Development and applications of tethered bis(8-quinolinolato) metal complexes (TBOxM)
Joshua P.
Abell†
and
Hisashi
Yamamoto
*
Department of Chemistry, The University of Chicago, 5735 South Ellis Avenue, Chicago, Illinois 60637, USA. E-mail: yamamoto@uchicago.edu
First published on 6th November 2009
Abstract
The tethered bis(8-quinolinolato) metal (TBOxM) complexes have found practical use in many asymmetric reactions. The development of highly enantioselective reactions using TBOxM is surveyed. General reaction schemes and examples using these highly active and selective catalysts are given and discussed in this tutorial review.
 Joshua P. Abell | Joshua Abell received his Bachelor’s degree from The University of Texas at San Antonio in 2004. He received his PhD from the University of Chicago under the direction of Professor Hisashi Yamamoto in 2009. During his PhD his focus was on the development and application of chiral tethered bis(8-quinolinolato) (TBOxH), specifically aluminium complexes. He is currently a post-doctoral fellow at Harvard University with Professor Eric Jacobsen. |
 Hisashi Yamamoto | Hisashi Yamamoto received his Bachelor’s degree from Kyoto and PhD from Harvard under Professor Nozaki and Professor Corey, respectively. In 1977 he became Assistant Professor at Kyoto University, and in 1977 Associate Professor of Chemistry at the University of Hawaii. In 1980 he became Professor at Nagoya, and from 2002 he has been Professor at the University of Chicago. His honors include: Prelog Medal in 1993, Le Grand Prix de la Fondation Maison de la Chimie in 2002, Tetrahedron Prize in 2006, The Japan Academy Prize in 2007, and the ACS Award in Synthetic Organic Chemistry in 2009. |
Introduction
The field of asymmetric catalysis has drastically matured, and chemists are coming ever closer to making any compound “at will”.1 Whether it is specific bond formation or stereochemistry, chemists are constantly developing newer, more reactive, and more selective catalysts which provide the requisite products on demand. New generations of catalysts have arisen from a number of inspirational platforms, including nature, previous scientific publications, and creativity.The continued efforts of the many research groups in asymmetric catalysis throughout the world have found a number of chiral catalysts which display unprecedented selectivity and reactivity.2–3 Of these, several chiral scaffolds have shown a broad range of applicability for a variety of unrelated asymmetric transformations. The ligands of these catalysts have been named “privileged” by virtue of the sheer number of asymmetric transformations they can catalyze with high levels of enantioselectivity and yield using only a catalytic amount.4 The organometallic complexes of these “privileged” ligands have paved the way for modern asymmetric catalysis and offer insight into what new methods of catalysis are yet to come.
In asymmetric catalysis, in particular metal-based catalysis, the chiral scaffold has received a significant amount of attention, covering structural features such as sterics as well as the electronic nature of the ligand. However, only recently has more attention been given to the metal center itself. The salen ligand and its reduced analogue, salan, for example, have been some of the leading pioneers in metal-based asymmetric catalysis. The ligand, when complexed to a metal center, has a slight step-wise secondary structure but is generally considered to have a flat topology while the asymmetric induction arises from the chiral diamine template from which it is derived (Fig. 1).
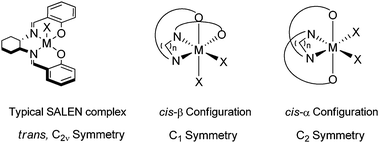 | ||
| Fig. 1 SALEN versus cis-β configuration. | ||
Tetradentate ligands around octahedral coordination spheres allow for two vacant sites for coordination. Intuitively, these two vacant sites allow for the simultaneous coordination of a nucleophile and electrophile, however, the coordination around the metal center is not so straightforward. The coordination can take one of three geometric inequivalent isomers, trans, cis-α, and cis-β (Fig. 1). The trans isomer, which is the typical coordination that metallosalen complexes adopt, has C2v symmetry and is not chiral-at-metal. However, the cis-α and cis-β isomers are chiral-at-metal with cis-α having a C2 symmetry and cis-β having a C1 symmetry. When enantiopure ligands are used to form either cis-α or cis-β isomers, diastereomers may be formed, either Λ or Δ depending on the way they twist around the metal center. One of the areas of interest within our research group has been the development of a new tethered ligand incorporating 8-hydroxyquinoline as the chelation motif. This ligand, tethered bis(8-quinolinolato) or TBOx, has been complexed with a variety of metals and the complexes have been fully characterized, and, as expected due to the rotational constraints of the binaphthyl backbone and quinoline motifs, many of them have the configuration of unique chiral cis-β geometry (Fig. 2). Moreover, the performance and selectivity of the TBOxM complexes have been unprecedented.5
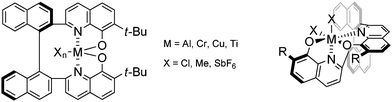 | ||
| Fig. 2 TBOxMXn complexes. | ||
New catalyst designs with new modes of activation are at the height of asymmetric catalysis today. The implications of chiral-at-metal complexes allow for the intimate interaction of nucleophile and electrophile and thus heighten the possibility of a stereospecific bond formation step. This configuration around the metal is not inherent to a ligand, as TBOxH has an overall C2-symmetry, but must be induced by a conformational constraint of the ligand or transition state. The idea behind the synthesis of TBOxH was such that complexation of a metal ion would not allow for a flat topology around the metal center. The enantiopure ligand of TBOxH provides a rigid platform to induce one helical conformation, either Λ or Δ, of the chiral-at-metal complex to be produced.
Chiral-at-metal complexes in asymmetric catalysis
In 1970, Bosnich first reported a cobalt complex with an enantiopure aminopyridine ligand that displayed a cis-β configuration around the metal with a tetradentate ligand (Fig. 3).6 This report described that cis-α complexes display little difference in the overall geometry of the complex, but cis-β offers a different environment for the substituents of the ligand and thus coordination sites. This complex not only provided a chiral ligand platform but also exhibited chirality at the metal center.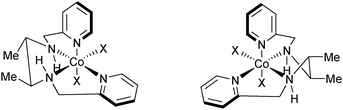 | ||
| Fig. 3 Tetradentate cobalt complex displaying the cis-β configuration. | ||
Several examples of chiral-at-metal complexes have been reported, however their application to asymmetric catalysis has only recently been explored in some detail.7–9 Of the various octahedral complexes with differing coordinated ligands, tetradentate ligands of the type N2O2 have been the greatest focus of chiral-at-metal complexes, particularly with respect to asymmetric catalysis. A representative sample of these highly active catalysts are briefly discussed (Scheme 1).
 | ||
| Scheme 1 Selected catalysts displaying the cis-β configuration. | ||
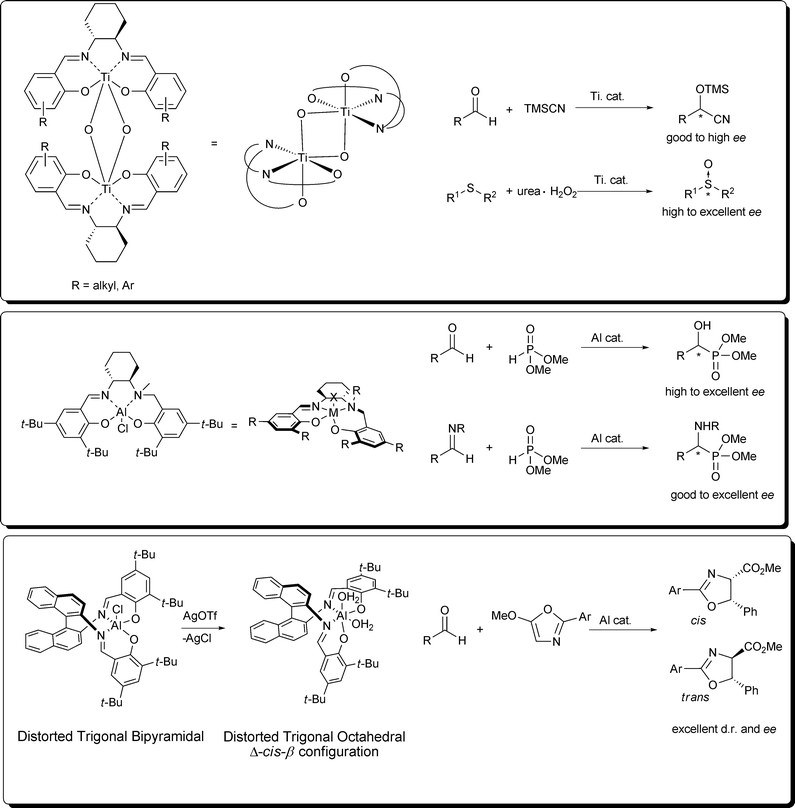
A great deal of the metallosalen complexes adopt a stepped or umbrella conformation though, however, are square planar around the metal center and have an overall C2-symmetric topology. Nevertheless, several transition metal complexes, as well as with the reduced salen and salalen, have shown severe distortions from this typical square planar structure, particularly with ancillary bidentate ligands or substrates that invoke one of the coordinating ligands to distort from the plane.9 These cis-β or pseudo cis-β structures have demonstrated unique reactivity, as well as stereoselectivity, for a variety of reactions.
A di-μ-oxo titanium salen complex was found to be applicable in asymmetric cyanohydrin synthesis and oxidation of sulfides.10 A half-reduced and alkylated aluminium salen complex was found to be particularly useful for the asymmetric Pudovik reaction of aldehydes and aldimines.11 Additionally an aluminium Schiff-base catalyst, which after counterion exchange revealed a highly active cis-β complex, was used for the synthesis of α-amino-β-hydroxyacid precursors.12 All of these reactions have demonstrated that cis-β complexes are extremely important for the generation of highly active and selective catalysts.
Tethered bis(8-quinolinolato) (TBOx) chromium complex and applications
Asymmetric pinacol coupling
Our introduction into the area of chiral-at-metal complexes was demonstrated by the high reactivity and selectivity of the chromium catalyzed pinacol coupling of aldehydes (Scheme 2).13 The presence of the chiral 1,2-diol motif is found ubiquitously in nature, and has found numerous practical advantages in chiral ligands and auxiliaries. Of the various methods to generate enantio-enriched 1,2-diols, the most direct method, asymmetric pinacol coupling of aldehydes, is particularly difficult as not only is discrimination of the prochiral faces required, but also diastereoselectivity must be controlled in a single bond formation step. Additionally, high stereoselectivity has remained problematic, even with the use of a stoichiometric amount of catalyst. However, the use of TBOxCrCl allowed for broad application to not only aromatic aldehydes, but also aliphatic aldehydes. 3 mol% was found to be the optimum amount of catalyst, and a reductant (Mn) and chlorosilane (TESCl) were needed to complete the catalytic cycle and regenerate the active catalyst. This result provided a solid framework in which to investigate other reactions.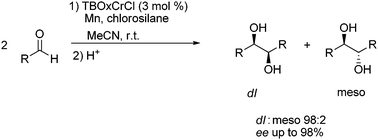 | ||
| Scheme 2 TBOxCrCl catalyzed asymmetric pinacol coupling. | ||
Asymmetric Nozaki–Hiyama allylation
Other asymmetric reactions have taken advantage of chromium’s ability to insert into conjugated carbon–halide bonds and generate a mild organometallic nucleophile towards aldehydes. In this regard, the asymmetric Nozaki–Hiyama allylation reaction14 was also found to provide the desired product with the TBOxCrCl catalyst for a wide range of aldehydes (Scheme 3).15 Previously, asymmetric variants of this reaction provided narrow substrate scopes, low reactivity and enantioselectivity for this highly important reaction. The allyl halide (X = Br, Cl) was found to largely unaffect the stereoselectivity of the reaction but in some cases the yield was affected. Furthermore, this report demonstrated the widest substrate scope to date, while providing all the products in excellent enantioselectivities using low catalyst loadings. A stereochemical model that could account for the outcome of the reaction was provided (Fig. 4). | ||
| Scheme 3 TBOxCrCl catalyzed asymmetric Nozaki–Hiyama allylation. | ||
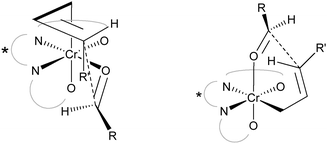 | ||
| Fig. 4 Stereochemical model accounting for selectivity. | ||
Asymmetric allenylation
In 2007, a paper on the allenylation of aldehydes using TBOxCr was reported (Scheme 4).16 Originally, it was expected propargylation would take place, but an unexpected allenylated product was obtained. This protocol represents an improvement over the current methods for the generation of enantio-enriched α-allenic alcohols, in terms of scope, yield, and ease of operational use with commercially available reagents. Moreover, the scope of the reaction was expanded to include terminally alkyl-substituted propargyl bromides to provide di-substituted α-allenic alcohols, which had never been achieved in high enantioselectivity with other chromium catalysts. The products are synthetically useful materials and can be further elaborated.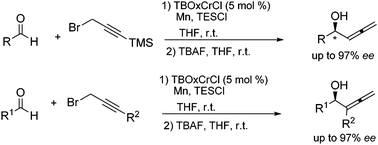 | ||
| Scheme 4 TBOxCrCl catalyzed asymmetric allenylation. | ||
Catalyzed asymmetric synthesis of 2-carbinol-1,3-dienes
The synthesis of 2-carbinol-1,3-dienes typically requires multi-step procedures requiring the use of organotin intermediates,17 where typically this is seen as a major drawback due to toxicity, limiting their use for further elaboration in organic synthesis. Other routes suffer from regioselectivity issues and low reactivity rendering some specialized products unobtainable by the typical procedure.18–19 The most recent report of the TBOxCr catalyst demonstrates a simple, straightforward protocol to obtain these elusive products in modest yield (Scheme 5).20 Additionally, good enantioselectivities were obtained for several aromatic aldehydes. An initial screening of propargylation showed promising results obtaining the secondary alcohol in 85% yield and 81% ee.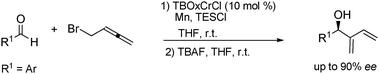 | ||
| Scheme 5 TBOxCrCl catalyzed asymmetric synthesis of 2-carbinol-1,3-dienes. | ||
These chromium catalyzed reactions have demonstrated the potential utility of this ligand with other metal ions which can catalyze a wide variety of asymmetric reactions. These seminal results led us to explore other metal ions that can coordinate in a similar cis-β, or pseudo cis-β conformation, and to explore their potential in asymmetric catalysis. Other chromium catalyzed reactions are currently under investigation using this high active and robust catalyst.
Tethered bis(8-quinolinolato) (TBOx) aluminium complex and applications
Asymmetric Mukaiyama–Michael reaction
Carbon–carbon bond formation reactions are undoubtedly some of the most important transformations in organic synthesis.21 The ability to build carbon framework complexity within organic compounds is a valuable tool chemists use in the construction of many natural products and bioactive molecules. On this basis, the Michael reaction, whereby a nucleophile is added to the β-carbon of an α,β-unsaturated carbonyl or related conjugated system is an extremely useful reaction regarding the aforementioned points. Of particular importance is the control of regioselectivity, especially when multiple functional groups are present. Lewis acids are known to promote 1,2-addition rather than 1,4-addition. Given this point, several Lewis acids, transition metal catalysts, as well as organocatalysts have been shown to catalyze this reaction with superior regioselectivity (1,4-adduct in preference to the 1,2-adduct).2 In 1974, Mukaiyama and co-workers, found a TiCl4/silyl enol ether system which undergoes this transformation under relatively mild reaction conditions.22 Since this seminal discovery, Lewis acid catalysis in this area has been the focus of several research groups to make highly enantioselective versions.Many of the approaches to enantioselective Mukaiyama–Michael type reactions use bidentate substrates and silyl ketene acetals. Our approach utilized acylphosphonates as the Michael acceptor and simple silyl enol ethers as the nucleophile. Using simple silyl enol ethers indicates the effective activation of the electrophile with our aluminium catalyst.23
A diverse array of silyl enol ethers were investigated, and the application was found to be quite broad. Moreover, the Michael acceptor could be changed at the β-position to include a bulky phenyl group which is typically problematic in Mukaiyama–Michael systems (Scheme 6).
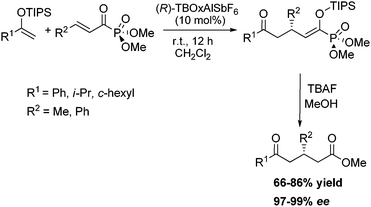 | ||
| Scheme 6 TBOxAlSbF6 catalyzed asymmetric Mukaiyama–Michael reaction. | ||
We further extended our system to the generation of all-carbon quaternary centers.23 Having successfully developed an effective catalyst for the conjugate addition of simple silyl enol ethers, the possibility of using sterically demanding tetrasubstituted enolates was investigated. The latter Michael donors would give rise to enantioenriched carbonyl products bearing α-quaternary stereocenters, whose preparation by catalytic means remains largely an unsolved, albeit highly important, problem.24 The enantioselective construction of quaternary centers in asymmetric catalysis has been a long standing issue due to the steric requirements on bond formation.25 Moreover, all-carbon enantioenriched stereocenters are even more challenging due to the similarities between the nucleophiles and electrophiles, which is exacerbated by the aforementioned issue.24,26–27 To overcome the reactivity aspect, a cyclic silyl enol ether was employed to prevent any issues related to steric constraints. To our delight, a range of tetrasubstituted trimethylsilyl enol ethers underwent the MM reaction and formed all-carbon quaternary stereocenters with high enantioselectivities (Scheme 7). Diastereoselectivities and yields varied from moderate to high ranges. Interestingly, triethoxy silyl enol ethers were found to be more reactive than corresponding trimethyl silyl enol ethers. The former nucleophiles underwent the conjugate addition reaction at 0 °C smoothly, leading to increased diastereoselectivities. To our knowledge, the present method represents the only catalytic asymmetric MM reaction that provides access to enantioenriched α-carbonyl all-carbon-substituted quaternary stereocenters.
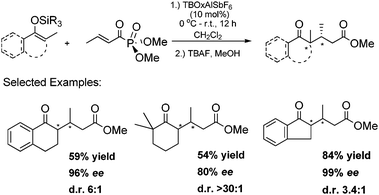 | ||
| Scheme 7 Asymmetric synthesis of all-carbon quaternary stereocenters. | ||
Asymmetric Friedel–Crafts alkylation with indoles
Indoles are known to be highly reactive towards electrophiles at the C-3 position.28 Electrophilic aromatic substitution, Friedel–Crafts alkylation (FCA), at the C-3 position of indoles is 1013 times more reactive than with benzene. The indole moiety is present in a number of naturally occurring alkaloids and pharmacophores, known to be a privileged platform. Given the aforementioned statement, a number of syntheses for producing optically active compounds possessing the indole motif have been reported, including addition to activated carbonyl compounds, α-dicarbonyl compounds, and imines.29 Due to the high nucleophilic character of the indole motif, a number of catalytic Friedel–Crafts alkylations of indole with α,β-unsaturated carbonyl compounds have been reported. Only recently have asymmetric versions of this highly important C–C bond formation reaction been reported. The asymmetric and non-asymmetric carbon–carbon bond formation between indoles and α,β-unsaturated carbonyl compounds or related compounds takes advantage of the inherent nucleophilic nature of the C-3 position of indoles.Using acyl phosphonates as the Michael acceptor, other protected and unprotected indoles were tested under our optimized conditions.23 Although the unprotected indole provided the product, the yield and enantioselectivity were significantly lower than that of the N-methyl indole (Scheme 8). This is presumably caused by the strong binding of the nitrogen atom of the indole to the catalyst due to the basic nature of the indole, rendering the catalyst inactive (also known as substrate/product inhibition).
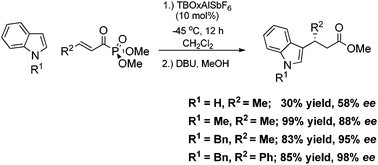 | ||
| Scheme 8 TBOxAlSbF6 catalyzed asymmetric Friedel–Crafts alkylation. | ||
Contrary to the unprotected indole, as expected the benzyl protected indole provided the product in high yield and enantioselectivity. Typically, when a large substituent at the β-position of the Michael acceptor is present, such as phenyl, most catalysts are unable to provide any product due to the steric constraints during the C–C bond formation step. Our aluminium catalyst is expected to be superior to other catalysts which display this problematic aspect. Accordingly, when a phenyl substituent was placed in the β-position of the acyl phosphonates, the product was again obtained in high yield and enantioselectivity. After hydrolysis of the enol phosphonates using DBU and MeOH, the corresponding methyl ester conjugate adduct was obtained cleanly.
Asymmetric Pudovik reaction with aldehydes and aldimines
Dialkylphosphites are said to exist in two equilibrium tautomers, the phosphonate and phosphite tautomers. This equilibrium is expected to exist entirely in the unreactive phosphonate tautomeric form, with 10−4% of the phosphite tautomer for dibutylphosphite.35 With such a low concentration of reactive nucleophile present, it would seem the utilization of dialkylphosphites as nucleophiles would be synthetically impossible. However, it is known that this equilibrium can be shifted to the more nucleophilic metallo-phosphite by use of a Lewis base or Lewis acid (Scheme 9).36
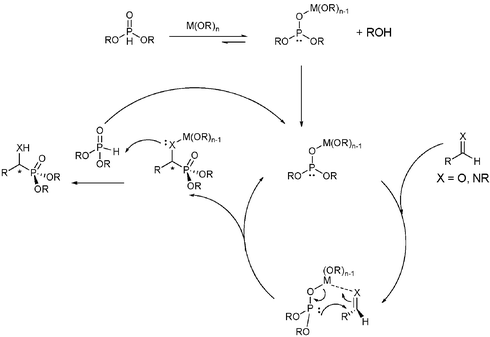 | ||
| Scheme 9 Pudovik reaction mechanism. | ||
There are several notable drawbacks to the current methodologies that exist to date for the enantioselective construction of α-hydroxyphosphonates. Catalyst loading among the aluminium catalysts remains quite high, typically >10 mol% in order to achieve turnover. Most of the current methodologies also require long reaction times, sometimes several days to achieve high yields. Furthermore, the reactions are typically done at temperatures lower than the ambient temperature to achieve high selectivities.
During investigations into the application of our newly developed ligand and examination of the intimate behavior of this reaction mechanism, we sought to use our aluminium catalyst as a new entry for this reaction. Aluminium catalysts have historically been particularly useful in this C–P bond formation reaction.31 Using various dialkylphosphites the reaction was examined using TBOxAlCl as the catalyst.37 While simple methyl and ethyl phosphate provided the product in low yield and enantioselectivity, the use of bis(2,2,2-trifluoroethyl) phosphite increased the yield to 94% and the enantioselectivity to 48% (Scheme 10). The actual mechanism has not been experimentally confirmed, it is believed that the first step is deprotonation of the phosphite to generate a more highly nucleophilic species (Scheme 9).36
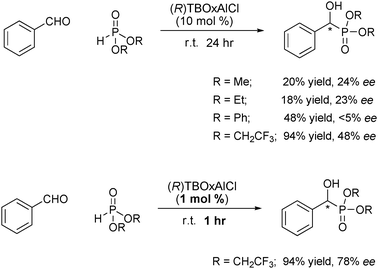 | ||
| Scheme 10 TBOxAlCl catalyzed asymmetric Pudovik reaction. | ||
With efforts now focused on increasing the enantioselectivity, increasing the size of the alkyl group only decreased the yield and selectivity, possibly due to decreased nucleophilicity of the phosphite and an increased steric environment. Decreasing the reaction temperature only decreased the reactivity while not increasing the enantioselectivity. Interestingly, after screening the reaction conditions, decreasing the catalyst loading of the reaction increased the enantioselectivity while maintaining the same reactivity and yield.
With the influence of the phosphite and solvent now established, the ligand structure was examined (Scheme 11). The ligand structure was found to be highly influential on the enantioselectivity of the product. Simply attaching a phenyl substituent at the 5,5′-position of the quinoline ring led to a dramatic increase in enantioselectivity. Pleased with this result we further examined other derivatives and found the catalyst with a mesityl group at the 5,5′-position to be the most reactive and selective.
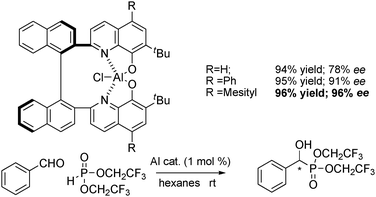 | ||
| Scheme 11 Effect of the 5,5′-position of TBOxAlCl. | ||
Having optimized the reaction conditions, the substrate scope of the reaction was explored (Scheme 12).37 Aromatic aldehydes with various substitution patterns were largely unaffected providing high enantioselectivity and yield. Additionally, aliphatic aldehydes could be employed with good enantioselectivity. All reactions proceeded very quickly and smoothly, with high yields and enantioselectivity. Furthermore, the catalyst loading could be decreased to 0.5 mol% with essentially no loss in enantioselectivity or yield with slightly prolonged reaction times.
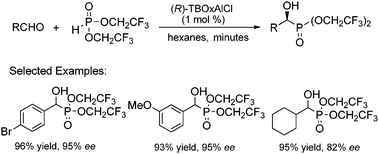 | ||
| Scheme 12 TBOxAlCl catalyzed asymmetric Pudovik reaction. | ||
Upon further investigation of this ligand system with previously synthesized ligands by our group, it was found that the substitution at the 5,5′-position of the quinoline ring had a dramatic increase on the rate of the reaction. To demonstrate the difference in reactivity, the enantiopure 5,5′-substituted ligand ((R)-Al cat) and an enantiopure unsubstituted oppositely stereo-configured ligand ((S)-Al cat) were added in equal catalytic amounts to a reaction vessel and subjected to the typical reaction conditions (Scheme 13). It was found that the product was favored for the substituted ligand in the same enantioselection as with the same ligand alone. This experiment proves that the substitution provided a more reactive catalyst system presumably by increasing the turnover rate of the catalyst.
 | ||
| Scheme 13 TBOxAlCl competition experiment. | ||
Various imines were synthesized and tested against our optimized conditions that were established for aldehydes. Various protecting groups on the imine were examined, however, the use of N-diphenylphosphinoyl imine provided the product in excellent yield and enantioselectivity (Scheme 14).37N-Diphenylphosphinoyl imines have been demonstrated previously as versatile electrophiles in a number of asymmetric reactions and are easily cleaved under mildly acidic conditions.38 Using the typical reaction conditions, the substrate scope of this reaction was established. A variety of imines are well suited for this catalyst system including a number of substituents and heteroatoms. Electron-rich aldimines showed higher reactivity, while electron-poor aldimines showed slightly diminished reactivity but still high yields of the protected amines. This high enantioselection using both aldehydes and aldimines has been shown previously, albeit with longer reaction times and higher catalyst loading.11,39–40
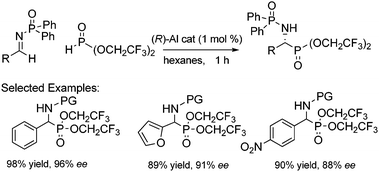 | ||
| Scheme 14 Asymmetric Pudovik reaction of imines. | ||
Simple acid hydrolysis was reported to be highly effective to generate the desired phosphonic acid (Scheme 15). Attempts to deprotect the product in methanolic concentrated HCl at room temperature resulted in no reaction but no loss in enantiopurity of the original substrate was observed. However, increasing the temperature to refluxing conditions provided the desired product almost quantitatively. Subsequent protection of the hydroxy/amino moiety and reprotection of the acid to the methyl phosphonates provided the fully reprotected methyl phosphonates. We were pleased to find that no erosion in enantioselectivity is observed during the hydrolysis of the bis(2,2,2-trifluoroethyl) phosphonate ester moiety. Another unique feature of this system is that with the protected amine in hand, both protecting groups can be removed in a single pot with acidic hydrolysis, or selective deprotection of the phosphinoyl moiety using mildly acidic conditions. In addition, it was observed that the absolute configuration of the hydroxyl and amino products were oppositely stereoconfigured, presumably due to the single coordination of the aldehyde and the double coordination of the aldimine to the aluminium catalyst.
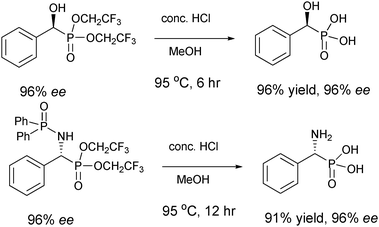 | ||
| Scheme 15 To α-hydroxy and α-amino phosphonic acids. | ||
Asymmetric Strecker reaction
The synthesis of α-amino acids by the Strecker reaction41 is the condensation of a carbonyl compound with ammonia to form an imine intermediate to which a cyanide nucleophile is then added to produce an α-amino nitrile. This nitrile is then hydrolyzed either in situ or later to provide the α-amino acids. This represents the oldest known method for producing these types of compounds.42 Later variants of this reaction included the preformation of imines and subsequent isolation or purification. These imines were then subjected to the classic conditions to give the protected α-amino nitrile. This represents the more widely used route to these types of compounds today.42The asymmetric catalytic synthesis of α-amino acids via hydrocyanation of imines has seen a large number reports since the first report slightly more than a decade ago.43 After this initial report several organocatalytic and metal catalyzed asymmetric Strecker syntheses were reported.42,44–47 Although there are a large number of enantioselective processes, there is still a need to further develop more efficient, and operationally simple, catalytic asymmetric protocols to produce α-amino acids.
The use of TMSCN or metal cyanide reagents has found extensive use in the asymmetric synthesis of α-amino nitriles. However, alternative, safer, and environmentally friendlier reagents which are less toxic than TMSCN or HCN are needed for this important transformation. In the context of enantioenriched cyanohydrins several alternative sources of cyanide have been found to be effective for the generation of functionalized cyanohydrins. The advantage is three-fold, the silylated α-hydroxynitriles generated by the reaction of TMSCN and aldehydes are unstable and need to be derivatized, thus the use of alternative reagents provides O-protected cyanohydrins. Secondly, these reagents are operationally simple to handle and strictly specialized techniques are not required. Furthermore, these reagents are typically more atom-economical than TMSCN and therefore more attractive from a cost standpoint.
Given aluminium SALEN’s historical perspective on the asymmetric Strecker reaction, we wanted to see if our catalyst could compete with other catalysts in terms of enantioselectivity, although through the dual activation mechanism. We have demonstrated that our TBOx aluminium catalyst is certainly able to activate a variety of carbonyl compounds and derivatives with complete control of facial selectivity.23,37 Moreover, although there are metal catalysts to date which are able to catalyze the asymmetric Strecker reaction, some of them display a very limited substrate scope.42
Gratifyingly, ethyl cyanoformate and ethyl cyanophosphonate provided the product in high yield and enantioselectivity. Acetone cyanohydrin provided no desired product and was not considered a viable cyanide source for our reaction. Since ethyl cyanoformate provided the product in almost quantitative yield, the use of this cyanide source was examined for further optimization.
Under the optimized conditions a variety of aldimines were subjected to this protocol (Scheme 16).48 Functional groups around the aromatic ring are well tolerated providing high enantiomeric excess in all cases. Electron deficient and larger aromatic aldimines showed reduced reactivity and yield requiring slightly prolonged reaction times. Typically problematic substrates, such as heteroaromatic imines, also provide α-amino protected hydronitrile products without deleterious effects on the enantioselectivity. Furthermore, unsaturated and aliphatic aldimines also demonstrated high enantiomeric excess under optimized conditions, although a more nucleophilic base, DMAP, was required.
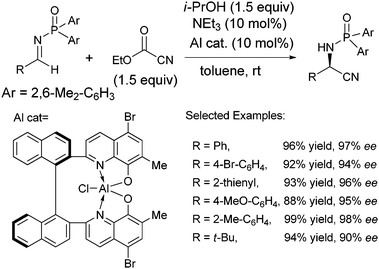 | ||
| Scheme 16 TBOxAlCl catalyzed asymmetric Strecker reaction with ethyl cyanoformate. | ||
We further extended this optimized methodology to ketimines, which provides access to enantiomerically enriched quaternary stereocenters (Scheme 17).24,26 Ketimines are inherently less reactive and more challenging substrates than their aldimine counterparts due to the steric demands during the formation of the carbon–carbon bond and discrimination of the prochiral faces. Although several notable and highly selective metal catalyzed and organocatalyzed variants of this reaction have been accomplished using TMSCN as the cyanide source, application with alternative sources has yet to be investigated.42,44,47 Typically, complications arise when changing substrates, such as the need to retune or reoptimize the reaction conditions, however we found that utilizing the same catalyst that was found to be optimal for aldimines was equally as effective for ketimines.
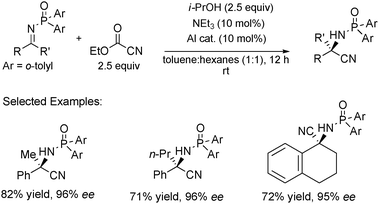 | ||
| Scheme 17 TBOxAlCl catalyzed asymmetric Strecker reaction of ketimines. | ||
On the basis of our previous results, we propose a possible transition state that accounts for our observed absolute stereochemistry (Fig. 5).37 Activation of the imine by coordination to the aluminium catalyst should take place such that the Si face is available for nucleophilic addition of cyanide. When screening ligands we found that as the steric size of R1 in the ligand increased, stark decreases in enantioselectivity were observed. Based on our model, R1 should block the Re face of the aldimine. However, increasing the steric size of R1 effectively blocks both the Re and Si faces of the aldimine equally, resulting in decreased discrimination of the prochiral faces. This observed facial selectivity is the same as with our hydrophosphonylation of imines.
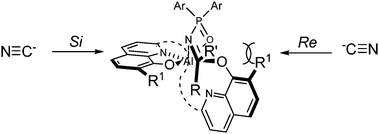 | ||
| Fig. 5 Proposed transition state. | ||
Conclusions
In this short review we have briefly described the advances we have made in the area of chiral-at-metal complexes using a newly designed tethered bis(8-quinolinolato) (TBOx) ligand. This ligand when complexed displays a cis-β configuration around the metal center, and is not generated through the reaction conditions. It is to this configuration that we attribute our catalyst’s high reactivity and stereoselectivity in the reactions we have demonstrated thus far. We expect to find other asymmetric reactions in which our ligand will find practical use when complexed to other metals. Improvements in many areas, particularly in catalyst loadings to find more active systems, are required in future generations of metal catalyzed reactions while maintaining high stereoselectivity. In the reactions shown in this review we feel we have made meaningful contributions to those areas in the area of substrate scope, reactivity, and selectivity. Modifications of the 8-hydroxyquinoline moiety of the ligand have shown stark increases in reactivity as well as selectivity. Currently investigations into other modifications are underway, as are other stereoselective reactions in which complex structure is generated rapidly.References
- R. Noyori, Nat. Chem., 2009, 1, 5 Search PubMed.
- E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, Berlin, New York, 1999 Search PubMed.
- I. Ojima, Catalytic Asymmetric Synthesis, Wiley-VCH, New York, 2nd edn, 2000 Search PubMed.
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS.
- H. Yamamoto and G. Y. Xia, Chem. Lett., 2007, 36, 1082–1087 CrossRef CAS.
- B. Bosnich and W. R. Kneen, Inorg. Chem., 1970, 9, 2191 CrossRef CAS.
- P. D. Knight and P. Scott, Coord. Chem. Rev., 2003, 242, 125–143 CrossRef CAS.
- U. Knof and A. von Zelewsky, Angew. Chem., Int. Ed., 1999, 38, 302–322 CrossRef.
- T. Katsuki, Chem. Soc. Rev., 2004, 33, 437–444 RSC.
- Y. N. Belokon, S. Caveda-Cepas, B. Green, N. S. Ikonnikov, V. N. Krustalev, V. S. Larichev, M. A. Moscalenko, M. North, C. Orizu, V. I. Tararov, M. Tasinazzo, G. I. Timofeeva and L. V. Yashkina, J. Am. Chem. Soc., 1999, 121, 3968–3973 CrossRef CAS.
- B. Saito, H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 1978–1986 CrossRef CAS.
- D. A. Evans, J. M. Janey, N. Magomedov and J. S. Tedrow, Angew. Chem., Int. Ed., 2001, 40, 1884–1888 CrossRef CAS.
- N. Takenaka, G. Y. Xia and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 13198–13199 CrossRef CAS.
- Y. Okude, S. Hirano, T. Hiyama and H. Nozaki, J. Am. Chem. Soc., 1977, 99, 3179–3181 CrossRef CAS.
- G. Y. Xia and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 2554–2555 CrossRef CAS.
- G. Y. Xia and H. Yamamoto, J. Am. Chem. Soc., 2007, 129, 496–497 CrossRef CAS.
- C. M. Yu, S. J. Lee and M. Jeon, J. Chem. Soc., Perkin Trans. 1, 1999, 3557–3558 RSC.
- M. M. Luo, Y. Iwabuchi and S. Hatakeyama, Synlett, 1999, 1109–1111 CrossRef CAS.
- M. M. Luo, Y. Iwabuchi and S. Hatakeyama, Chem. Commun., 1999, 267–268 RSC.
- M. Naodovic, G. Xia and H. Yamamoto, Org. Lett., 2008, 10, 4053–4055 CrossRef CAS.
- R. L. Augustine, Carbon–Carbon Bond Formation, M. Dekker, New York, 1979 Search PubMed.
- K. Narasaka, K. Soai and T. Mukaiyam, Chem. Lett., 1974, 1223–1224 CAS.
- N. Takenaka, J. P. Abell and H. Yamamoto, J. Am. Chem. Soc., 2007, 129, 742–743 CrossRef CAS.
- E. J. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388–401 CrossRef.
- K. Fuji, Chem. Rev., 1993, 93, 2037–2066 CrossRef CAS.
- J. Christoffers and A. Mann, Angew. Chem., Int. Ed., 2001, 40, 4591 CrossRef CAS.
- C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363–5367 CrossRef CAS.
- R. J. Sundberg, in The Chemistry of Indoles, Academic Press, New York, 1970, pp. 78–83 Search PubMed; L. Marion, in The Alkaloids. Chemistry and Physiology, Academic Press, New York, 1952, vol. 2, pp. 371–481 Search PubMed.
- T. B. Poulsen and K. A. Jørgensen, Chem. Rev., 2008, 108, 2903 CrossRef CAS.
- A. N. Pudovik and I. V. Konovalova, Synthesis, 1979, 81–96 CrossRef CAS.
- P. Merino, E. Marques-Lopez and R. P. Herrera, Adv. Synth. Catal., 2008, 350, 1195–1208 CrossRef CAS.
- O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 2005, 16, 3295–3340 CrossRef CAS.
- O. I. Kolodiazhnyi, E. V. Grishkun, S. Sheiko, O. Demchuk, H. Thoennessen, P. G. Jones and R. Schmutzler, Tetrahedron: Asymmetry, 1998, 9, 1645–1649 CrossRef CAS.
- J. A. Ma, Chem. Soc. Rev., 2006, 35, 630–636 RSC.
- G. O. Doak and L. D. Freedman, Chem. Rev., 1961, 61, 31–44 CrossRef CAS.
- J. P. Duxbury, J. N. D. Warne, R. Mushtaq, C. Ward, M. Thornton-Pett, M. L. Jiang, M. J. R. Greatrex and T. P. Kee, Organometallics, 2000, 19, 4445–4457 CrossRef CAS.
- J. P. Abell and H. Yamamoto, J. Am. Chem. Soc., 2008, 130, 10521 CrossRef CAS.
- S. M. Weinreb and R. K. Orr, Synthesis, 2005, 1205–1227 CrossRef CAS.
- T. Arai, M. Bougauchi, H. Sasai and M. Shibasaki, J. Org. Chem., 1996, 61, 2926–2927 CrossRef CAS.
- H. Sasai, S. Arai, Y. Tahara and M. Shibasaki, J. Org. Chem., 1995, 60, 6656–6657 CrossRef CAS.
- A. Strecker, Justus Liebigs Ann. Chem., 1850, 75, 27–45.
- H. Groger, Chem. Rev., 2003, 103, 2795–2827 CrossRef.
- M. S. Iyer, K. M. Gigstad, N. D. Namdev and M. Lipton, J. Am. Chem. Soc., 1996, 118, 4910–4911 CrossRef.
- C. Spino, Angew. Chem., Int. Ed., 2004, 43, 1764–1766 CrossRef CAS.
- L. Yet, Angew. Chem., Int. Ed., 2001, 40, 875–877 CrossRef CAS.
- N. U. H. Khan, R. I. Kureshy, S. H. R. Abdi, S. Agrawal and R. V. Jasra, Coord. Chem. Rev., 2008, 252, 593–623 CrossRef CAS.
- P. Merino, E. Marques-Lopez, T. Tejero and R. P. Herrera, Tetrahedron, 2009, 65, 1219–1234 CrossRef CAS.
- J. Abell and H. Yamamoto, J. Am. Chem. Soc., 2009, 131, 15118–15119 CrossRef CAS.
Footnote |
| † Current address: Harvard University, Department of Chemistry & Chemical Biology, 12 Oxford Street, Cambridge, MA 02138, USA |
| This journal is © The Royal Society of Chemistry 2010 |