Supramolecular n/p-heterojunction photosystems with oriented multicolored antiparallel redox gradients (OMARG-SHJs)
Rajesh Bhosale, Jiří Míšek, Naomi Sakai* and Stefan Matile*
Department of Organic Chemistry, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch; Web: http://www.unige.ch/sciences/chiorg/matile/ Fax: +41 22 379 5123; Tel: +41 22 379 6523
First published on 7th September 2009
Abstract
Directional electron and hole transport is essential in photosynthesis. Applied to molecular optoelectronics such as organic solar cells, these lessons from nature call for oriented supramolecular n/p-heterojunctions (SHJs) that contain various chromophores and antiparallel redox gradients (OMARGs). In this tutorial review, we summarize recent progress made toward the construction of OMARG-SHJs. This conceptually innovative twist added to a timely topic should appeal to the synthetic organic, supramolecular, biological, physical, analytical and materials chemist as well as to the expert in energy and environmental sciences.
 Naomi Sakai, Rajesh Bhosale, Stefan Matile and Jiří Míšek | Naomi Sakai gained her BS from Keio University (1987) and her PhD from Tokushima Bunri University (1994). After a postdoc with Professor Koji Nakanishi at Columbia University (1994–1996), she has focused on the creation of supramolecular functional nanoarchitectures for applications such as solar cells and biosensors, first in Washington DC (Georgetown University, 1996–1999), then in Geneva (1999 to present). Rajesh Bhosale was born in 1977 in Udgir Latur, India. He received his MSc from SRTM, University Nanded, India. In 2005, he moved to the University of Geneva, Switzerland, where he is currently carrying out his PhD under the supervision of Professor S. Matile. His research is focused on the design, synthesis and evaluation of artificial photosystems. Stefan Matile received a Diploma (1989) and PhD (1994) from the University of Zurich under the supervision of Wolf Woggon. After a postdoc with Koji Nakanishi at Columbia in New York (1994–1996), he joined the faculty of Georgetown University, Washington DC before he moved to Geneva in 1999. His research interests are at the interface of organic, biological and supramolecular materials chemistry, with emphasis on supramolecular multifunctional architectures such as photosystems, sensors and ion channels. Jiří Míšek received his MSc (2004) and PhD (2008) from the Charles University in Prague and the Institute of Organic Chemistry and Biochemistry in Prague (Czech Republic). During his PhD in the group of Dr I. Starý he was synthesizing azahelicenes and studying their properties and potential applications. Currently he is a Postdoctoral Roche Fellow in the group of Professor S. Matile in Geneva focusing on various supramolecular approaches to functional photosystems. |
1. Introduction
In biological photosystems, excitation of the special pair of chlorophylls (a1, Fig. 1(a)) is followed by directional, ultrafast electron transfer cascade along sophisticated redox gradients to the remote ubiquinone acceptor (a6, Fig. 1(a)).1,2 The hole left behind is transferred in the other direction along an orthogonal redox gradient to ultimately oxidize water. These cascade electron/hole transfer pathways are essential for the efficiency of photosynthesis, and held together in antiparallel manner by a large protein embedded in the bilayer membrane.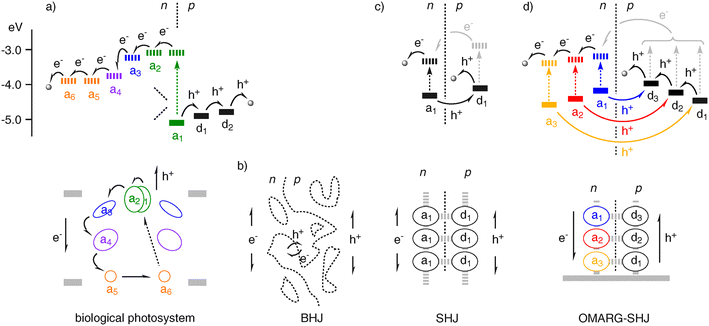 | ||
| Fig. 1 Energetics (top; solid, HOMO; dashed, LUMO) and architectures (bottom) of electron (e−; a, electron acceptors) and hole (h+; d, electron donors = hole acceptors) transfer cascades to the final destinations (e.g., electrodes; grey circles in a, d) in (a) biological photosystems (a1/a2, chlorophyll special pair; a5/a6, ubiquinone), (b) BHJs in organic solar cells, (c) SHJs, and (d) OMARG-SHJs. In OMARG-SHJs, excitation of acceptors an of different color can be followed by donor–acceptor n/p-charge separation and electron and hole transfer down antiparallel redox gradients toward their final destination; donor excitation followed by electron (rather than energy) transfer to acceptors an (grey) leads to the same situation and is thus additive. | ||
In organic solar cells, a macroscopic layer of electron- or n-semiconducting material is placed next to a hole- or p-semiconducting layer. In such a bilayer configuration,3 photogenerated excitons dissociate at the donor–acceptor interface into electrons and holes that move to their respective final destination and generate photocurrent. Increases of active interface by blending the two conductors resulted in dramatically increased efficiencies of photosystems. Reminiscent of the maximized surface area of thylakoid membranes in chloroplasts, the interlaced donor/acceptor-layers in organic solar cells are usually referred to as bulk n/p-heterojunctions (BHJs) (Fig. 1(b)).4 Maximized exciton dissociation achieved at maximized interfaces in BHJ solar cells comes at the cost of reduced hole and electron mobility in poorly organized, increasingly discontinuous donor and acceptor domains. Top-down approaches to improve the organization, and thus the conductivity include the use of differential solubilizers, dendritic polymers, covalent dyad systems, mixed crystals, and so on.4–9 Increasing organization of BHJs ultimately leads to the situation where hole (h+) and electron (e−) transporting channels are separated and aligned on the molecular level, that is supramolecular n/p-heterojunctions (SHJs) (Fig. 1(c)). The bottom-up nature of this approach allows for the rational design of ordered nanostructures not only to achieve high conductivity but also to implement complexity in the system. However, lessons from nature call for much more, namely SHJ architectures that come in colors everywhere to capture as much light as possible and quickly funnel electrons and holes in opposite directions to the electrodes along oriented co-axial molecular h+ and e− transporting channels. In the following, this bioinspired ideal SHJ architecture is called OMARG-SHJ, that is an SHJ with oriented multicolored antiparallel redox gradients (Fig. 1(d)).
The creation of OMARG-SHJs is far from possible today because of the exceptional challenges involved. Recent progress made toward OMARG-SHJs is summarized in this review. Before focusing on surface architectures, we briefly summarized methods of characterization and highlight pioneering approaches toward SHJs in solution. The discussion of oriented SHJs on surfaces begins with selected examples for the “fuzzy” architectures obtained from layer-by-layer (LBL) assembly. From there, we zoom in on recent pioneering approaches to reorient LBL-architectures in polymer brushes or to install interdigitating inter- and intralayer recognition motifs in LBL architectures to build oriented SHJs on solid grounds. The following chapter on photosystems with redox gradients discusses first covalent approaches with self-assembled monolayers SAMs, including examples for the usefulness of biological scaffolds such as DNA duplexes or α-helices to address specific topics. From there, pioneering breakthroughs with 3D architectures with oriented redox gradients are summarized, covering increasingly organized LBL systems and coordinative covalent capture approaches. After a brief summary of rainbow systems, we combine all separate topics to close with emerging approaches toward OMARG-SHJs.
2. Characterization methods
The characterization of synthetic supramolecular photosystems covers many different aspects. Essential insights come from the determination of HOMO–LUMO levels by cyclic voltammetry and absorption spectroscopy, ultrafast photophysics to characterize charge separation (CS) and charge recombination (CR) kinetics or structural studies with various spectroscopic, microscopic, crystallographic and acoustic methods.10 However, the heart of the matter are J–V and J–L curves, plots or profiles: They reveal the essential characteristics of the functional system by direct functional feedback (Fig. 2).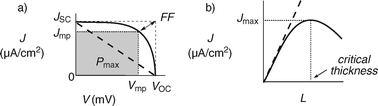 | ||
| Fig. 2 “Ohmic” (dashed) and “non-ohmic” (solid) J–V curve (a) and linear (dashed) and bell-shaped (solid) J–L curves (b) with indication of key parameters. Ideal OMARG-SHJs should have non-ohmic J–V with high JSC, VOC and FF as well as linear J–L with unsaturable Jmax and critical thickness. | ||
J–V curves describe the dependence of the photocurrent density (J) on the applied voltage (V). Key parameters in J–V curves include the short circuit current density JSC, the open circuit voltage VOC, and most importantly, maximal output power Pmax (the grey area in Fig. 2(a)) with the corresponding Jmp and Vmp. The fill factor FF = Jmp×Vmp/JSC×VOC describes the squareness of the J–V curve, and is a commonly used parameter to describe Pmax (= FF×JSC×VOC). Linear (“ohmic”) J–V curves have a low FFmin = 0.25. Optimized BHJ solar cells have FFBHJ∼ 0.61.5 The power conversion efficiency is defined as ηeff = Pmax/Pin× 100% where Pin is the irradiance. Leading BHJ solar cells have ηeff∼ 5%.
With the increasing organization of h+ and e− transporting channels,11 OMARG-SHJs are expected to have high FF. At high FF, VOC is determined by the energy difference between HOMO of donor and LUMO of acceptor and strongly influenced by other parameters such as device connection in series. OMARG-SHJs could have high VOC because it is presumably not the smallest but either the proximal12 or perhaps even the maximal HOMO–LUMO difference in a redox gradient that matters. Because they collect much light, OMARG-SHJs should have comparably high JSC.
J–L curves describe the dependence of the photocurrent density on the layer thickness or number of layers deposited on the surface. In general, the higher the absorbance of the layer, the more charges are generated and thus, the higher J. Therefore, at the beginning, J–L curves are usually linear (Fig. 2(b)). At a critical thickness, J–L curves can saturate at Jmax and flatten out or even decrease with increasing thickness (Fig. 2(b), bold). If evidence for continuing growth after the photocurrent saturation is secured, then the critical thickness is of highest interest because it indicates the distance holes and electrons can travel without meeting each other to recombine. In OMARG-SHJs, the generated charges should be immediately driven away from each other, and thus very large critical thickness could be expected.
Important information beyond J–V and J–L curves comes from action spectra, quantifying the photocurrents generated by individual chromophores, and AFM images, describing the surface roughness of the artificial photosystems (smooth surfaces have been associated with highly ordered structures and thus high efficiencies).6
Comparison of different systems has to be done with caution, as different conditions can in general affect the outcome with supramolecular functional systems dramatically.
3. SHJs
Common aromatics (e.g., benzenes) are π-basic and characterized by a negative quadrupole moment (e.g., Qzz = −8.5 B). Their electron-rich surfaces allow cation-π interactions and formation of p-semiconducting π-stacks (e.g., hexabenzocoronenes, HBCs).13 The much more exotic π-acidic aromatics (e.g., hexafluorobenzene) have a positive quadrupole moment (e.g., Qzz = +9.5 B) and an electron-poor surface to attract anions, and their π-stacks can be n-semiconductors (e.g., naphthalenediimides, NDIs). SHJs are a big challenge because the programmed assembly of architectures such as 1 with co-axial h+ and e− transporting π-stacks from π-basic and π-acidic aromatics is unfavorable (Fig. 3). Oligomers of complementary aromatics such as π-acid 2 and π-base 3 prefer to undergo aromatic donor–acceptor interactions and form charge-transfer complexes such as double helix 4 instead.14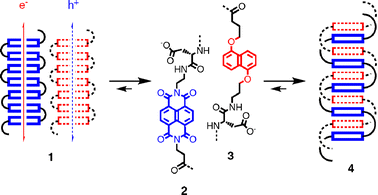 | ||
| Fig. 3 Programmed assembly of π-acidic and π-basic oligomers 2 and 3 into charge-transfer duplex 4 rather that the notional SHJ architecture 1. | ||
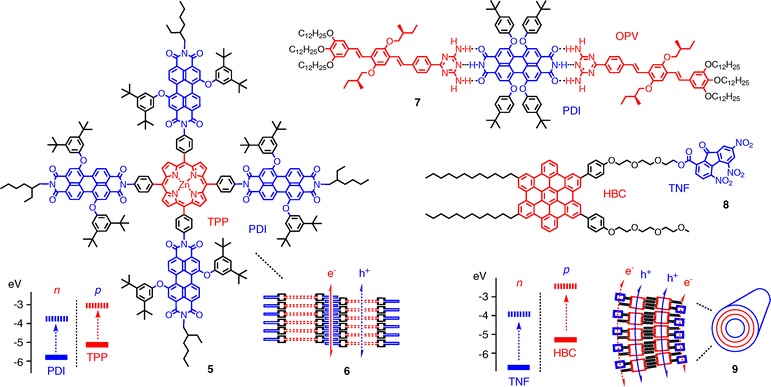
The multichromophoric hybrids 5 self-assemble into π-stack nanoarchitectures 6 with SHJ-like characteristics (Fig. 4).15 Hybrids 5 are composed of a central tetraphenylporphyrin (TPP) donor that is surrounded by four perylenediimide (PDI) acceptors. To self-assemble into nanoparticles 6, PDIs from neighboring monomers are thought to interdigitate and form continuous face-to-face π-stacks. This places the zinc porphyrins far enough apart to retain their ability to coordinate small ligands such as pyridine. PDI stacks are of interest because they are known as excellent e− transporting channels.16,17
 | ||
| Fig. 4 Solution approaches toward SHJ architectures. | ||
The supramolecular trimer 7 self-assembles into left-handed helices that further self-assemble into right-handed supercoils.18 Trimer 7 contains a PDI acceptor in the middle. The acceptor-donor–acceptor (ADA) hydrogen-bonding motif of imides is used to bind a complementary DAD 2,4-diamino-1,3,5-triazine. This DAD domain is linked to a minimalist oligophenylvinyl (OPV) donor. Important for self-assembly, ADA–DAD hydrogen-bonding places the OPV donors in plane with the PDI acceptor. Support for SHJ architectures with π-stacked co-planar OPV–PDI–OPV arrays can be seen in the unusually fast CR in superhelical assemblies of 7. Thermal denaturation slows down CR, and less favorable assemblies obtained from covalent and deplanarized OPV–PDI–OPV analogs recombine slower in assembled and disassembled form. Poor diode behavior found upon spin coating of 7 on the conducting surface was attributed to the wrong lateral orientation of the helices relative to the surface.
Hexabenzocoronene (HBC) stacks rank among the best p-semiconductors (Fig. 4).17 Amphiphilic HBCs such as 8 self-assemble under carefully optimized conditions into bilayer nanotubes such as 9.19 In nanotube 9, the trinitrofluorene (TNF) acceptors are added at inner and outer surface and form co-axial n-channels next to the HBC channel in the tube. This formal SHJ architecture generates photocurrent when spin-coated onto electrodes. Contrary to expectations for SHJs, mixed nanotubes with reduced TNF content give higher photocurrents.20 Covalent cross-linking by ring-opening olefin metathesis (ROMP) is applicable to stabilize the active architectures.21
4. Oriented SHJs
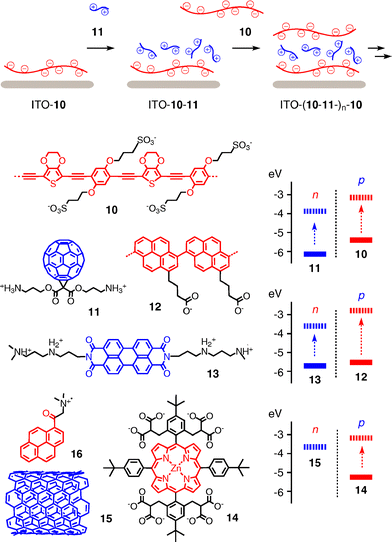
Compared to self-assembly in solution, the creation of oriented SHJ architectures on solid substrates is much more challenging. The deposition of preformed systems by “top-down” approaches that operate without molecular-level resolution lack directionality and are intrinsically incompatible with the creation of oriented architectures, not to speak of the installation of antiparallel redox gradients.Layer-by-layer (LBL) assembly is the classical “bottom-up” approach to oriented surface architectures (Fig. 5).22 In this approach, solid substrates such as ITO (indium tin oxide) are dipped first into a solution of polyanions such as 10.23 Cooperative multivalent ion pairing attracts sufficient polyanion 10 not only to produce a polyanionic layer but also to cause surface charge inversion. The resulting anionic ITO–10 system is then dipped into solutions of di- or polycations such as 11. The obtained, charge-inverted bilayers ITO–10–11 are incubated with polyanions 10, anionic ITO–10–11–10 with polycations 11, and so on. This process can be repeated many times to give thick films with ITO–(10–11–)n LBL architectures.
 | ||
| Fig. 5 Selected LBL assembly of donor–acceptor architectures. | ||
LBL assembly has been used extensively for the creation of synthetic photosystems. Most popular are conjugated polymers as donors, including polyphenylethynyls, polythiophenes or mixed systems such as 10.23 Often, they are combined with charged fullerenes such as 11 as universal acceptors. The combination of oligopyrene donors 12 and PDI acceptors 13 is representative for possible variations that have been realized with the LBL theme.24 Combined with standard TPP donors 14, a recent breakthrough with LBL photosystems introduces carbon nanotubes 15 as acceptors with exquisite conductivity.25,26 Carbon nanotubes are solubilized and charged for LBL assembly with cationic pyrene donors 16 that bind to their aromatic surface by strong π,π-interactions.
The bell-shaped J–L curve of LBL architectures ITO–(10–11–)n reveal an outstanding critical thickness of ∼100 layers. The maximal ηeff = 0.041%, claimed to be the best among LBL photosystems, is mainly limited by a disappointing FF = 0.31. This value is consistent with poor organization11 as expected for LBL architectures22 and as supported by characteristic nodular features on surfaces with a roughness up to 2.5 nm (rms). For comparison, LBL architectures ITO–(12–13–)n feature a critical thickness of 10 layers and a similarly bumpy surface with a roughness that, characteristically, increases from 2.6 to 4.7 nm (rms) with increasing thickness (ηeff and FF were not reported).24 LBL architectures ITO–(14–15/16–)n have a linear J–L curve up to 17 layers (J–V characteristics were not reported).25
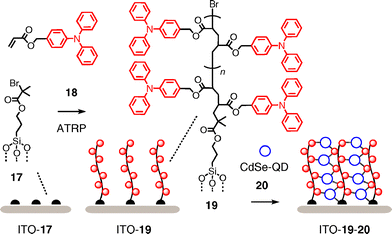
Easy accessibility to LBL architectures comes at the cost of poor organization and horizontal orientation of the adjacent h+ and e− transporting layers. Polymer brushes represent an inviting concept to reorient polymer backbones and achieve the vertical alignment desired for better charge collection at the electrodes (Fig. 6).27,28 Polymer brushes are prepared by surface-initiated polymerization. The obtained close packing forces the polymers to align vertically to the surface. In the pioneering example focusing on SHJ architectures, ATRP (atom transfer radical polymerization) is used as most popular method to make the brushes. Initiator 17 is grafted onto ITO surfaces to polymerize the triphenylamine acrylate 18 directly from ITO–17 in the presence of the standard Cu catalysts. The obtained ITO–19 brushes are then dipped into solutions of CdSe nanocrystals 20. Displacement of some of their pyridine ligands by triphenylamines captures the CdSe nanocrystal acceptors along single polymer brush donors.
 | ||
| Fig. 6 Notional SHJ photosystems made from polymer brushes. | ||
In polytriphenylamineacrylate brushes, the current density normal to the substrate is up to three orders of magnitude higher than in spin-coated films. Another increase in the current density by three orders of magnitude in the presence of nanocrystals in ITO–19–20 is due to the high conductivity of the latter and supports their complete infiltration into the brushes. Although high dark currents hinder standard characterization as formal SHJ photosystem, the general nature of the approach has high potential for future developments toward OMARG-SHJs.
Electropolymerization can be considered as special route toward polymer brushes.29,30 Electropolymerization of thiophenes together with semiconductor nanoparticles is expected to produce SHJ-type hybrid architectures that is very similar to the ATRP-derived ITO–19–20.29
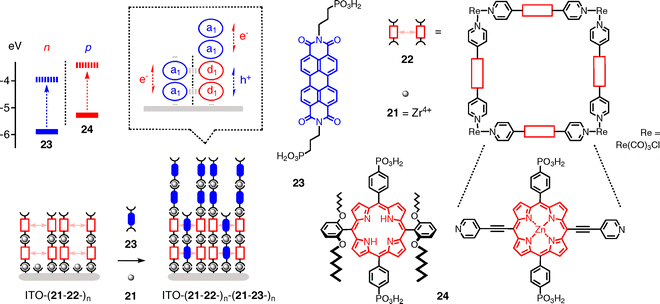
A very elegant and explicit approach toward oriented SHJ architectures uses zirconated ITO–21 surfaces to capture the four phosphonates at the bottom of porphyrin squares 22 (Fig. 7).31 Zirconation of the four phosphonates prepares for the binding of another porphyrin square. This ordered LBL process is repeatable and yields porous ITO–(21–22–)n multilayers. The pores in ITO–(21–22–)n are then filled by repetitive LBL addition of PDI acceptors 23 and zirconium cations 21 for reactivation of the terminal phosphonates. During the LBL infiltration of PDIs into the porphyrin squares, PDI acceptors are simultaneously captured on top of the porphyrin donors. Only the lower part of the obtained ITO–(21–22–)n–(21–23–)n architecture thus represents a formal SHJ motif, where PDI donors are lined up vertically next to co-axial TPP acceptors, whereas the PDIs on top add a non-SHJ redox gradient.
 | ||
| Fig. 7 Formal SHJ architecture obtained with porous arrays of porphyrin donors filled with PDI acceptors. | ||
Elegant experiments have been conceived to support the existence of the designed architecture. Discontinuous PDI reducibility, for instance, provides excellent support that the PDIs really penetrate the porphyrin squares. Replacement of squares 22 with crowded porphyrins 24 gives similar architectures. Both architectures afford linear J–L curves up to 8–10 layers. Action spectra demonstrate that both PDIs and TPPs contribute to photocurrent generation. J–V curves were not reported.
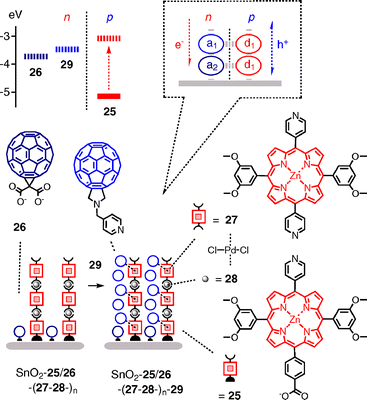
A recent ambitious approach combining aspects of several previous systems begins with carboxylate linker on tin oxide to produce a mixed monolayer of porphyrin donors 25 and fullerene acceptors 26 (Fig. 8).32 The porphyrins in SnO2–25/26 are activated by adding palladium propagators 27 to the pyridine ligand. Incubation of the obtained SnO2–25/26–27 with dipyridine porphyrin 28 followed by reactivation with Pd 27 gives SnO2–25/26–27–28–27. Repeated ordered LBL incubation with 28 and 27 produces supramolecular metalloporphyrin brushes SnO2–25/26–(27–28–)n. To infiltrate a coaxial string of donors, fullerenes 29 are added with the expectation that their pyridine tail acts as axial ligand to the zinc porphyrins. The elegant combination of peripheral and central porphyrin coordination chemistry in SnO2–25/26–(27–28–)n–29 affords a formal SHJ architecture with strings of porphyrin acceptors that grow from the surface and are lined by coaxial strings of fullerene acceptors. The lowered LUMO of fullerene 26 compared to fullerene 29 adds a formal redox gradient to the n-channel.
 | ||
| Fig. 8 Notional SHJ photosystems made from supramolecular polymer brushes. | ||
J–L curves of SnO2–25/26–(27–28–)n–29 reveal a critical thickness of five layers with an incident photon-to-current efficiency IPCE = 21% at Jmax. Photocurrent decrease after more than five layers of depositions coincides with inhibited growth of the system. This suggests that reduced growth mainly accounts for photocurrent decrease. At Jmax with five layers, the addition of pyridine fullerenes 29 nearly doubles the photocurrent density, whereas fullerenes with phenyls in place of pyridines cause only an increase of about 10%. AFM images reveal reasonably smooth surfaces, with surface roughness increasing with increasing number of layers in the presence and the absence of fullerenes (J–V characteristic were not reported).
Zipper assembly of rod-stack architectures such as Au–30–(31–32–)n has been introduced as metal-free, all-organic approach to SHJ photosystems (including OMARG-SHJs, see below).33 Zipper architectures are assembled from multichromophoric donor–acceptor hybrids beginning with the short POP–B initiator 30 (Fig. 9. POP: p-oligophenyl). The disulfide at one end is introduced to bind covalently to gold, negatively charged blue NDIs B are placed along the short rigid-rod POP scaffold to avoid non-specific interaction with gold. The formal Au–30 monolayer is then exposed to POP–B propagator 31. Driven by π,π-stacking, intrastack hydrogen bonding and interstack ion pairing,34,35 the lower half of the blue and cationic NDIs of 31 are expected to π-stack with the anionic NDI acceptors of 30, whereas the upper half remains free as “sticky end” to zip up with the complementary anionic POP–B propagator 32. Repeated incubation with cationic 31 and anionic 32 gives zipper Au–30–(31–32–)n.
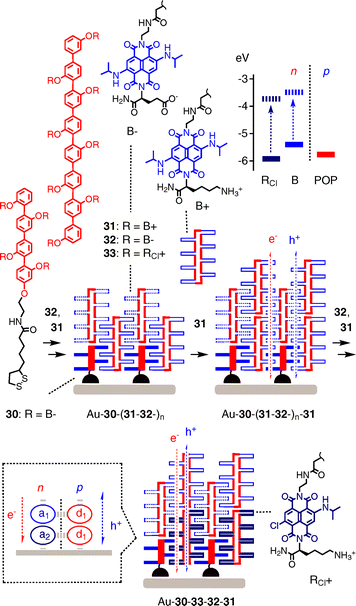 | ||
| Fig. 9 Zipper assembly of notional SHJ architectures. | ||
Au–30–(31–32–)n is designed to feature π-stacks formed by interdigitating aromatic planes from neighboring scaffolds along strings of interdigitating rigid rods. This architecture excels with operational inter- and intralayer recognition motifs as well as the co-axial alignment of e− transporting NDI stacks next to h+ transporting POP rods. However, photo induced CS (PCS) between NDI stacks and POP rods is not possible with Au–30–(31–32–)n because the HOMO of blue NDIs B is above that of POPs. As with the special pair of chlorophylls in the heart of biological photosystems, this gives a donor–acceptor-free system with symmetry-breaking PCS between blue NDIs B as only way to proceed, one B being reduced to B−. and one B being oxidized to B+., with minimal losses of energy.35,36 In this situation, both hole and electron would move in adjacent NDI stacks, with POPs serving as rigid-rod scaffolds only without any functional role.
According to the kinetics of the transient absorption of the NDI−. radical anion, monomeric 31 is capable of ultrafast, symmetry-breaking PCS. PCS lives for 60 ps in monomeric 31 and 400 ps in self-assembled tetramers in lipid bilayer membranes.35,36 Zipper architectures Au–30–(31–32–)n exhibit linear J–L curves up to 10 layers.33 Capping of the sticky ends in Au–30–31 with short 30 cleanly inhibits further increases in photocurrent generation, as does replacement of the cationic propagator 31 with disorganizing polylysines. Mini-zippers with biphenyl initiators and p-quaterphenyl propagators generate less photocurrent and have bell-shaped J–L curves with Jmax at 10 layers, corresponding to five layers with standard Au–30–(31–32–)n zippers.37 Consistent with high organization of zipper architectures, the “non-ohmic”J–V curve of Au–30–31–32–31 was characterized by FF = 0.42, a quite remarkable value for gradient-free architectures with an expected thickness of ∼6 nm only.38
To activate both h+ and e− transporting channels in notional rod–stack POP–NDI zipper SHJ architectures, POP–RCl donor–acceptor hybrids were included.38 With the HOMO of RCl lying below that of POP, PCS by rod–stack electron transfer from POP donors to RCl acceptors is possible in this case. PCS with POP–RCl dyads 33 is correspondingly long-lived, with decay components reaching into the ns range. Consistent with rod–stack PCS, that is the formation of notional SHJ architectures, the action spectra of mixed zippers show that photocurrent generation by RCl exceeds that by B dramatically. The J–V curve of Au–30–33–32–31 excels with high VOC and an outstanding FF = 0.55, whereas the isomeric Au–30–31–32–33 with the reversed redox gradient in the NDI stack has a nearly “ohmic”FF = 0.31 and lower VOC. This difference in FF between the system with constructive redox gradients to guide the electrons to the gold surface and that with destructive ones driving the electron away from the surface demonstrate how B–RCl gradients in POP–NDI zipper architectures matter for function. Au–30–33–32–31, containing NDIs of red and blue color, can thus be considered as minimalist prototype of a formal OMARG-SHJ photosystem.
An inspired approach toward all-organic SHJ architectures is under development with systems such as Au–34–35/36 (Fig. 10).39 This approach has much future potential because it introduces the rich supramolecular chemistry of hydrogen-bonding network architectures. Best known for the self-assembly of supramolecular rosettes and tapes,40,41 barbiturate derivatives as in fullerene acceptors 35 are popular sectors that offer two linear acceptor–donor–acceptor (ADA) triads in a 60° angle. They pair with the matching DAD triads in the 60° sector of melamines in oligothiophenes 36. For their surface-initiated supramolecular polymerization, both components are deposited together on gold coated with monolayers of barbiturate derivative 34. Photocurrents generated with 35 and 36 are higher in the presence of initiator 34 than in the presence of methylated analogs and barbiturate initiators.
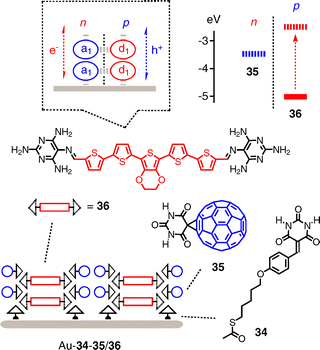 | ||
| Fig. 10 Toward SHJ systems with H-bonding network architectures. | ||
5. Redox gradients
In ideal OMARG-SHJs, oriented SHJs would be equipped with antiparallel redox gradients to move electrons and holes in opposite directions to their final destination. Although today’s small collection of formal SHJs contains examples with traces of one unidirectional two-component gradient, significant SHJ cascades remain to be realized, not to speak of two antiparallel gradients in n- and p-channels. However, several photosystems with quite significant redox gradients have been prepared by top-down approaches.For example, vapor deposition of layers of first tetraceno[2,3-b]thiophene donors followed by bifunctional copper phthalocyanines and fullerene acceptors gives trilayer photovoltaic cascade devices with increased VOC.12 Full increase in VOC occurs only with phthalocyanine layers thicker than 5 nm. This finding is very important because it demonstrates that VOC in cascade architectures is determined not by the smallest HOMO–LUMO differences but by either neighboring or perhaps even maximal HOMO–LUMO differences. Redox gradients can thus be considered as promising approach to increase VOC.
The covalent construction of redox gradients has been explored quite extensively with self-assembled monolayers (SAMs).42,43 An excellent example for an advanced system is the formation of mixed SAMs with fullerene–porphyrin–ferrocene triad 37 and boron dipyrrin 38 as energy donor on the gold surface (Fig. 11).
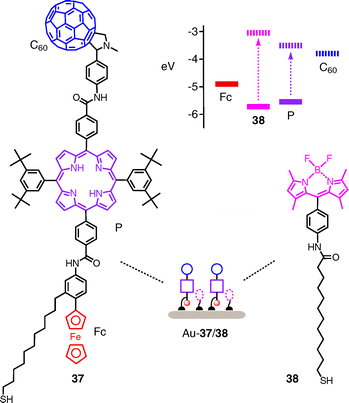 | ||
| Fig. 11 Mixed SAM photosystems to combine redox gradients with light harvesting. | ||
SAMs are simple enough to explore the more complex systems accessible with biological scaffolds. Judicious multiple cystein mutation, for example, can produce SAMs with bioengineered photosystems for photocurrent generation.44 SAMs from DNA double helices such as 39 and 40 are perfect to explore the importance of h+ transporting efficiency of the π-stack for photocurrent generation (Fig. 12).45 To do so, duplexes 39 and 40 are terminated with a naphthaleneimide (NI) acceptor on one side and a thiol on the other side for covalent capture on gold.
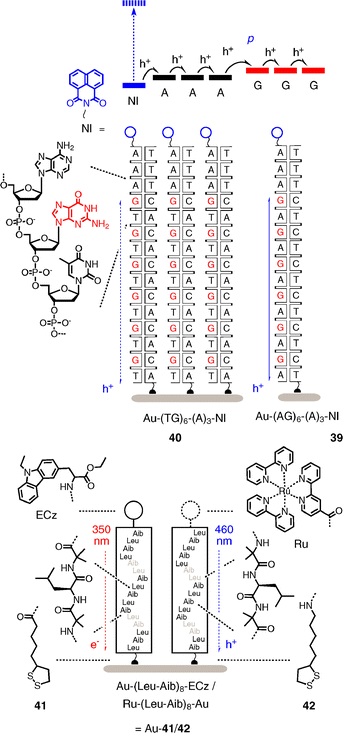 | ||
| Fig. 12 DNA duplexes and peptide α-helices as bioinspired scaffolds to elucidate the importance of charge mobility and macrodipoles for photocurrent generation, respectively. | ||
With constant A3 spacers between NI acceptors and G donors, DNA 39 with GA repeats generates significant photocurrent, whereas DNA 40 with GT repeats is inactive. This difference originates from the difference in charge mobility with GA and GT repeats, the former being sufficient to remove the hole before charge recombination of the NI−˙–G+˙ ion pair occurs. Elongation of the A3 to A4 spacers results in nearly the same photocurrent generation with GT and GA as charge transporters, whereas shortening to A2 spacers inactivates also the systems with GA as charge transporter. Single pair mismatches in the h+ transporting part of the DNAs are detectable as reduction in photocurrent.
α-Helical peptide scaffolds are perfect to explore the usefulness of macrodipoles to control the directionality of electron transfer.46,47 The minimalist helices 41 and 42 are made from a simple hydrophobic Leu-Aib repeat to minimize interference from competing secondary structures. In helix 41, a disulfide is added at the N-terminus for covalent linking to gold and an ECz chromophore is added at the C-terminus. In helix 42, however, the disulfide is at the C-terminus and a Ru chromophore at the N-terminus. In mixed SAMs on gold, the macrodipole of helix 41 points toward the ECz chromophore, whereas the macrodipole of helix 42 points away from the Ru chromophore. As a result, excitation of the ECz chromophore at 350 nm causes electron transfer to the gold surface, whereas excitation of the Ru chromophore at 460 nm causes hole transfer to the gold surface. The extraordinary result is a photodiode switch, where, due to the antiparallel orientation of the α-helical macrodipole, irradiation at 460 nm reversibly generates cathodic and irradiation at 350 nm anodic photocurrents.
Despite relatively low charge mobility due to the lack of molecular level organizations, LBL assembly remains the supramolecular approach of choice to photosystems with more significant redox gradients (Fig. 13). In a pioneering model, anionic fullerene dendrimers 43 on a ITO surface are covered with cationic porphyrin acceptors 44, which in turn are covered with anionic zinc porphyrin donors and cationic ferrocenes as final donors.48 Action spectra demonstrate that ITO–43–44 produces very little, ITO–43–44–14 more and ITO–43–44–14–45 most photocurrent. At the absorption maximum of the porphyrins, the final system is with an IPCE = 1.6% 108-times more active than the simple fullerene–porphyrin bilayer Au–43–44.
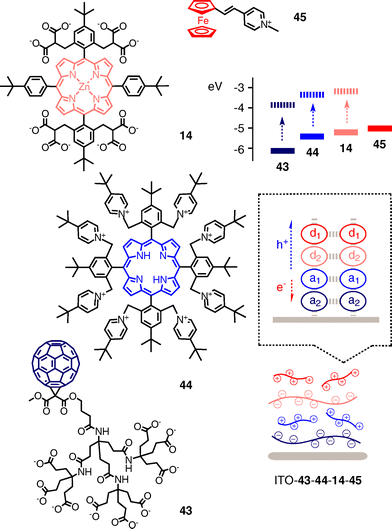 | ||
| Fig. 13 All-organic LBL photosystems with oriented redox gradients. | ||
An early and seminal breakthrough toward ordered, SHJ-compatible (Fig. 8) coordination networks with porphyrins focuses on the programmed assembly of oriented redox gradients (Fig. 14).49 Dithiolated zinc porphyrin donors 46 are deposited on gold and activated by coordination to copper 47 for the repetitive ordered LBL build up of a donor domain Au–(46–47)n. On top of this domain, a bifunctional domain is added by repetitive exposure to dithiolate porphyrin acceptors 48 and copper 47. On the resulting Au–(46–47–)n–(48–47–)n, the alkyl converter 49 with a thiol at one and a phosphonate at the other end is added. The phosphonate at the surface of Au–(46–47–)n–(48–47–)n–49 is the starting point to build an acceptor layer by repetitive incubation in solutions of zirconium 50 and diphosphonated viologen acceptor 51.
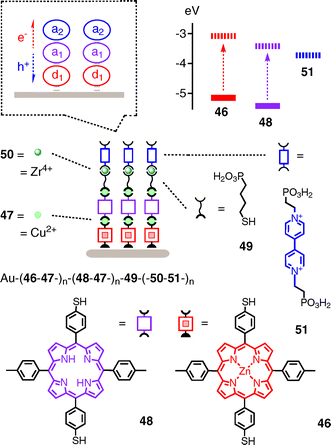
Compared to similar systems, the J–V curves of Au–(46–47–)n–(48–47–)n–49–(50–51–)n shows outstanding characteristics. Fill factors increase from FF = 0.31 for Au–(48–47–)3–49–(50–51–)3 with a weak redox gradient to FF = 0.58 for Au–(46–47–)3–49–(50–51–)3 with a strong redox gradient and a similar FF for the full cascade in Au–(46–47–)2–(48–47–)1–49–(50–51–)3. The VOC of full cascades Au–(46–47–)2–(48–47–)1–49–(50–51–)3 is as large as that of Au–(48–47–)3–49–(50–51–)3 with a weak redox gradient and larger than that of Au–(46–47–)3–49–(50–51–)3 with a strong gradient. This observation is in agreement with the earlier speculation that VOC in redox cascades are determined by neighboring or maximal rather than minimal HOMO–LUMO differences. As a result, the overall charge-separation quantum yield for full redox cascade is with 3.5% larger than with photosystems missing one component (2.4%).
An elegant approach focusing on the combination of surface-initiated supramolecular polymerization by interdigitating mutual porphyrin coordination with covalent capture addresses the challenge of how to build thick films without losing the long-range organization (Fig. 15).50 The construction begins with the deposition of the doubly thiolated zinc porphyrin initiator 52 on gold. To Au–52, the dimeric porphyrin 53 is added. One methylimidazole ligand of propagator 53 binds as axial ligand to the zinc porphyrin 52, whereas the methylimidazole ligand of initiator 52 back-binds as axial ligand of the zinc porphyrin 53. This mutual, doubly coordinative binding not only strengthens the interaction but also firmly orients propagator 53 with regard to initiator 52. The second zinc porphyrin and methylimidazole ligand of propagator 53 remains free at the surface of Au–52–53 to capture the first porphyrin of the next propagator 53, and so on. Once the surface-initiated supramolecular polymerization (SISP) stops, Grubbs catalyst is added to crosslink all porphyrins by ring-closing olefin metathesis (RCM). After RCM, SISP with propagator 53 is operational again, and RCM and SISP with propagator 53can be repeated to build, according to linear SPR J–L curves, films as thick as 7 nm in five cycles. Coordinative capping with porphyrin 54 carrying a terminal fullerene acceptor more than doubles the generated photocurrent. J–V curves show nearly ohmic behavior.
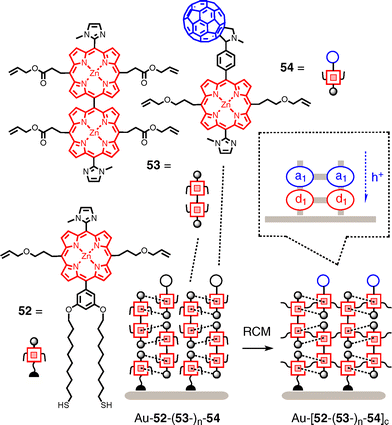 | ||
| Fig. 15 Combining coordinative surface-initiated supramolecular polymerization (SISP) with covalent capture by ring-closing olefin metathesis (RCM) to stabilize thick and ordered porphyrin architectures with terminal fullerene acceptors. | ||
6. Multicolor systems
In ideal OMARG-SHJs, the coaxial h+ and e− transporting channels are not only equipped with antiparallel redox gradients but also contain chromophores that absorb light over and beyond the entire visible range. For the programmed assembly of multicolor architectures on surfaces, this modulation of HOMO/LUMO energy differences must occur without global changes in structure. Multicolor systems are conceivable with many chromophores. The compatibility of existing rainbow collections such as AlexaFluor series, fluoresceins, BODIPYs, cyanines, azobenzenes, porphyrinoids or triangular dehydrobenzo[12]annulenes with OMARG-SHJ architectures remains to be explored.51–57 As long as multicolor systems are used for energy transfer only, HOMO–LUMO energy differences matter but absolute frontier orbital energies are irrelevant. Such energy cascades have been built in many variations to harvest light. Highlights reach from filled and oriented zeolites56 to bioengineered tobacco mosaic viruses.57 Tandem solar cells are simple engineering approach to multicolor photosystem.58 Beyond supramolecular chemistry, quantum dots appear attractive for energy transfer in multicolor photovoltaics, whereas their use to construct redox gradients seems less promising because decreasing HOMO with increasing LUMO levels produce low-energy electron traps.59,60For compatibility with OMARG-SHJs, multicolor chromophore collections must possess many challenging characteristics. NDIs61–65 are ideal candidates because they offer the spectacular combination of (1) decreasing HOMO–LUMO energies with increasing HOMO–LUMO energy differences,10,35,62–64 (2) n-semiconductivity,16,65 (3) π-acidity,13 (4) planarity, (5) global structural preservation, (6) compactness (“atom efficiency”) and (7) synthetic accessibility (Fig. 16).
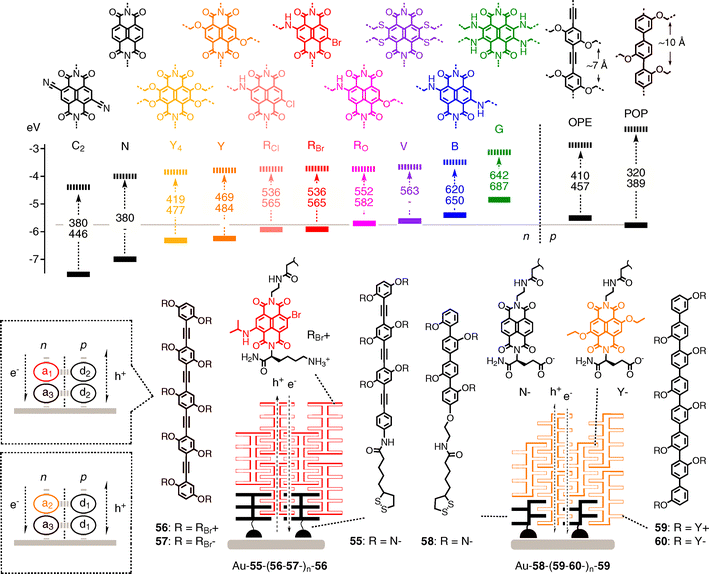 | ||
| Fig. 16 Frontier orbital energy levels of NDIs (general structures C2–G), OPEs and POPs (solid lines, HOMO; dashed lines, LUMO; dashed arrows, absorption of light; with wavelength (nm) of maximal absorption (top) and emission (bottom) and structure of POP–Y and OPE–RBr dyads 55–60 as well as their zipper assemblies. | ||
NDIs cover the primary colors by replacing only a few atoms.61–64 Introduction of two alkoxy substituents into the NDI core converts the colorless parent structure N into the yellow, green-emitting fluorophores Y. Formal substitution of one oxygen atom in Y with a nitrogen atom gives the red, orange-emitting RO. Substitution of the second alkoxy with an alkylamino π-donor gives the blue, red-emitting minimalist chlorophyll mimic B. Many more HOMO–LUMO energy differences are accessible with NDIs having two to four electron-donating (amines, ethers, sulfides, halogens, etc.) and accepting substituents (cyano, etc) in the core.61–64 Among other NDI characteristics, the fact that their face-to-face π-stacks account for one of the few air-stable molecular n-semiconductors deserves particular attention.16,17 Although easier to synthesize, the homologous PDI66 series appears less suitable to build OMARG-SHJs because core substitution twists the aromatic plane, may hinder ordered stacking and thus reduce the charge mobility of potential MRG stacks. In addition, the planar NDIs are more compact and provide access to high-energy photons.
7. Toward OMARG-SHJs
Rod-stack zipper assembly could potentially serve as a platform to build OMARG-SHJ architectures (Fig. 9 and 16). Toward this end, oligophenylethynyls (OPEs) are examined as alternative and accompanying rigid-rod scaffolds to the original POPs.67 Compared to POPs, OPEs are attractive because they can be planarized, transport holes better and absorb more visible light. Moreover, their repeat distance matches with ∼7 Å the repeat of π-stacks. The mismatched 10 Å repeat of POPs has been shown to cause hyperbolidal collapse into more helical architectures.35 Most importantly for OMARG-SHJs, the HOMO of OPEs lies above that of POPs.Zipper assembly of OPE–NDI architectures Au–55–(56–57–)n, is achieved by repetitive dip-wash procedures as described above for POP zippers.67 OPE–NDI zippers Au–55–(56–57–)n excel with FF = 0.61, a critical thickness of 20 layers, increasing (rather than decreasing) surface smoothness with increasing thickness, and perfect responsiveness to functional tests such as zipper capping and LBL assembly without initiator 55. The mismatched POP–NDI zipper homologs have a critical thickness of ∼8 layers, rougher surfaces and, in the case of brominated NDIs only, poor responsiveness to functional probes. These differences provide experimental support that zipper architectures exist and that topological matching matters.
Action spectra of final Au–55–(56–57–)n indicate that OPEs are planarized and contribute to photocurrent generation. Consistent with the formation of SHJs, transient absorption spectroscopy demonstrates that OPE excitation is followed by electron (rather than energy) transfer to the NDI acceptors in the femtosecond timescale.
The yellow POP–Y zippers Au–58–(59–60–)n complement red OPE–RBr zippers Au–55–(56–57–)n.10 In POP–Y zippers Au–58–(59–60–)n, the optoelectronic matching of Y acceptors with POP donors is correctly reflected in maximal photocurrent generation compared to the less favorable red POP–RCl and blue POP–B zippers. Long-lived PCS in POP–Y dyads in the nanosecond timescale is in agreement with this interpretation. High π-acidity, very smooth surfaces, high FF = 0.53 in J–V curves and high critical thickness of ∼20 layers in J–L curves of POP–Y zippers Au–58–(59–60–)n reveal origin and existence of highly ordered rod–stack SHJ architectures.
With unsubstituted NDI acceptors near the gold surface, both POP–Y zipper Au–58–(59–60–)n and OPE-RBr zipper Au–55–(56–57–)n have a formal redox gradient in the e− transporting channel. Placed on top of each other, mixed POP–Y and OPE–RBr zippers would give formal OMARG-SHJs, where Y acceptors in the NDI stack can accept electrons from RBr donors and direct them to the bottom of the multilayer architecture, whereas h+ transporting strings of OPEs can accept holes from strings of POPs underneath and direct them to the top.
8. Epilogue
This tutorial review introduces the concept of OMARG-SHJ architectures as bioinspired ideal photosystems and summarizes recent progress made to get there. The overall impression is that the decisive challenge concerns the question of how to build “bottom-up” and with molecular-level precision on solid grounds. Whereas a broad palette of parameters influences the final phenotype, the key to success thus seems to be at the interface of synthetic organic, supramolecular, polymer and analytical surface chemistry. Promising and emerging methods employed and conceived to tackle this challenge include LBL assembly, zipper assembly, surface-initiated polymerization (polymer brushes), surface-initiated supramolecular polymerization, electropolymerization and covalent capture. Much potential can be seen in the development of multicolor donor–acceptor systems beyond current preferences such as porphyrins and fullerenes. Expectations such as high critical thickness, fill factors and open circuit voltage for OMARG-SHJ architectures are supported by several examples. Access to these characteristics would find broad applicability in molecular optoelectronics, including high-efficiency solar cells or logic devices. Although it remains to be seen whether or not OMARG-SHJs ultimately live up to these expectations, the road to get there promises in any case to be everything else than boring.Acknowledgements
This work was supported by the University of Geneva and the Swiss NSF. J. M. is a fellow of the Roche Research Foundation. Sincere apologies to all colleagues whose work could not be covered because of limitations in space and scope.References
- J. Deisenhofer and H. Michel, Science, 1989, 245, 1463–1473 CrossRef.
- N. Nelson and A. Ben-Shem, Nat. Rev. Mol. Cell Biol., 2004, 5, 971–982 CrossRef CAS.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef.
- C. Ma, M. Fonrodona, M. C. Schikora, M. M. Wienk, R. A. J. Janssen and P. Bäuerle, Adv. Funct. Mater., 2008, 18, 3323–3331 CrossRef CAS.
- J.-F. Eckert, J.-F. Nicoud, J.-F. Nierengarten, S.-G. Liu, L. Echegoyen, N. Armaroli, F. Barigelletti, L. Ouali, V. Krasnikov and G. Hadziioannou, J. Am. Chem. Soc., 2000, 122, 7467–7479 CrossRef CAS.
- V. Balzani, M. Venturi and A. Credi, Molecular Devices and Machines, Wiley-VCH, Weinheim, 2003 Search PubMed.
- R. S. K. Kishore, O. Kel, N. Banerji, D. Emery, G. Bollot, J. Mareda, A. Gomez-Casado, P. Jonkheijm, J. Huskens, P. Maroni, M. Borkovec, E. Vauthey, N. Sakai and S. Matile, J. Am. Chem. Soc., 2009, 131, 11106–11116 CrossRef CAS.
- F. Yang, M. Shtein and S. Forrest, Nat. Mater., 2005, 4, 37–41 CrossRef CAS.
- S. Sista, Y. Yao, Y. Yang, M. L. Tang and Z. Bao, Appl. Phys. Lett., 2007, 91, 223508/1–223508/3 CAS.
- J. Mareda and S. Matile, Chem.–Eur. J., 2009, 15, 28–37 CrossRef CAS.
- G. J. Gabriel and B. L. Iverson, J. Am. Chem. Soc., 2002, 124, 15174–15175 CrossRef CAS.
- T. Van der Boom, R. T. Hayes, Y. Zhao, P. J. Bushard, E. A. Weiss and M. R. Wasielewski, J. Am. Chem. Soc., 2002, 124, 9582–9590 CrossRef.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 15259–15278 CrossRef CAS.
- J. M. Warman, M. P. de Haas, G. Dicker, F. C. Grozema, J. Piris and M. G. Debije, Chem. Mater., 2004, 16, 4600–4609 CrossRef CAS.
- F. Würthner, Z. Chen, F. J. M. Hoeben, P. Osswald, C.-C. You, P. Jonkheijm, J. Herrikhuyzen, A. P. H. J. Schenning, P. P. A. M. van der Schoot, E. W. Meijer, E. H. A. Beckers, S. C. J. Meskers and R. A. J. Janssen, J. Am. Chem. Soc., 2004, 126, 10611–10618 CrossRef.
- Y. Yamamoto, T. Fukushima, Y. Suna, N. Ishii, A. Saeki, S. Seki, S. Tagawa, M. Taniguchi, T. Kawai and T. Aida, Science, 2006, 314, 1761–1764 CrossRef CAS.
- Y. Yamamoto, T. Fukushima, A. Saeki, S. Seki, S. Tagawa, N. Ishii and T. Aida, J. Am. Chem. Soc., 2007, 129, 9276–9277 CrossRef CAS.
- T. Yamamoto, T. Fukushima, Y. Yamamoto, A. Kosaka, W. Jin, N. Ishii and T. Aida, J. Am. Chem. Soc., 2006, 128, 14337–14340 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- J. K. Mwaura, M. R. Pinto, D. Witker, N. Ananthakrishnan, K. S. Schanze and J. R. Reynolds, Langmuir, 2005, 21, 10119–10126 CrossRef CAS.
- L. Zhao, T. Ma, H. Bai, G. Lu, C. Li and G. Shi, Langmuir, 2008, 24, 4380–4387 CrossRef CAS.
- D. M. Guldi, G. M. A. Rahman, M. Prato, N. Jux, S. Qin and W. Ford, Angew. Chem., Int. Ed., 2005, 44, 2015–2018 CrossRef CAS.
- D. M. Guldi, J. Phys. Chem. B, 2005, 109, 11432–11441 CrossRef CAS.
- G. L. Whiting, H. J. Snaith, S. Khodabakhsh, J. W. Andreasen, D. W. Breiby, M. M. Nielsen, N. C. Greenham, R. H. Friend and W. T. S. Huck, Nano Lett., 2006, 6, 573–578 CrossRef CAS.
- H. J. Snaith, G. L. Whiting, B. Sun, N. C. Greenham, W. T. S. Huck and R. H. Friend, Nano Lett., 2005, 5, 1653–1657 CrossRef CAS.
- R. C. Shallcross, G. D. D'Ambruoso, B. D. Korth, H. K. Hall, Z. Zheng, J. Pyun and N. R. Armstrong, J. Am. Chem. Soc., 2007, 129, 11310–11311 CrossRef CAS.
- E. Hwang, K. M. N. de Silva, C. B. Seevers, J.-R. Li, J. C. Garno and E. E. Nesterov, Langmuir, 2008, 24, 9700–9706 CrossRef CAS.
- A. B. F. Martinson, A. M. Massari, S. J. Lee, R. W. Gurney, K. E. Splan, J. T. Hupp and S. T. Nguyen, J. Electrochem. Soc., 2006, 153, A527–A532 CrossRef CAS.
- A. Kira, T. Umeyama, Y. Matano, K. Yoshida, S. Isoda, J. K. Park, D. Kim and H. Imahori, J. Am. Chem. Soc., 2009, 131, 3198–3200 CrossRef CAS.
- N. Sakai, A. L. Sisson, T. Bürgi and S. Matile, J. Am. Chem. Soc., 2007, 129, 15758–15759 CrossRef CAS.
- P. Talukdar, G. Bollot, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2005, 127, 6528–6529 CrossRef CAS.
- S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84–86 CrossRef CAS.
- N. Banerji, A. Fürstenberg, S. Bhosale, A. L. Sisson, N. Sakai, S. Matile and E. Vauthey, J. Phys. Chem. B, 2008, 112, 8912–8922 CrossRef CAS.
- M. Lista, N. Sakai and S. Matile, Supramol. Chem., 2009, 21, 238–244 CrossRef CAS.
- A. L. Sisson, N. Sakai, N. Banerji, A. Fürstenberg, E. Vauthey and S. Matile, Angew. Chem., Int. Ed., 2008, 47, 3727–3729 CrossRef CAS.
- C.-H. Huang, N. D. McClenaghan, A. Kuhn, G. Bravic and D. M. Bassani, Tetrahedron, 2006, 62, 2050–2059 CrossRef CAS.
- J. M. Kerckhoffs, M. G. ten Cate, M. A. Mateos-Timoneda, F. W. van Leeuwen, B. Snellink-Ruel, A. L. Spek, H. Kooijman, M. Crego-Calama and D. N. Reinhoudt, J. Am. Chem. Soc., 2005, 127, 12697–12708 CrossRef CAS.
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS.
- H. Imahori, Y. Mori and Y. Matano, J. Photochem. Photobiol. C: Photochem. Rev., 2003, 4, 51–83 CrossRef CAS.
- S. Fukuzumi, Bull. Chem. Soc. Jpn., 2006, 79, 177–195 CrossRef CAS.
- I. Carmeli, L. Frolov, C. Carmeli and S. Richter, J. Am. Chem. Soc., 2007, 129, 12352–12353 CrossRef CAS.
- T. Takada, C. Lin and T. Majima, Angew. Chem., Int. Ed., 2007, 46, 6681–6683 CrossRef CAS.
- S. Yasutomi, T. Morita, Y. Imanishi and S. Kimura, Science, 2004, 304, 1944–1947 CrossRef CAS.
- S. Kimura, Org. Biomol. Chem., 2008, 6, 1143–1148 RSC.
- D. M. Guldi, I. Zilbermann, G. Anderson, A. Li, D. Balbinot, N. Jux, M. Hatzimarinaki, A. Hirsch and M. Prato, Chem. Commun., 2004, 726–727 RSC.
- F. B. Abdelrazzaq, R. C. Kwong and M. E. Thompson, J. Am. Chem. Soc., 2002, 124, 4796–4803 CrossRef CAS.
- M. Morisue, S. Yamatsu, N. Haruta and Y. Kobuke, Chem.–Eur. J., 2005, 11, 5563–5574 CrossRef CAS.
- Y. Rio, M. S. Rodriguez-Morgade and T. Torres, Org. Biomol. Chem., 2008, 6, 1877–1894 RSC.
- K. Tahara, T. Fujita, M. Sonoda, M. Shiro and Y. Tobe, J. Am. Chem. Soc., 2008, 130, 14339–14345 CrossRef CAS.
- M. Lopalco, E. N. Koini, J. K. Cho and M. Bradley, Org. Biomol. Chem., 2009, 7, 856–859 RSC.
- O. Sadovski, A. A. Beharry, F. Zhang and G. A. Woolley, Angew. Chem., Int. Ed., 2009, 48, 1484–1486 CrossRef CAS.
- R. P. Haugland, The Handbook. A Guide to Fluorescent Probes and Labeling Techniques, 10th edn, Invitrogen, Eugene, Oregon, USA, 2005 Search PubMed.
- R. Koeppe, O. Bossart, G. Calzaferri and N. S. Sariciftci, Sol. Energy Mater. Sol. Cells, 2007, 91, 986–995 CrossRef CAS.
- R. A. Miller, A. D. Presley and M. B. Francis, J. Am. Chem. Soc., 2007, 129, 3104–3109 CrossRef CAS.
- J. Y. Kim, K. Lee, N. E. Coates, D. Moses, T.-Q. Nguyen, M. Dante and A. J. Heeger, Science, 2007, 317, 222–225 CrossRef CAS.
- E. A. Weiss, V. J. Porter, R. C. Chiechi, S. M. Geyer, D. C. Bell, M. G. Bawendi and G. M. Whitesides, J. Am. Chem. Soc., 2008, 130, 83–92 CrossRef CAS.
- A. Kongkanand, K. Tvrdy, K. Takechi, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2008, 130, 4007–4015 CrossRef CAS.
- S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331–342 RSC.
- C. Röger and F. Würthner, J. Org. Chem., 2007, 72, 8070–8075 CrossRef.
- A. Blaszczyk, M. Fischer, C. von Hänisch and M. Mayor, Helv. Chim. Acta, 2006, 89, 1986–2005 CrossRef CAS.
- S. Chopin, F. Chaignon, E. Blart and F. Odobel, J. Mater. Chem., 2007, 17, 4139–4146 RSC.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- R. Bhosale, A. Perez-Velasco, V. Ravikumar, R. S. K. Kishore, O. Kel, A. Gomez-Casado, P. Jonkheijm, J. Huskens, P. Maroni, M. Borkovec, T. Sawada, E. Vauthey, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2009, 48, 6461–6464 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |