Q. Chen and Z. K. Liu, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 214202 CrossRef.
- J. D. Bernal, E. Diatlova, I. A. Kazarnovskii and S. Reichstein, Z. Kristallogr., 1935, A32, 344.
- E. R. Vance, R. Roy, J. G. Pepin and D. K. Agrawal, J. Mater. Sci., 1982, 17, 947 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |