Responsive magnetic resonance imaging contrast agents as chemical sensors for metals in biology and medicine
Emily L.
Que
a and
Christopher J.
Chang
*ab
aDepartment of Chemistry, University of California, Berkeley, CA 94720, USA
bThe Howard Hughes Medical Institute, University of California, Berkeley, CA 94720, USA. E-mail: chrischang@berkeley.edu
First published on 7th October 2009
Abstract
This tutorial review highlights progress in the development of responsive magnetic resonance imaging (MRI) contrast agents for detecting and sensing biologically relevant metal ions. Molecular imaging with bioactivatable MRI indicators offers a potentially powerful methodology for studying the physiology and pathology of metals by capturing dynamic three-dimensional images of living systems for research and clinical applications. This emerging area at the interface of inorganic chemistry and the life sciences offers a broad palette of opportunities for researchers with interests ranging from coordination chemistry and spectroscopy to supramolecular chemistry and molecular recognition to metals in biology and medicine.
 Emily L. Que | Emily Que was born in Ithaca, New York, and grew up in St. Paul, Minnesota. She attended the University of Minnesota, Twin Cities (B.S. 2004) where she was an undergraduate researcher in the laboratories of Prof. Lawrence Que and Prof. Andreas Stein. Emily received a Barry Goldwater scholarship for her accomplishments in undergraduate research. Emily recently received her PhD (2009) in the laboratory of Prof. Chris Chang at the University of California, Berkeley. Future plans include a postdoctoral position in the laboratory of Prof. Tom O’Halloran at Northwestern University. Her general research interests lie in the field of bioinorganic chemistry. |
 Christopher J. Chang | Chris Chang received his BS and MS degrees from Caltech, in 1997, working with Prof. Harry Gray. After spending a year as a Fulbright scholar in Strasbourg, France, with Dr Jean-Pierre Sauvage, Chris received his PhD from MIT in 2002 under the supervision of Prof. Dan Nocera. He stayed at MIT as a postdoctoral fellow with Prof. Steve Lippard and then began his independent career at UC Berkeley in July 2004. Chris’ research laboratory currently uses a combination of inorganic chemistry, organic chemistry, chemical biology, and molecular biology approaches to study problems in neuroscience, immunology, energy research, and green chemistry. |
1. Introduction
Bioinorganic chemistry is a field at the interface between inorganic chemistry and the life sciences that spans many traditional disciplines of study. Metals are essential elements for the growth and development of all living organisms, but improper regulation of metal ion stores is also connected to acute and long-term diseases including heart disease, cancer, diabetes, and neurodegeneration. Likewise, the unique chemical properties of metals and other elements across the periodic table can be exploited for human benefit through the development of novel therapeutics and diagnostics.An emerging intersection between metals in biology and metals in medicine is molecular imaging, which uses chemistry to visualize living biological systems from the molecule to cell to tissue to organism level. In this regard, a particularly powerful, clinically-used modality for molecular imaging is magnetic resonance imaging (MRI), a technique that offers the ability to capture three-dimensional images of living specimens with exquisite anatomical resolution.1 An exciting frontier for MRI is the development of responsive contrast agents that can be used to report chemical species and reactions of interest in living biological systems. In this tutorial review, we will summarize advances in the creation of new responsive MRI contrast agents for detecting and sensing biologically relevant metal ions. Rather than providing a comprehensive list of molecular probes, our goal is to showcase opportunities where expertise and interest in coordination chemistry and spectroscopy, supramolecular chemistry and molecular recognition, and metals in biology and medicine can make important contributions to this growing field. We note elegant work outside the scope of this review on “smart” MRI contrast agents for detecting enzymatic activity, pH, pO2, glucose, lactate, nitric oxide, etc.2,3 We will first outline principles for designing effective metal-responsive MRI contrast agents, followed by a selection of specific examples of MRI-based sensors for detecting s-block (potassium, magnesium, calcium) and d-block (zinc, copper, and iron) metals. We will then close with a discussion on current challenges and future directions for exploration.
2. MRI contrast agents and relaxivity
Magnetic resonance imaging is a technique used widely in the medical field for imaging anatomical structure. With MRI, three-dimensional images of entire live specimens can be obtained with high resolution and without the use of ionizing radiation. The most common contrast observed in MR images is derived from protons in water molecules, with different environments leading to different signal intensities. Other NMR-active nuclei (e.g.19F, 13C, and 129Xe) are also emerging additions to this chemical toolbox and outside the scope of this review.In up to 50% of all MRI scans in the clinic,1,4 contrast agents are used to enhance image contrast. These contrast agents contain paramagnetic metal ions that increase the relaxation rate of interacting water protons. A number of different paramagnetic metal ions can offer contrast enhancement, including Mn2+, Mn3+, Fe3+, Cu2+, and Gd3+. Gd3+, with 7 unpaired electrons (S = 7/2), has emerged as an important player in the field of MR contrast agents owing to its large number of unpaired electrons and its ability to efficiently relax nearby nuclei. As such, the majority of contrast agents discussed in this review are small-molecule Gd3+-based contrast agents, though one Mn3+-chelate, one Eu3+-chelate, and one superparamagnetic iron oxide nanoparticle (SPION) system will be discussed as well.
The efficiency with which a contrast agent enhances the relaxation rate is termed relaxivity (ri) and is defined by eqn (1).1 In this equation, Ti refers to the relaxation time, (1/Ti) refers to the 1/Ti of the contrast agent solution, (1/Ti)0 refers to the 1/Ti of the contrast agent-free solution, ri refers to the relaxivity, and [CA] is the millimolar concentration of the contrast agent. Contrast agents enhance both longitudinal (1/T1) and transverse (1/T2) relaxation rates. Gd3+-based contrast agents affect longitudinal and transverse relaxivities to a similar degree and are better suited for T1-weighted imaging, whereas iron nanoparticle agents increase transverse relaxivity to a greater degree and thus are better suited for T2-weighted imaging.
| (1/Ti) = (1/Ti)0 + ri[CA] | (1) |
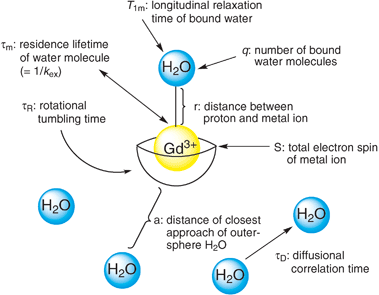 | ||
| Fig. 1 Variables contributing to contrast agent relaxivity. | ||
3. Sensors for the MRI modality by activating changes in relaxivity
The signal intensity in a proton-based MR image depends on the concentration of water and the T1 and T2 relaxation times. As such, by designing contrast agents that change T1/T2 relaxation times of the water protons in response to a given analyte or reaction of interest, one can create chemical and biological sensors using the MRI modality. Because MRI is relatively insensitive compared to other modalities such as optical imaging or PET, the magnitude of this relaxivity change is critical for devising useful chemosensors. For calibration, as little as a 10% change in 1/T1 can be observed using MRI at clinically relevant field strengths,1 but larger dynamic ranges are obviously more desirable.In terms of contrast agent design, a variety of factors govern their relaxivity, and as such, many approaches can be exploited for sensing purposes; common routes to metal ion sensing using MRI contrast agents are depicted in Fig. 2. Most prevalently, contrast agent sensors display a change in q in response to a particular analyte. Meade et al. pioneered this concept in the development of EGad, a reporter for β-galactosidase that represents the first bioactivatable MRI contrast agent.8 In addition to q-modulation, another factor that can be exploited is τR, with a larger τR value giving a higher relaxivity. Finally, one can also envision tuning water exchange rate to invoke a relaxivity change.
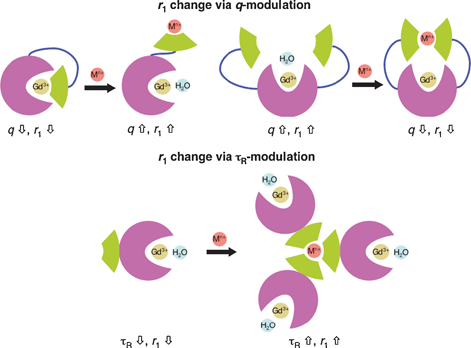 | ||
| Fig. 2 MR-based metal ion sensing via modulation of q or τR. | ||
4. Design criteria for creating metal-responsive MRI contrast agents
The most important factor in designing metal-responsive MRI contrast agents for research and clinical applications is chemical selectivity for a metal ion of interest over other metal ions in the presence of various anions, small organic molecules, and macromolecules. This type of molecular recognition is particularly challenging as biological media are polar, protic, and of high ionic strength. A wide range of metal ions is present in biological systems, often at high mM concentrations (i.e. Na+, K+, Mg2+, and Ca2+). Useful MRI sensors must therefore respond only to a particular metal ion of interest so that false signals are not observed, and not be muted by this high background of cations. Selectivity is governed by the architecture of the response element of the contrast agent. Characteristics such as donor type (hard/soft, neutral/charged, etc.), number of donors, cavity size, and geometry can all be tailored to match a metal ion of choice. Binding affinity is also critical in designing metal ion sensors and Kd values should be modulated to match a system of interest. High relaxivity values and large modulations in relaxivity can minimize the amount of contrast agent needed for imaging applications, which in turn minimizes any potential artifacts that arise from introduction of these external indicators. Finally, MRI probes must be compatible with biological systems. Practical properties include contrast agents that are water-soluble, non-toxic, and able to image extracellular and/or intracellular spaces. The next section will highlight specific examples of MRI-based metal sensors. A summary of the metal-sensing properties of contrast agents discussed is presented in Table 1.| Contrast agent | 1H frequency, temperature | r 1 | K d | |
|---|---|---|---|---|
| No Mn+ | Mn+ | |||
| a Transverse relaxivity values (r2). b EC50 value. c Measured using the fluorescence response of the free ligand (DPA–C2)2–TPPS3. d Responds equally to Cu+ and Cu2+. | ||||
| Ca 2+ -sensitive agents | ||||
| DOPTA–Gd, Gd-1 | 500 MHz, 25 °C | 3.26 | 5.76 | 0.96 μM |
| Gd-2a | 500 MHz, 25 °C | 3.35 | 3.85 | 13 mM |
| Gd-2b | 500 MHz, 25 °C | 2.6 | 2.9 | 2.0 mM |
| Gd-3 | 500 MHz, 25 °C | 4.7 | 5.4 | 0.25 mM |
| Gd-4 | 500 MHz, 25 °C | 5.4 | 7.1 | 0.40 mM |
| Gd-5a | 500 MHz, 25 °C | 4.05 | 6.86 | 0.20 mM |
| Gd-5b | 500 MHz, 25 °C | 3.44 | 6.29 | 20 μM |
| Gd-6 | 400 MHz, 27 °C | 3.5 | 6.9 | 11 μM |
| 3C : 1M | 200 MHz | 220a | 45a | 1.4 μMb |
| 3C : 1R | 200 MHz | 200a | 34a | — |
| K + -sensitive agent | ||||
| KMR–K1, Gd-7 | 20 MHz, 40 °C | 5.05 | 4.78 | 0.45 mM |
| Mg 2+ -sensitive agent | ||||
| KMR–Mg, Gd-8 | 20 MHz, 40 °C | 4.98 | 3.95 | 4.1 mM |
| Zn 2+ -sensitive agents | ||||
| Gd-9 | 300 MHz, 25 °C | 6.06 | 3.98 | — |
| Gd-10 | 300 MHz, 25 °C | 4.8 | 3.4 | — |
| Gd–daa3, Gd-11 | 60 MHz, 37 °C | 2.3 | 5.1 | 0.24 mM |
| Gd–apa3, Gd-12 | 60 MHz, 37 °C | 3.4 | 6.9 | — |
| Mn3+–(DPA–C2)2–TPPS3, Mn-13 | 200 MHz, 25 °C | 8.70 | 6.65 | 12 nMc |
| Eu(dotampy), Eu-14 | — | — | — | 26 nM |
| Gd–DOTA–BPEN, Gd-15 | 23 MHz, 37 °C | 6.6 | 17.4 | 33.6 nM |
| Fe 2+ -sensitive agents | ||||
| Gd-16 | 30 MHz, 25 °C | 20.2 | 32.2 | — |
| Gd-18 | 20 MHz, 25 °C | 6.2 | 11 | — |
| Cu 2+ -sensitive agents | ||||
| CG1, Gd-22 | 400 MHz, 25 °C | 3.76 | 5.29 | 167 μM |
| CG6d, Gd-27 | 60 MHz, 37 °C | 2.2 | 3.8 | 0.99 fM |
| Cu + -sensitive agents | ||||
| CG2, Gd-23 | 60 MHz, 37 °C | 1.5 | 6.9 | 0.26 pM |
| CG3, Gd-24 | 60 MHz, 37 °C | 1.5 | 6.9 | 0.037 pM |
| CG4, Gd-25 | 60 MHz, 37 °C | 1.7 | 6.6 | 14 pM |
| CG5, Gd-26 | 60 MHz, 37 °C | 1.2 | 2.3 | 32 pM |
5. Survey of MRI-based metal sensors
5.1 MRI contrast agents for calcium detection
The average human contains over 1 kg of calcium, with the majority of this metal stored in mineral form within bones. However, approximately 1% of the body’s calcium circulates in fluid solution, with μM Ca2+ concentrations inside the cell and mM concentrations outside the cell. Ca2+ is a critical second messenger in a wide range of cell signaling processes throughout the body. Upon activation with various stimuli, normal intracellular Ca2+ concentrations (∼100 nM) can rise as much as four orders of magnitude (1–2 mM), initiating signaling cascades that control cellular events from fertilization to development to division to apoptotic death.9Selected Ca2+-responsive MRI contrast agents are displayed in Fig. 3. Meade and co-workers developed the first Ca2+-sensitive MRI contrast agent DOPTA–Gd (Gd-1).10,11 DOPTA–Gd contains two DO3A–Gd chelates that are linked by a modified BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N′N′-tetraacetic acid) moiety, a known selective binder of Ca2+. The iminodiacetate groups of the BAPTA linker bind to the Gd3+ centers in the absence of Ca2+ resulting in a relaxivity of 3.26 mM−1 s−1. Upon addition of Ca2+ ions, these groups preferentially bind Ca2+ and coordination sites around the Gd3+ centers are opened up to solvent water, leading to a ca. 80% relaxivity increase to 5.76 mM−1 s−1. Binding of Ca2+ is in the micromolar range (apparent Kd = 0.96 μM) and the modified BAPTA moiety maintains its preference for Ca2+ over Mg2+. In a biological context, the selectivity and Ca2+ binding affinity of DOPTA–Gd make this agent potentially suitable for monitoring intracellular Ca2+ fluctuations. Further investigation into the mechanism of Ca2+ sensing by DOPTA–Gd confirmed a change in the number of inner-sphere water molecules upon introduction of Ca2+ as suggested by luminescence lifetime measurements of DOPTA–Tb.
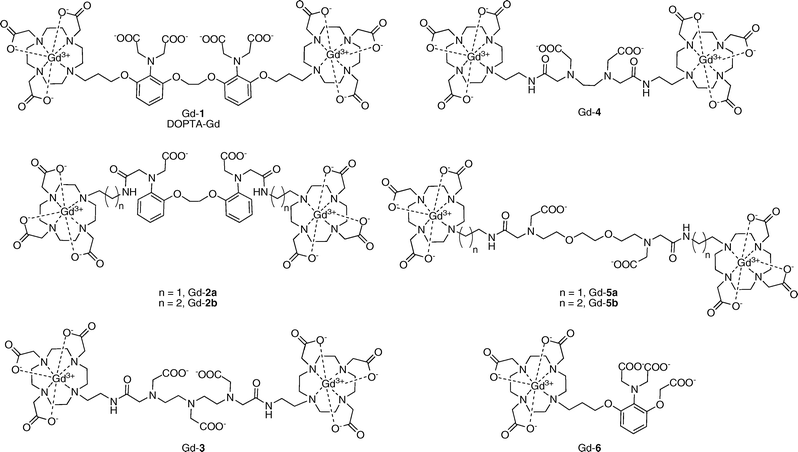 | ||
| Fig. 3 Calcium-activated MRI contrast agents. | ||
Logothetis, Tóth, and co-workers have pursued Ca2+-binding domains designed to possess lower affinity for Ca2+ with the aim of developing agents suitable for studying fluctuations in extracellular Ca2+ levels. One report describes two bismacrocyclic Gd3+ chelates in which the two Gd–DO3A cores are joined with a BAPTA–bisamide linker (Gd-2a and Gd-2b).12 These agents have modest increases in relaxivity in response to excess Ca2+ (15% and 10%), consistent with q-modulation as determined by luminescence measurements with Eu-2a and Eu-2b. The hydration states of Eu-2a were further investigated by high-resolution UV-vis spectroscopy. Replacement of two of the carboxylate groups in BAPTA for amide groups in Gd-2a and Gd-2b effectively decreases the Ca2+ binding affinity (Kd = 13 and 2.0 mM, respectively) while maintaining selectivity for Ca2+ over Mg2+.
Other low affinity MRI contrast agents for calcium sensing have been developed that contain EDTA, DTPA, and EGTA linkers between two Gd–DO3A cores.13,14 DTPA-derivative Gd-3 exhibits a 15% increase in relaxivity in response to Ca2+ (r1 = 4.7 to 5.4 mM−1 s−1) and has millimolar affinity for Ca2+ (Kd = 0.25 mM).13 This agent is also responsive to Mg2+ (Kd = 2.0 mM). Luminescence lifetime measurements with Eu-3 indicate that the relaxivity change is not due to a change in the coordination environment at the Gd3+ center as no change in q is observed. EDTA-derivative Gd-4 has a 32% increase in relaxivity in response to Ca2+.13 Converting from the tricarboxylate DTPA-derivative to the dicarboxylate EDTA-derivative appears to reduce the Mg2+ binding affinity enough to make the response to Mg2+ negligible while still maintaining good affinity for Ca2+ (Kd = 0.40 mM).
EGTA-derivatives Gd-5a and Gd-5b also respond to Ca2+ ions and are selective for Ca2+ over Mg2+, again demonstrating the importance of carboxylate denticity for discriminating Ca2+versus Mg2+ because the sensor portions of these ligands contain only two carboxylates as opposed to three as in Gd-3.14 Gd-5a, where the EGTA group is connected to the DO3A moieties via an ethylene linker, displays a 69% increase in relaxivity when 1 equiv. of Ca2+ is added and has a Kd of 0.20 mM for the ion. Propylene-linked Gd-5b also binds to Ca2+ but has a higher response (a 83% increase in relaxivity with addition of 1 equiv. of Ca2+) and a tighter binding affinity (Kd = 20 μM). The differences between these two compounds demonstrate how even subtle changes to the contrast agent architecture can lead to sensors with altered properties. Encouraged by the favorable properties of these complexes, experiments were performed to determine whether these agents could be used to sense Ca2+ fluctuations in biological settings. In addition to being able to sense Ca2+ in simple buffer solutions, these two agents also respond to Ca2+ in a DMEM–F-12 medium with a GIBCO™ N-2 supplement, an approximation of the brain extracellular medium (BEM). In this buffer system, both sensors exhibit ∼50% increases in relaxivity in response to Ca2+ and maintain similar Ca2+ binding affinities (Kd = 50 μM for Gd-5a and 25 μM for Gd-5b).
A Ca2+-sensitive contrast agent containing only one Gd–DO3A core has been reported recently.15 Gd-6 has the highest turn-on response among the small-molecule MRI-based Ca2+ sensors, going from r1 = 3.5 mM−1 s−1 in the absence of Ca2+ to r1 = 6.9 mM−1 s−1 with 1 equiv. of Ca2+ (a 97% relaxivity increase). The binding affinity for Ca2+ is in the micromolar range (Kd = 11 μM), but use of this more compact metal-chelating moiety also influences Ca2+ specificity, as Mg2+ and Zn2+ can also trigger relaxivity increases for this contrast agent. This MRI sensor was also tested in artificial cerebrospinal fluid (ACSF) and artificial extracellular matrix (AECM) at 37 °C to simulate biological conditions and displayed 36% and 25% relaxivity enhancements, respectively, in response to Ca2+.
In addition to small-molecule Gd3+-chelates, superparamagnetic iron oxide (SPIO) nanoparticles have been exploited for MRI-based Ca2+ detection.16 SPIO nanoparticles are T2 relaxation agents and their ability to increase the T2 relaxation rate is dependent on a number of factors, including the aggregation state of the particles. Jasanoff and co-workers exploit this property for Ca2+ sensing using calmodulin (CaM), a classic calcium-signaling protein system that binds target peptides in a manner that is dependent on Ca2+ levels. In this design, two types of SPIOs will aggregate upon calcium addition by crosslinking between CaM and signal peptides, causing a change in T2 relaxivity. Specifically, streptavidin-modified SPIO nanoparticles were conjugated to biotinylated-CaM or biotinylated target peptide sequences M13 and RS20. Different ratios of CaM- and peptide-decorated SPIO nanoparticles were tested for Ca2+ sensitivity, and light-scattering and atomic force microscopy experiments show that a 3 : 1 CaM–peptide ratio yielded optimal nanoparticle aggregation properties. T2-weighted relaxivity measurements revealed large relaxivity changes in response to Ca2+ for both the 3 : 1 CaM–M13 (3C : 1M, r2 = 220 to 45 mM−1 s−1) and the 3 : 1 CaM–RS20 (3C : 1R, r2 = 200 to 34 mM−1 s−1) SPIO nanoparticle mixtures. The EC50 value for 3C : 1M was 1.4 μM, a suitable range for intracellular Ca2+ detection. The introduction of mutant CaM proteins onto the nanoparticle surfaces can provide EC50 values ranging from 0.8 to 10 μM.
5.2 Potassium detection by MRI
For normal muscle and nerve functioning, the body must exert strict homeostatic control over its intracellular and extracellular K+ concentrations. The uneven distribution of K+ between the extracellular (3.8–5 mM) and intracellular (120–140 mM) milieu creates a membrane potential that is exploited to control important physiological functions such as neurotransmission and muscle contraction.17One Gd3+ contrast agent has been reported by Suzuki’s group whose relaxivity depends on K+ concentration (KMR–K1, Gd-7, Fig. 4).18 KMR–K1 incorporates two 15-crown-5 ethers into a Gd–DTPA core. A slight 5% decrease in relaxivity is observed when K+ is added (r1 = 5.05 to 4.78 mM−1 s−1). Binding of K+ is millimolar (Kd = 0.45 mM) and the agent does not respond to Na+, Mg2+, or Ca2+.
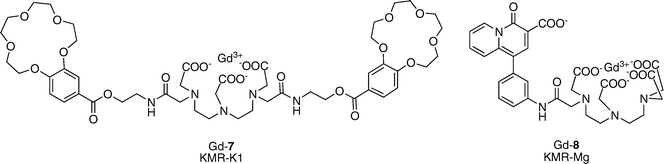 | ||
| Fig. 4 Potassium- and magnesium-activated contrast agents. | ||
5.3 Magnesium detection by MRI
Mg2+ is an electrolyte present in high concentrations throughout the body (0.5 mM intracellular, 0.7 mM extracellular) and is an essential cofactor in over 300 enzymatic reactions. Interactions between Mg2+ and phosphate anions are prevalent in biology, playing a significant role in the stabilization of DNA and RNA biomolecules. Magnesium deficiencies are associated with a number of cardiovascular disorders.19A Gd–DTPA-derivative modified with one charged β-diketone functionality (KMR–Mg, Gd-8, Fig. 4) has been reported to respond to changes in Mg2+ levels.18 The r1 of KMR–Mg decreases from 4.98 to 3.95 mM−1 s−1 when 1 equiv. of Mg2+ is added (a 21% decrease). A 1 : 1 KMR–Mg/Mg2+ binding stoichiometry is observed and the Kd was measured to be 4.1 mM. This agent also has some sensitivity to Ca2+, though it binds Ca2+ more weakly (Kd = 8.3 mM). Na+ and K+ do not significantly affect the relaxivity values observed for KMR–Mg.
5.4 MRI contrast agents for zinc sensing
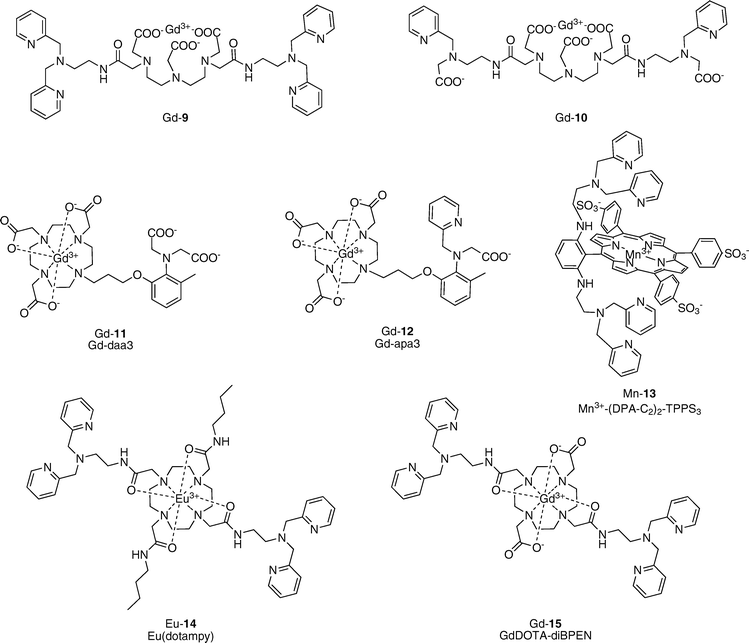
Zn2+ is a cofactor for all six major classes of proteins, including zinc finger protein transcription factors that make up 1–3% of the human proteome. Zn2+ has also emerged as an important signaling ion in recent times, with Zn2+ concentrations reaching as high as 300 μM in vesicles of certain types of glutamatergic neuronal, prostate, and pancreatic cells.20 Elevated Zn2+ levels are also observed in plaques formed in patients with Alzheimer’s disease, whereas zinc deficiency is linked to prostate disease and diabetes.A series of MR-based Zn2+ sensors are depicted in Fig. 5. Nagano’s group reported the first two MRI-based sensors for Zn2+, both relying on a Gd–DTPA scaffold.21,22 The initial sensor Gd-9 utilizes two dipicolylamine (DPA) units for selective Zn2+ binding.21 Addition of 1 equiv. of Zn2+ results in a 33% decrease in relaxivity, going from r1 = 6.06 to r1 = 3.98 mM−1 s−1. The high initial relaxivity value indicates that in the absence of Zn2+ ions, the DPA groups do not bind to the Gd3+ center, thus allowing water binding to the metal center. Addition of 1 equiv. of Zn2+ brings both DPA groups together, creating a shield around the open coordination sites of the Gd3+ center with concomitant reductions of water access and proton relaxivity. However, when 2 equiv. of Zn2+ are added, the relaxivity increases back up to its original value. A possible explanation for the observed behavior is that when more than 1 equiv. of Zn2+ is added, each DPA unit binds one Zn2+ and the Gd3+ center is no longer shielded. This unusual behavior provides complications for sensing applications, as the signals from 0.5 and 1.5 equiv. of added Zn2+ are comparable. To circumvent this issue, a second sensor Gd-10 was designed in which two of the picolyl moieties were replaced with carboxylic acid groups.22 In this case, r1 decreases from 4.8 to 3.4 mM−1 s−1 when 1 equiv. of Zn2+ is added (a 30% decrease). Additional equivalents of Zn2+ do not elicit a change in this final relaxivity value. The Zn2+ selectivities for both these sensors are very good, as they do not respond to Na+, K+, Mg2+, or Ca2+.
 | ||
| Fig. 5 Zinc-activated contrast agents. | ||
Two Gd–DO3A-based contrast agents that respond to Zn2+ were described by Meade and co-workers.23,24 Gd–daa3 (Gd-11) contains an iminodiacetate group for Zn2+ binding.23 A 122% increase in relaxivity is observed for this agent (r1 = 2.3 to 5.1 mM−1 s−1). The Kd for Zn2+ is 240 μM. Gd–daa3 is selective for Zn2+ over Ca2+ and Mg2+, with some response to Cu2+. The Zn2+ turn-on is associated with a change in q as shown by luminescence lifetime experiments with Tb–daa3. Direct evidence for binding of the iminodiacetate group to the Eu3+ center in the absence of Zn2+, and ligand exchange from Eu3+ to Zn2+ upon addition of Zn2+, was obtained by NMR spectroscopic measurements on the 13C-labeled Eu–daa3 complex. A second contrast agent, Gd–apa3 (Gd-12), contains one pyridine and one acetate arm for Zn2+ binding and has a 103% increase in r1 in response to Zn2+ (r1 = 3.4 to 6.9 mM−1 s−1).24 Both sensors Gd-11 and Gd-12 are selective for Zn2+ over cellular abundant Na+, K+, Mg2+, or Ca2+ cations.
Lippard’s laboratory described a Mn3+–porphyrin complex with Zn2+-dependent relaxivity.25 This alternative scaffold was chosen for its ability, unlike Gd–DOTA or Gd–DTPA, to permeate cell membranes. Additionally, the unmetalated porphyrins are fluorescent, affording the potential possibility for bimodal imaging using this platform. The water-soluble sulfonated TPPS3 porphyrin was modified with DPA units and evaluated for its fluorescence properties as the free porphyrin ((DPA–C2)2–TPPS3) and its relaxivity properties as the Mn3+ complex (Mn3+–(DPA–C2)2–TPPS3, Mn-13). (DPA–C2)2–TPPS3 is indeed a selective fluorescent sensor for Zn2+ and the porphyrin can enter cells and detect fluctuations in Zn2+ concentration therein. Mn-13 experiences a 24% decrease in r1 upon addition of 1 equiv. of Zn2+, going from r1 = 8.70 mM−1 s−1 to r1 = 6.65 mM−1 s−1. Changes in T2 relaxivity were also observed at high ionic strength. HEK-293 cells were incubated with Mn-13 and differences in both T1 and T2 values were seen between cells with and without Zn2+ treatment. Unlike the solution measurements, cells incubated with Zn2+ showed lower T1 values than those that were not exposed to Zn2+, indicating that in a cellular context, Mn-13 may show a Zn2+-triggered relaxivity increase. A relaxivity increase associated with Zn2+ was also observed in T2-weighted images of these cells. This report marks the first cell-permeable MRI sensor for metal ions.
Sherry and co-workers have developed a PARACEST (paramagnetic chemical exchange saturation transfer) contrast agent for selective sensing of Zn2+.26 In PARACEST, paramagnetic contrast agents induce shifts in proximal NMR-active nuclei.27 Exchangeable protons, such as water molecules bound to the paramagnetic center, or nearby –NH or –OH protons, can be detected and their shifts can be modulated depending on the coordination environment of the paramagnetic center. Eu(dotampy) (Eu-14) is a Eu3+-chelate whose CEST profile is Zn2+ dependent. Two DPA units for Zn2+ binding are present on a cyclen–tetraamide core and the relative CEST magnitude changes from ∼28 to ∼14. A 1 : 1 Zn2+–Eu(dotampy) complex is formed (Kd = 26 nM) and no change in CEST effect is seen with Mg2+ or Ca2+ addition.
Recently, a new Zn2+-sensitive contrast agent (Gd-15, Gd–DOTA–BPEN) possessing a similar scaffold to Eu(dotampy) was reported.28 Gd–DOTA–BPEN displays a modest increase in r1 in response to Zn2+ in Tris buffer (r1 = 5.0 to 6.0 mM−1 s−1, a 20% increase). A similar response is also observed for Cu2+, which indicates that the dipicolylamine receptors can also bind this metal ion. When the same titration is performed in the presence of human serum albumin (HSA), a much larger relaxivity response is observed upon addition of Zn2+ (r1 = 6.6 to 17.4 mM−1 s−1, a 165% increase). Fluorescent displacement experiments to characterize the binding interactions between HSA and apo or Zn2+-bound Gd–DOTA–BPEN provide evidence for binding of the Zn2+-bound complex to site 2 of subdomain IIIA of HSA (Kd = 48 μM), whereas no specific interaction was observed between HSA and the apo complex. The authors suggest that the significantly larger r1 value observed for the Zn2+-bound complex results from a longer τR value due to interactions with HSA.
5.5 Iron-sensitive MRI contrast agents
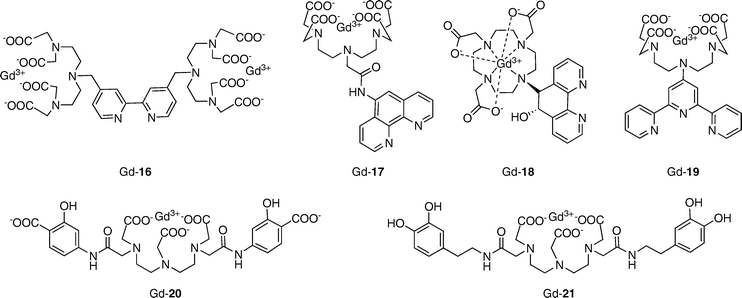
Iron is the most abundant transition metal in the body, with the average adult human possessing ca. 5 g of this essential element. The most common oxidation states observed in biological systems are ferric (Fe3+) and ferrous (Fe2+) ions. Both iron excess and iron deficiency are detrimental to human health.29 In particular, with aging, iron levels in the brain increase and are linked to neurodegenerative Alzheimer’s and Parkinson’s diseases. Ferritin and related iron-proteins provide endogenous contrast observed in MR images where external contrast agents are not used.30Several heterobimetallic Gd–Fe complexes have been reported in the literature; selected examples are given in Fig. 6.31–36 These supramolecular assemblies were designed as high-relaxivity contrast agents, with one Fen+ center linking multiple Gd3+-chelates to produce larger complexes with correspondingly longer rotational correlation times and higher relaxivity values. Although for the most part these agents have not been characterized as iron-responsive MRI indicators, they provide a good starting point for future sensor designs. For example, both DTPA- and DOTA-based supramolecular complexes have been reported. For Fe2+ chelation, 2,2′-bipyridine (Gd-16),31 phenanthroline (Gd-17, 18),32,33 and terpyridine (Gd-19)34 derivatives have been employed. Siderophore-derived binding groups including salicylic acid (Gd-20)35 and catechol (Gd-21)36 have been utilized for Fe3+ chelation. All of these complexes exhibit higher relaxivity values upon addition of iron ions. Bipyridine compound Gd-16 gives a 59% increase in effective mass relaxivity with addition of 1/3 equiv. of Fe2+ (r1eff = 20.2 to 32.2 mM−1 s−1),31 whereas phenanthroline compound Gd-18 displays a 77% increase in relaxivity upon introduction of 1/3 equiv. of Fe2+ (r1 = 6.2 to 11 mM−1 s−1).33 As no selectivity data for Fen+ ions are described in these reports, further investigation is needed to determine if these types of agents will provide practically useful MRI-based iron sensors.
 | ||
| Fig. 6 Iron-binding MRI contrast agents. | ||
5.6 MRI-based copper sensors
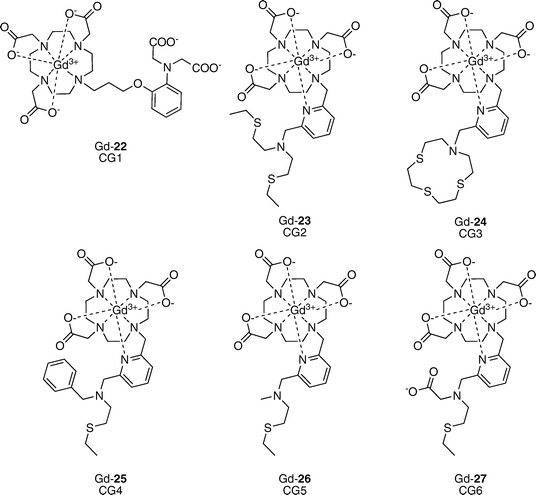
Copper is a required metal nutrient for all living organisms, serving as a redox-active cofactor in many essential eukaryotic enzymes in the body, including cytochrome c oxidase, superoxide dismutase, and tyrosinase. Cupric (Cu2+) and cuprous (Cu+) ions are the prevalent oxidation states for copper in biological systems. Because of its potent redox activity, misregulation of copper homeostasis is implicated in a number of severe diseases.37 Prominent examples include Wilson’s disease, where excess copper accumulation is observed in the liver, and neurodegenerative Alzheimer’s and prion diseases, where elevated copper levels occur in protein-derived extracellular plaques with concomitant loss of copper from intracellular stores.Our laboratory has initiated a broad-based program to study the contributions of copper to physiological and pathological situations using molecular imaging,38–42 and MRI-based copper sensors from these efforts to date are presented in Fig. 7. Our initial copper-activated MRI contrast agent, Copper–Gad 1 (CG1, Gd-22), is a Gd–DO3A-based compound that utilizes an iminodiacetate moiety for Cu2+ binding.39 CG1 exhibits a 41% increase in relaxivity in response to Cu2+, going from r1 = 3.76 in the apo form to 5.29 mM−1 s−1 in the holo form. The observed Cu2+ binding affinity is in the micromolar range (Kd = 167 μM). CG1 features good Cu2+ selectivity in HEPES or PBS buffer, binding to Cu2+ more tightly than to other biologically relevant metal ions as expected from Irving–Williams series considerations. One drawback of this first-generation design is that the Cu2+ response is partially muted in the presence of 10-fold excess Zn2+.
 | ||
| Fig. 7 Copper-activated MRI contrast agents. | ||
In second-generation designs, we sought to improve the metal ion selectivity of CG-type sensors, particularly with regard to Zn2+, achieve greater dynamic range for turn-on responses to copper, and devise probes that would signal Cu+ and/or Cu2+. In this context, thioether ligands show a preference for binding of copper ions in aqueous solution and we took advantage of this design feature to create the suite of sensors CG2–CG6 (Gd-23–Gd-27).42 CG2–CG5 incorporate two nitrogen and one to three thioether donors for selective binding of Cu+. Two of these sensors, CG2 and CG3, display markedly improved turn-on values when 1 equiv. of Cu+ is added, going from r1 = 1.5 mM−1 s−1 to r1 = 6.9 mM−1 s−1 (a 360% increase in relaxivity). The observed Cu+ binding affinities were systematically tuned by varying the number of donor atoms in the sensor portion of the molecule, resulting in Kd values spanning three orders of magnitude from 0.037 to 32 pM. Moreover, the thioether-rich receptors confer high copper specificity; CG2-5 do not respond to other biologically relevant metal ions including Na+, K+, Ca2+, Mg2+, Fe2+, Fe3+, Cu2+, and Zn2+, and none of these ions interfere with copper-induced turn-on signals. No response to Zn2+ is observed even when 10 equiv. are added, indicating that the thioether donors effectively impart selectivity for Cu+ over Zn2+. Dysprosium-induced 17O shift experiments revealed an increase in the number of inner-sphere water molecules with addition of Cu+, and in the cases of CG2–4, the largest turn-on sensors, a Δq of 2 was observed. These studies were further supported through fitting of NMRD profiles. CG6 is a unique sensor in this series. A combination of N, S, and O donors in the sensing portion of CG6 results in relaxivity increases upon addition of both Cu+ and Cu2+. A 73% increase in r1 is observed upon addition of 1 equiv. of either Cu+ or Cu2+ (r1 = 2.2 to 3.8 mM−1 s−1). Although CG6 responds to addition of both oxidation states of copper, the UV-vis spectrum of the Cu+–CG6 complexes quickly transforms into that of the Cu2+–CG6 complex, indicating that the cupric form is preferred under ambient aqueous conditions. Binding of Cu2+ is tight, with an apparent Kd of 0.99 fM. As with CG2–CG5, this thioether-containing scaffold confers selectivity for copper ions over a range of biologically relevant metal ions including Zn2+. Finally, we established the ability of the second-generation CG probes to detect changes in aqueous copper levels by MRI at clinical field strengths.
6. Conclusions and future prospects
The combination of metal-based MRI contrast agents with metal-selective molecular recognition elements provides a promising new class of chemosensors for molecular imaging in biological systems. To date, responsive MRI contrast agents have been developed for a number of essential s-block and d-block metal ions, and many of these sensors possess promising in vitro properties. The next grand challenge is to develop and apply these types of reagents for MRI in live specimens ranging from cells to tissue to whole in vivo organisms, a translation that will require advances that adhere to stringent chemical and biological criteria.Many opportunities are available to meet the ultimate potential of this frontier area of research, which is still in its infancy. Developing contrast agents with even higher relaxivity changes is vital owing to the low detection limit of MRI. Another important consideration when moving towards in vivo MRI is maintaining chemical selectivity in complex environments, as heterogeneous biological specimens can be drastically different from buffered aqueous solutions. For example, high concentrations of anions and interfering organic molecules are present in both the intracellular and extracellular milieu, and can potentially interfere with mechanisms that rely on modulating coordination spheres of paramagnetic metal centers. Other frontier opportunities involve target/analyte validation and quantification. Quantitative measurements using MRI as a single imaging modality are challenging because the local concentration of contrast agent is unknown. Combining MRI with alternative techniques such as optical emission, PET, CT, etc. can provide multimodal imaging agents to meet this need. A final frontier is the creation of “theranostic” agents, which combine imaging and therapeutics in a single deliverable probe.43 Since metals can neither be created nor destroyed in living organisms, theranostics that can specifically bind and transport metal ions of interest offer an attractive area for research and clinical applications. The rich basic science challenges for chemists interested in coordination chemistry, supramolecular chemistry, and molecular recognition, along with the wealth of opportunities for exploring and understanding metals in biology and medicine, presages an exciting future for molecular imaging.
Acknowledgements
We apologize in advance if references to contributions were omitted due to journal space constraints. We thank the University of California, Berkeley, the Dreyfus, Beckman, Packard, and Sloan Foundations, the Hellman Faculty Fund (UC Berkeley), the National Science Foundation (CAREER CHE-0548245), the National Institute of General Medical Sciences (NIH GM 79465), Amgen, and the Howard Hughes Medical Institute for funding this work. E.L.Q. acknowledges a Branch graduate fellowship from UC Berkeley.References
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293 CrossRef CAS.
- B. Yoo and M. D. Pagel, Front. Biosci., 2008, 13, 1733 CrossRef CAS.
- J. L. Major and T. J. Meade, Acc. Chem. Res., 2009, 42, 893 CrossRef CAS.
- E. J. Werner, A. Datta, C. J. Jocher and K. N. Raymond, Angew. Chem., Int. Ed., 2008, 47, 8568 CrossRef CAS.
- I. Solomon, Phys. Rev., 1955, 99, 559 CrossRef CAS.
- N. Bloembergen, J. Chem. Phys., 1957, 27, 572 CrossRef CAS.
- N. Bloembergen and L. O. Morgan, J. Chem. Phys., 1961, 34, 842 CrossRef CAS.
- R. A. Moats, S. E. Fraser and T. J. Meade, Angew. Chem., Int. Ed. Engl., 1997, 36, 726 CrossRef CAS.
- M. J. Berridge, P. Lipp and M. D. Bootman, Nat. Rev. Mol. Cell Biol., 2000, 1, 11 CrossRef CAS.
- W.-H. Li, S. E. Fraser and T. J. Meade, J. Am. Chem. Soc., 1999, 121, 1413 CrossRef CAS.
- W.-H. Li, G. Parigi, M. Fragai, C. Luchinat and T. J. Meade, Inorg. Chem., 2002, 41, 4018 CrossRef CAS.
- K. Dhingra, P. Fousková, G. Angelovski, M. Maier, N. Logothetis and É. Tóth, JBIC, J. Biol. Inorg. Chem., 2008, 13, 35 CAS.
- A. Mishra, P. Fousková, G. Angelovski, E. Balogh, A. K. Mishra, N. K. Logothetis and É. Tóth, Inorg. Chem., 2008, 47, 1370 CrossRef CAS.
- G. Angelovski, P. Fousková, I. Mamedov, S. Canals, É. Tóth and N. K. Logothetis, ChemBioChem, 2008, 9, 1729 CrossRef CAS.
- K. Dhingra, M. E. Maier, M. Beyerlein, G. Angelovski and N. K. Logothetis, Chem. Commun., 2008, 3444 RSC.
- T. Atanasijevic, M. Shusteff, P. Fam and A. Jasanoff, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14707 CrossRef CAS.
- J. H. Youn and A. A. McDonough, Annu. Rev. Physiol., 2009, 71, 381 CrossRef CAS.
- H. Hifumi, A. Tanimoto, D. Citterio, H. Komatsu and K. Suzuki, Analyst, 2007, 132, 1153 RSC.
- J. G. Gums, Am. J. Health Syst. Pharm., 2004, 61, 1569 CAS.
- C. J. Frederickson, J. Y. Koh and A. I. Bush, Nat. Rev. Neurosci., 2005, 6, 449 CrossRef CAS.
- K. Hanaoka, K. Kikuchi, Y. Urano and T. Nagano, J. Chem. Soc., Perkin Trans. 2, 2001, 1840 RSC.
- K. Hanaoka, K. Kikuchi, Y. Urano, M. Narazaki, T. Yokawa, S. Sakamoto, K. Yamaguchi and T. Nagano, Chem. Biol., 2002, 9, 1027 CrossRef CAS.
- J. L. Major, G. Parigi, C. Luchinat and T. J. Meade, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13881 CrossRef CAS.
- J. L. Major, R. M. Boiteau and T. J. Meade, Inorg. Chem., 2008, 47, 10788 CrossRef CAS.
- X. Zhang, K. S. Lovejoy, A. Jasanoff and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10780 CrossRef CAS.
- R. Trokowski, J. Ren, F. K. Kálmán and A. D. Sherry, Angew. Chem., Int. Ed., 2005, 44, 6920 CrossRef CAS.
- S. Zhang, M. Merritt, D. E. Woessner, R. E. Lenkinski and A. D. Sherry, Acc. Chem. Res., 2003, 36, 783 CrossRef CAS.
- A. C. Esqueda, J. A. López, G. Andreu-de-Riquer, J. C. Alvarado-Monzón, J. Ratnakar, A. J. M. Lubag, A. D. Sherry and L. M. De León-Rodríguez, J. Am. Chem. Soc., 2009, 131, 11387 CrossRef CAS.
- L. Zecca, M. B. H. Youdim, P. Riederer, J. R. Connor and R. R. Crichton, Nat. Rev. Neurosci., 2004, 5, 863 CrossRef CAS.
- Y. Gossuin, R. N. Muller and P. Gillis, NMR Biomed., 2004, 17, 427 CrossRef CAS.
- J. B. Livramento, É. Tóth, A. Sour, A. Borel, A. E. Merbach and R. Ruloff, Angew. Chem., Int. Ed., 2005, 44, 1480 CrossRef CAS.
- T. N. Parac-Vogt, L. V. Elst, K. Kimpe, S. Laurent, C. Burtéa, F. Chen, R. V. Deun, Y. Ni, R. N. Muller and K. Binnemans, Contrast Media Mol. Imaging, 2006, 1, 267 Search PubMed.
- J. Paris, C. Gameiro, V. Humblet, P. K. Mohapatra, V. Jacques and J. F. Desreux, Inorg. Chem., 2006, 45, 5092 CrossRef CAS.
- R. Ruloff, G. van Koten and A. E. Merbach, Chem. Commun., 2004, 842 RSC.
- S. Aime, M. Botta, M. Fasano and E. Terreno, Spectrochim. Acta, Part A, 1993, 49, 1315 CrossRef.
- T. N. Parac-Vogt, K. Kimpe and K. Binnemans, J. Alloys Compd., 2004, 374, 325 CrossRef CAS.
- D. J. Waggoner, T. B. Bartnikas and J. D. Gitlin, Neurobiol. Dis., 1999, 6, 221 CrossRef CAS.
- L. Zeng, E. W. Miller, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 10 CrossRef CAS.
- E. L. Que and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 15942 CrossRef CAS.
- D. W. Domaille, E. L. Que and C. J. Chang, Nat. Chem. Biol., 2008, 4, 168 CrossRef CAS.
- E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517 CrossRef CAS.
- E. L. Que, E. Gianolio, S. L. Baker, A. P. Wong, S. Aime and C. J. Chang, J. Am. Chem. Soc., 2009, 131, 8527 CrossRef CAS.
- S. Warner, Scientist, 2004, 18, 38 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |