Stereocontrolled ring-opening polymerization of cyclic esters: synthesis of new polyester microstructures
Christophe M. Thomas*
Ecole Nationale Supérieure de Chimie de Paris, UMR CNRS 7223, 11 rue Pierre et Marie Curie, 75231 Paris Cedex 05, France. E-mail: christophe-thomas@enscp.fr
First published on 28th September 2009
Abstract
Synthesis of aliphatic polyesters has been studied intensively due to their biocompatible and biodegradable properties and their potential applications in medical and agricultural fields. There has been particular emphasis over the past decade on the synthesis of discrete, well-characterized complexes that are active polymerization initiators for the ring-opening polymerization (ROP) of lactide (LA) and β-butyrolactone (BBL) to give, respectively, poly(lactide) (PLA) and poly(3-hydroxybutyrate) (PHB). These recent advances in catalyst design have led to a variety of polyester microstructures. This tutorial review focuses on the use of metal-based complexes for the stereoselective ROP of rac-LA and rac-BBL.
 Christophe M. Thomas | Christophe Thomas obtained his PhD degree in 2002 working under the supervision of Professor Süss-Fink. He then joined Professor Coates’ group at Cornell University as a postdoctoral fellow supported by the Swiss National Science Foundation (2002–2003). After spending one year in Professor Ward’s laboratories, he was appointed as Assistant Professor at the University of Rennes in 2004. In 2008, he was promoted to full professor at the Ecole Nationale Supérieure de Chimie de Paris. His research interests comprise the study of fundamental processes in organometallic chemistry with an emphasis on catalytic transformations and the control of stereochemistry. |
Introduction
Of the many commercial synthetic polymers, polyolefins are by far the most important class of materials.1 A combination of factors, including monomer availability and cost, synthetic ease, and excellent properties, have fueled the widespread use of these materials. Despite the numerous advantages of these materials, two major drawbacks remain to be solved, namely, the use of non-renewable resources in their production and the ultimate fate of these large-scale commodity polymers. In addition, the extensive use of synthetic polymeric materials has created another important and troubling problem; despite increasing popularity of plastic recycling, disposal of these undegradable materials has led to serious environmental pollution. In an effort to solve these growing problems, a wealth of research has centered on synthesis of biodegradable polymers and the spectacular advances achieved over the last 30 years in the synthesis, manufacture, and processing of these materials have given rise to a broad range of practical applications from packaging to more sophisticated biomedical devices. Of the variety of biodegradable polymers known, linear aliphatic polyesters have a leading position since hydrolytic and/or enzymatic chain cleavage yields ω-hydroxyacids, which in most cases are ultimately metabolized.2Polyesters are commonly produced by polycondensation reactions from a diol and a diacid or by ring-opening polymerization (ROP) of cyclic esters.3 Although the application of condensation polymerization techniques certainly allows access to a broader range of polymer structures than ROP through a higher accessibility of the monomers involved, the most convenient and efficient method to obtain aliphatic polyesters is the ROP of cyclic esters. In contrast to polyesterification, ROP of cyclic monomers uses mild reaction conditions and avoids the formation of small molecule byproducts. The thermodynamic driving force for ROP processes is the relief of ring strain, which enables the entropy, unfavorable in all polymerizations, to be overcome.4 A range of simple lactones of varying ring size and strain have been investigated over the past decades and two cyclic monomers have recently attracted much attention for this process: lactide and β-butyrolactone.4,5 The corresponding polymers, namely poly(lactide) (PLA) and poly(3-hydroxybutyrate) (PHB), are the most prominent synthetic aliphatic polyesters and numerous studies have been devoted to the syntheses, properties, and processing of these biodegradable thermoplastics.6
Among the various ROP processes, including anionic, cationic, organocatalytic and coordination–insertion, the latter has gained increasing attention.2 It is now commonly accepted that the most efficient method for the production of well-controlled polyesters in terms of molecular weight, composition and microstructure is the ROP with metal-coordination initiators. Therefore, a large number of investigations have been directed towards synthesizing efficient metal-based initiators and studying their reactivities. The first generation of active initiators was mainly constituted by simple homoleptic metal complexes,7 principally the industrially relevant tin(II) bis(2-ethylhexanoate), zinc(II) lactate and aluminium(III) isopropoxide (AlOiPr3).8,9 These metal-based initiating systems were widely used for the controlled ring-opening polymerization of cyclic esters and brought important contributions for the mechanism understanding. Dittrich and Schulz were the first to define a three-step coordination–insertion mechanism for the ROP of cyclic esters.10 Then, Kricheldorf et al. and Teyssiéet al. have beautifully demonstrated that such a mechanism was involved in the Al(OiPr)3-initiated polymerization of different lactones.7a,7b The coordination–insertion mechanism of lactide polymerization proceeds via coordination of the monomer to the metal center, followed by insertion into the metal-alkoxide species through the acyl–oxygen bond with retention of configuration (Scheme 1).7 This results in a new metal-alkoxide species capable of further insertion.
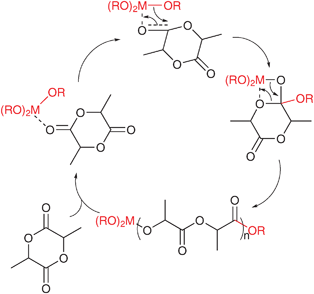 | ||
| Scheme 1 Coordination–insertion mechanism of lactide polymerization using metal-alkoxide catalysts. | ||
However, although tin, aluminium and zinc-based systems proved to be quite convenient in the preparation of aliphatic polyesters, several drawbacks such as complicated equilibria phenomena and multiple nuclearities have limited the control of the polymerization and the structural influence on catalyst activity.2 In particular the homoleptic nature of these species results in detrimental side reactions such as intramolecular and intermolecular transesterifications. These competition reactions, in which cleavage and reformation of polymer chains lead to a broadening of the molecular weight distribution, become significant at higher temperatures and have been described as a function of the monomer conversion.11 In addition there is also no possible control of stereochemistry.
Efforts to obviate these problems focused on the development of supporting ligands designed to control the structure of the corresponding heteroleptic complexes and their polymerization reactivity. Therefore, a second generation of discrete, well-defined metal complexes with fine ligand adjustment attracted interest in order to achieve better control, activity, and selectivity of the polymerization reaction. Furthermore, the application of readily available stereopure monomers in association with stereocontrolled ROP has enabled the facile manipulation of the tacticity of the resultant polymers, considerably affecting their properties. In this respect, coordination complexes with tuning of ligands play an important role not only in molecular weight and molecular weight distribution but also in the production of stereoregular polyesters.
In this tutorial review, we will describe selected published efforts to achieve these research goals using discrete, structurally well-characterized metal complexes with particular ligand frameworks. We will focus primarily on polymerization studies of LA, but also include some discussion of work with β-butyrolactone.
Poly(lactide)s: stereochemistry and microstructure
In terms of stereochemistry, many types of lactide feedstocks are accessible. Diastereomers (S,S)-lactide, (R,R)-lactide, and meso-lactide are available in pure form, as well as the racemic mixture rac-lactide (50 : 50 (S,S)-LA and (R,R)-LA) (Scheme 2). Many polymer microstructures (i.e. atactic, isotactic, heterotactic and syndiotactic) can be constructed from this basic set of monomers. For example, polymerization of (R,R)-lactide with a typical (homoleptic or heteroleptic) catalyst gives isotactic poly((R)-lactide), a crystalline polymer with a high melting transition (Tm = 170–180 °C). Atactic PLA, an amorphous polymer with a random distribution of stereocenters along the polymer backbone, can be prepared using a non-stereoselective metal-based catalyst and rac-lactide. Despite the recent development of achiral and chiral complexes for the ROP of lactide,12 relatively few well-defined metal catalysts are capable of achieving high stereochemical control in the ROP of meso- or rac-lactide.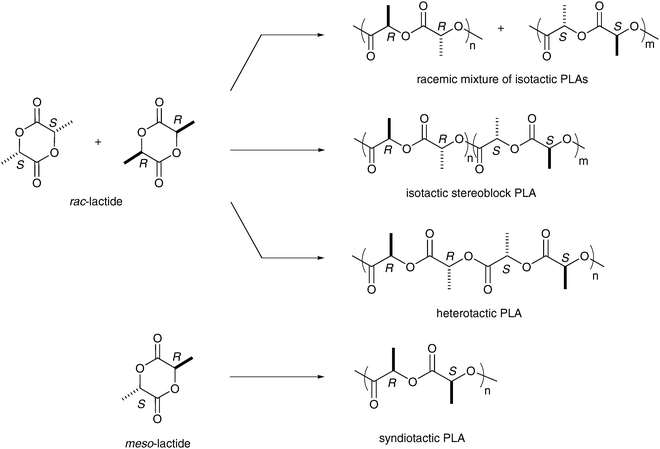
Synthesis of syndiotactic PLA from meso-lactide
To date, the only example of an efficient catalyst for the stereoselective preparation of highly syndiotactic poly(lactide) was reported by Coates and Ovitt.13 Syndiotactic, semicrystalline poly(lactide) with enantiotopic selectivity >85% was formed by ROP of a series of meso-lactide in the presence of the chiral aluminium isopropoxide complex 1a, which preferentially ring-opened one acyl–oxygen site, resulting in a highly alternating arrangement of stereocenters in the polymer. Due to the high degree of stereoregularity, the highest syndiotactic PLA (96%) obtained was found to exhibit a melting temperature at 152 °C.Synthesis of isotactic PLA from rac-lactide
Spassky et al. were the first to demonstrate that Schiff-base (SALEN type) aluminium complexes are highly selective initiators for the polymerization of racemic lactide (rac-LA).14 As early as 1996, his group reported the kinetic resolution polymerization of rac-LA using an aluminium methoxide complex bearing a binaphthyl Schiff-base ligand 1b (Scheme 3). The chiral nature of the catalyst led to the highly selective ring-opening of (R,R)-LA to give isotactic PLA through an enantiomorphic site-control mechanism; the (S,S)-LA was left largely unreacted.14 At 70 °C, the catalyst exhibited a 20 : 1 preference for the polymerization of (R,R)-lactide over (S,S)-lactide (kRR/kSS = 20).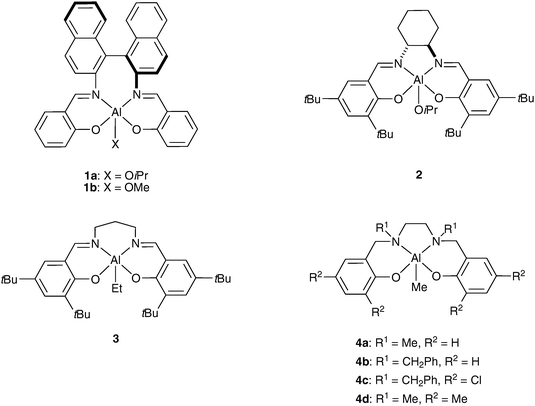 | ||
| Scheme 3 Stereoselective aluminium-based systems for the ROP of rac-lactide. | ||
Inspired by this pioneering work on the isotactic polymerization of rac-LA, the next reported initiators were essentially achiral and chiral aluminium Schiff-base systems (Scheme 3). All these aluminium-based systems proceed in highly stereoselective manner and with a good control of the molecular weights. In comparison with Spassky’s catalyst, Feijen et al. reported efficient and reverse stereocontrol with analogous aluminium chiral complex 2 based on (R,R)-cyclohexanediamine Schiff base, which preferentially polymerizes (S,S)-LA (kSS/kRR = 14).15 In both cases, the chiral ligand induces the enantiomorphic site control of LA polymerization. More, PLA stereocomplexes and PLA stereoblocks, which show enhanced thermal stability compared to the homochiral PLAs (Tm up to 210 °C), were also prepared with these initiators. Racemic aluminium catalysts 1a16 and 215 have been used for the parallel stereocontrolled ROP of (R,R)- and (S,S)-LA from rac-LA to obtain isotactic stereoblock PLA. The formation of a stereoblock PLA was explained by a polymer exchange mechanism where growing chains switch between (R)- and (S)-species.16a,17 Nomura et al. reported the first achiral Al-based system 3 which is able to generate a highly stereoblock PLA from rac-lactide via chain-end control mechanism.18a The formation of isotactic stereoblock PLA (Pm up to 0.79) was also demonstrated by Gibson et al. with tetradentate N,N′-disubstituted bis(aminophenoxide) (designated as SALAN, a saturated version of Schiff-base SALEN ligand) aluminium complexes 4a and 4b.18b Remarkably, it was demonstrated that a wide range of microstructures are accessible using these aluminium initiators. In particular it was shown that isotactic PLAs are produced in the case of complexes bearing unsubstituted phenoxide groups whereas heterotactic PLAs (vide infra) are obtained when the phenoxide units of the SALAN ligand contain substituents in the 3 and 5 positions (e.g. for complexes 4c and 4d). It was also demonstrated that the tacticity is significantly influenced by the substituents R1 attached to the nitrogen atoms. For example, the benzylamine derivative 4b affords higher isotacticity (Pm = 0.79) than its methylamine analogue 4a (Pm = 0.68).
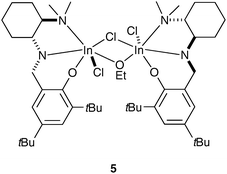
Moving towards a different metal-based system, Mehrkhodavandi and co-workers have recently reported a chiral alkoxy-bridged dinuclear indium catalyst capable of active, living, and selective ROP of lactide (Scheme 4).19 By using this indium system, the authors demonstrated that there is a possibility of functional-group-tolerant polymerization and catalyst recovery. Furthermore, although the racemic catalyst 5 showed modest isoselectivity in LA polymerization, the enantiopure catalyst 5 revealed significantly decreased enantioselectivity in LA ROP, thus highlighting the importance of a site-control mechanism.
Recently Arnold et al. demonstrated that a racemic mixture of two homochiral C3-symmetric yttrium complexes can be used as initiators for the formation of isotactic polylactide.20 These complexes provide a new class of very active initiators for the polymerization of rac-lactide into stereocomplex PLA with retention of stereochemical control even at high monomer conversions and high molecular weights.
In 2009, Otero et al. described the synthesis and characterization of a new enantiopure heteroscorpionate neodymium complex which acts as an efficient single-site initiator for the controlled ring-opening polymerization of rac-lactide, producing isotactic polyester.21 This neodymium complex showed a homosteric preference for one of the two enantiomers at low conversions (e.g. Pm = 0.61 at 10% conversion) but at high conversions a significant decrease of isotacticity was observed (Pm = 0.29 at 52% conversion).
Synthesis of heterotactic PLA from rac-lactide
In the past years, Coates et al. described a new class of β-diiminate zinc complexes for ROP of lactides.22a These catalysts, such as complexes 6 (Scheme 5), were found to act as single site, living initiators for the polymerization of (S,S)-lactide, rac-lactide and meso-lactide to PLA. In particular, these achiral complexes featured high activities and selectivities, affording highly heterotactic PLA (Pr up to 0.94% at 0 °C) from rac-LA, by incorporating the (R,R)- and (S,S)-LA in an alternating fashion. The molecular structures of complexes 6, determined by X-ray diffraction, revealed dimeric species in the solid state where isopropoxide ligands bridge distorted tetrahedral zinc centers (Fig. 1). On the basis of solution exchange experiments between related methoxide complexes,22b the authors have elegantly demonstrated that all three alkoxide catalysts retain their dimeric structures in solution. In addition it was shown that the substituents on the β-diiminate ligand exert a significant influence upon the course of the polymerizations, affecting both the degree of stereoselectivity and the rate of polymerization. Interestingly, complex 6c exhibited the highest activity and stereoselectivity (Pr = 0.90 at RT) of the zinc complexes studied for the polymerization of rac-lactide to PLA. For instance, changing the ligand substituents from isopropyl to ethyl groups resulted in a decrease in heterotacticity (Pr = 0.79). Similarly, substitution with n-propyl groups lowers the heterotacticity (Pr = 0.76). | ||
| Scheme 5 Stereoselective systems for the heterotactic ROP of rac-lactide. | ||
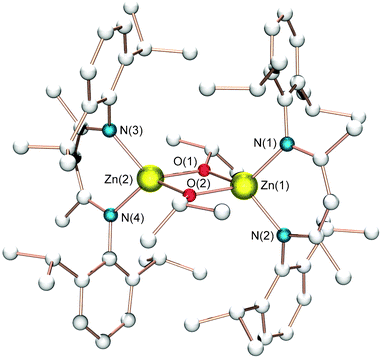 | ||
| Fig. 1 Molecular structure of complex 6c; all hydrogen atoms and solvent molecules are omitted for clarity. | ||
In 2004, Chisholm et al. prepared and investigated a series of well-defined tris-pyrazolyl borate (TpR, R = iPr, tBu, 9-BBN-pz2) calcium complexes for lactide polymerization (Scheme 5).23 The monomeric amide or aryloxide complexes of the form (TptBu)CaX (7a–b) were shown to be highly active and stereoselective, leading to heterotactic PLA (Pr = 0.90). The use of bulky substituent as seen in the TptBu ligand (Fig. 2) is necessary to confer single-site living polymerization behavior and stereoselectivity in the ring-opening event. Interestingly, the less sterically demanding TpiPr ligand does not confer any stereocontrol.
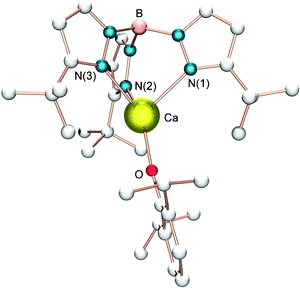 | ||
| Fig. 2 Molecular structure of complex 7b; all hydrogen atoms and solvent molecules are omitted for clarity. | ||
High levels of heterotacticity were also reported by Gibson and co-workers with SALAN-aluminium-based initiators (e.g. complexes 4c and 4d, Scheme 3).18b The microstructures of the poly(lactide)s obtained were found to be dependent upon the ancillary ligand substituents, ranging from isotactic for unsubstituted phenoxide units (vide supra) to highly heterotactic with phenoxide groups containing Me and tBu substituents, (Pr up to 0.96). As already mentioned for the synthesis of isotactic PLA, it was shown that the size of the nitrogen substituents R1 plays a crucial role in the tacticity of the polymer produced using these aluminium initiators. On the basis of the single-crystal X-ray analysis of complex 4d (Fig. 3), Gibson proposed that the alkylamino backbone substituents can closely approach the site of polymer chain growth and thereby influence monomer selectivity.
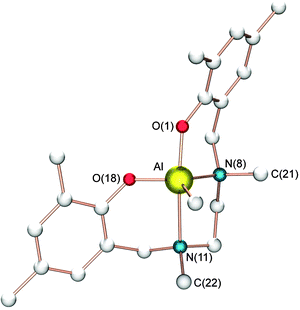 | ||
| Fig. 3 Molecular structure of complex 4d; all hydrogen atoms and solvent molecules are omitted for clarity. | ||
Despite the fact that organometallic germanium-based compounds are generally non-toxic and less likely to undergo undesirable transesterification reactions than tin complexes,24 the first example of a single-site germanium initiator for the ROP of LA was only reported by Davidson and co-workers in 2007 (Fig. 4).25 The new germanium isopropoxide 8 was obtained after the reaction of Ge(OiPr)4 with a C3-symmetric amino-tris(phenol) ligand, which bears methyl groups in the ortho positions of the phenol rings. Preliminary catalytic experiments showed that the resulting germanium-based system was active for the solvent-free ROP of rac-LA, to provide heterotactic-enriched PLA (up to 0.82) in the bulk. Having identified an active catalyst which displays promising selectivity, the authors decided to investigate different germanium complexes with the aim of optimizing the selectivity and activity of Ge-based single-site initiators for ROP of LA. Unfortunately, the synthetic route used for 8 proved to be unsuitable for other Ge-OiPr complexes (for example, where aryl substituents are tBu or Cl rather than Me); thus far, the authors have been unsuccessful in preparing other germanium complexes than 8.
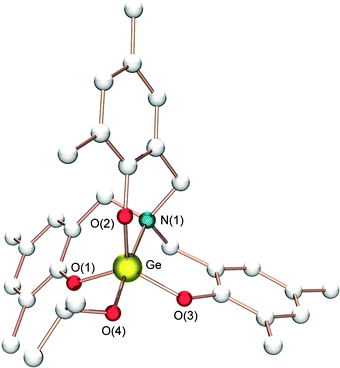 | ||
| Fig. 4 Molecular structure of complex 8; all hydrogen atoms and solvent molecules are omitted for clarity. | ||
A major breakthrough has very recently been achieved by Hillmyer et al. who prepared highly heterotactic PLA (0.86 < Pr < 0.94 at 25 °C, Pr = 0.97 at 0 °C) from rac-LA using a catalyst prepared in situ from indium trichloride, benzyl alcohol and triethylamine.26 The resulting robust system was found to operate under a variety of reaction conditions to yield heterotactic PLA with controlled molecular weight and a narrow molecular weight distribution. In contrast to other stereoselective catalysts which contain a sterically bulky and/or chiral ligand that is critical to obtain highly tactic PLA, this system was able to induce stereocontrol without an added directing multidentate ligand.
McLain et al. were the first to demonstrate the high potential of rare-earth complexes for the synthesis of poly(lactide) with an homoleptic complex described as “Y(OCH2CH2NMe2)3”, which was shown to polymerize rac-lactide in a rapid and controlled manner.27 Inspired by this work, Coates and Ovitt decided to investigate the catalytic behavior of a new heteroleptic yttrium alkoxide complex, using the same chiral Schiff-base ligand that enabled aluminium complexes 1 to produce isotactic poly(lactide).28 However, although this complex revealed relatively higher activity than the parent aluminium derivative, no stereoselectivity was observed for the polymerization of rac-LA.
From this early study, other research groups anticipated that well-defined rare-earth metal complexes supported by multidentate bis(phenolate) ligands would be of interest in order to achieve effective ROP of rac-LA.29 For instance, Bonnet et al. published the synthesis of new lanthanoid borohydride complexes supported by a diamino-bis(phenolate) ligand.30 Single-crystal X-ray analysis of the lanthanum derivative revealed a dimeric species with μ-bridging borohydrides. These complexes proved to be active initiators for the ROP of rac-LA. The highest selectivity was obtained with the yttrium compound, which was found to favor the formation of heterotactic-enriched PLA (Pr = 0.87). A mechanism occurring via insertion of the monomer into the metal–borohydride bond was speculated by analogy with the results obtained in ε-caprolactone polymerization.31
The synthesis and reactivity of well-defined rare-earth metal (i.e. yttrium, lanthanum and neodymium) complexes supported by amino-bis(phenolate) ligands were also recently investigated.32 Among these, the yttrium amido and alkoxo derivatives 9a–d (Scheme 6) have shown interesting performance for the heterotactic living polymerization of rac-LA. It was demonstrated that slight changes in the ligand architecture resulted in dramatic enhancements in polymerization selectivities. In particular the steric bulk of the substituents on the aromatic rings, and particularly the ortho-phenolate substituents, plays a crucial role in achieving high heteroselectivity for the chain-end controlled polymerization of rac-lactide. Complex 9a, which bears tert-butyl groups in the ortho positions of the phenolates, was able to produce heterotactic-enriched polymer (Pr = 0.80), whereas complex 9d gave low selectivity (Pr = 0.56) for heterotactic PLA, probably due to the higher tendency to form aggregated species. By using bulky and conformationally flexible cumyl (α,α-dimethylbenzyl) groups as R1 substituents, the PLA produced by 9b–c was found to be substantially more heterotactic, with a Pr of 0.90. Subsequently analogous alkyl yttrium derivatives 9e and 9f were synthesized and displayed higher stereoselectivity for the polymerization of rac-lactide to give heterotactic poly(lactide) with a Pr ranging from 0.97 to 0.99.33 Interestingly, it was shown that amino-bis(phenolate) yttrium systems 9a–c are also able to act as catalytic-like species in the presence of excess alcohol, producing large quantities of highly heterotactic PLA with small amounts of catalyst, thus optimizing productivity and minimizing contamination of polymer with metal residues.34
 | ||
| Scheme 6 Stereoselective systems for the heterotactic ROP of rac-lactide. | ||
Okuda and co-workers reported the synthesis of several lanthanoid complexes such as 10 and 11 supported by 1,ω-dithiaalkanediyl-bridged bis(phenolate) ligands (Scheme 7).35 The ancillary ligand proved to be critical for polymerization control and selectivity. In addition, among these dichalcogen-bridged bis(phenolate) derivatives, scandium complexes 10a and 11a show high heterotactic selectivity (Pr up to 0.95) during the ROP of rac-LA. The authors showed that the sterics of the ortho substituents of the bis(phenolate) ligands are not crucial for the selectivity and attributed the high selectivity to a dynamic monomer-recognition process involving interconversion of the ligand configuration.
 | ||
| Scheme 7 Lanthanide complexes supported by dichalcogen-bridged bis(phenolate) ligands. | ||
Stereoselective ROP of rac-β-butyrolactone
Poly(3-hydroxybutyrate) (PHB) is an isotactic, highly crystalline thermoplastic polyester produced by bacteria and other living organisms. This biodegradable and biocompatible natural polymer is isotactic with all stereocenters in the (R) configuration. Owing to its high crystallinity (Tm = 180 °C) and low thermostability, melt processing of PHB is difficult, thereby limiting its industrial importance. Therefore, one of the most convenient and promising synthetic routes to PHB uses metal initiators to effect the ring-opening polymerization of β-butyrolactone (BBL). Unlike bacteria-mediated polymerization, which gives only isotactic PHB, controlled ROP of BBL allows access to a variety of PHB microstructures. Although most initiating systems are extremely slow and produce low molecular weight PHB, Coates et al. described in a recent report that β-diiminate zinc alkoxide complexes, which have shown high catalytic activities and stereoselectivities for the ROP of lactide (6, Scheme 5), are also able to efficiently polymerize rac-BBL under mild conditions to produce PHBs in a controlled manner.22b Unfortunately, all the PHBs produced were found to be atactic. Despite recent advances in this topic, stereospecific metal-based initiators remain rare.36In 1989, the group of Spassky reported the isospecific polymerization of rac-BBL using chiral initiators obtained from the reaction of organometallic derivatives (ZnEt2, CdMe2, AlEt3) with (R)-3,3-dimethylbutane-1,2-diol to give optically active PHB (Scheme 8).37 With zinc and aluminium systems, the (R)-enantiomer was preferentially incorporated in the polymer chain whereas the (S)-enantiomer was polymerized with the cadmium initiator. The zinc-based catalyst was found to be the most selective system at low conversions: the 46% enantiomeric excess of the unreacted monomer confirmed that the (R)-BBL was preferentially incorporated in the polymer, leading to the high like diad proportion (kR/kS = 1.6).
 | ||
| Scheme 8 Isospecific polymerization of rac-BBL. | ||
Recently, Rieger and co-workers demonstrated that chromium(III) salophen complexes are active initiators for the stereoselective ROP of racemic β-butyrolactone (Scheme 9).38 These achiral Cr(III) initiators (12a–d) combine high activity with high molecular weight and interesting isotacticities of the PHB products (0.63 < Pm < 0.66 at 100 °C). In addition, the catalyst performance can be influenced by varying the aryl substituents on the salophen framework, thus allowing improved control of the polymer properties by catalyst design.
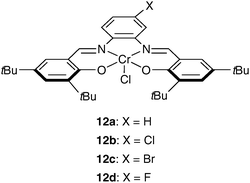 | ||
| Scheme 9 Salophen Cr(III) complexes used for the polymerization of rac-BBL. | ||
Syndiotactic PHB of various molecular weights can be generated with different tin(II,IV)-based catalysts.39 The first demonstration of syndiospecific ROP of rac-BBL was reported by Gross et al.40 The polymerization was carried out with Bu3SnOMe to give low molecular weight PHB enriched in syndiotactic diads (Pr = 0.70). Dialkyltin derivatives (Bu2Sn(OMe)2, BuSn(OMe)3, (Bu3Sn)2O, (Ph3Sn)2O) were also reported to catalyze the syndiospecific polymerization of rac-BBL.41 In all cases, the polymerization reactions result in low molecular weight PHBs. More recently, two distannoxane catalysts 13a–b, which allow a better control of the polymerization, were demonstrated to produce a predominantly syndiotactic PHB (Pr = 0.67) by chain-end control mechanism (Scheme 10).39a
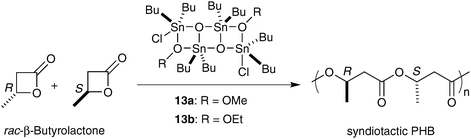 | ||
| Scheme 10 ROP of rac-BBL with distannoxane initiators. | ||
As previously demonstrated for the ROP of rac-lactide, yttrium complexes supported by dianionic aminoalkoxy-bis(phenolate) ligands proved to be efficient initiators in the ROP of rac-β-butyrolactone to give highly syndiotactic PHB. The most notable feature of these initiators is to exhibit high polymerization activity and productivity, combined in some cases with high stereoselectivity. The most sterically crowded complexes42 in the series (Scheme 11) were found to be the most selective catalysts for the syndiospecific ROP of rac-BBL. Highly syndiotactic PHB were prepared using catalysts 14 (Pr = 0.94 at 20 °C) or 9b–c (Pr = 0.90 at 20 °C, Pr = 0.94 at −20 °C) (Fig. 5).
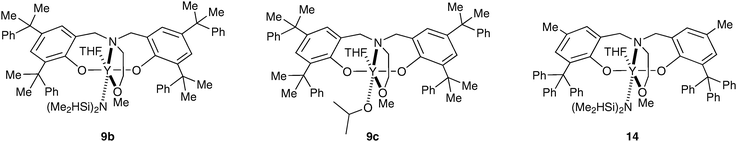 | ||
| Scheme 11 Amino-bis(phenolate) yttrium-based complex used for the ROP of rac-BBL. | ||
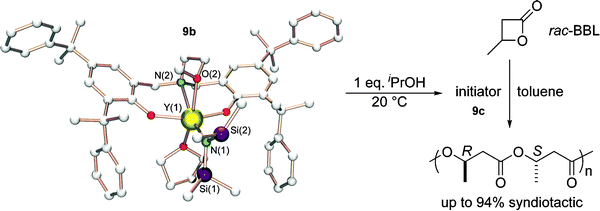 | ||
| Fig. 5 Synthesis of catalyst 9c and syndiospecific polymerization of rac-BBL. | ||
The mechanism of the ring-opening polymerization of rac-β-butyrolactone using these amino-bis(phenolate) yttrium complexes as initiators has been investigated by NMR and shown to occur via a coordination–insertion pathway.43 The microstructure of the resulting syndiotactic PHBs has been studied by 13C NMR spectroscopy, enabling a detailed assignment of resonances at the diad and triad levels. On this basis, a statistical Bernoullian analysis has evidenced that syndioselectivity originates from a chain-end control. Some thermal properties of these PHBs have shown to be markedly affected by the syndiotacticity degree. This is particularly the case for the melting temperature which rises up to 183 °C for Pr = 0.94, a temperature higher than that of natural isotactic PHB (ca. 180 °C).
Conclusion
In the area of enantioselective ring-opening polymerization of lactide and β-butyrolactone, promising results have been obtained using organometallic complexes as initiators. However, despite recent significant advances in ROP of cyclic esters, some major points remain to be addressed and improved.Although research efforts have been dedicated to ROP initiating systems and processes, the number of active, productive and selective initiators remains limited. Indeed the activity and productivity of the organometallic initiators are quite low compared to those obtained in the coordination polymerization processes of olefins. This might be improved most likely by designing new initiating systems, more reactive and more robust toward large amounts of monomer. In addition, polymerization reactions under homogeneous conditions result in polymer contamination by metal residues and in loss of catalyst. Moreover, the synthesis of organometallic complexes is usually a time-consuming and costly process. As a result, it can be anticipated that supported heterogeneous catalysts would be highly valuable for ROP of cyclic esters and would represent an attractive approach for the design of cleaner processes.
One important challenge in the ROP of cyclic esters is the one-pot copolymerization of monomers with different polymerizabilities. Current catalysts allow the synthesis of alternated polymers in select systems in which cross-propagation rate constants are large and block copolymers via the sequential addition of monomers to a living system. It would be of great interest to develop new catalysts that allow higher order sequence control of this latter type, which will be used to create macromolecular structures with advanced properties, stemming from alternated or block structures. This should allow the synthesis of new types of improved innovative (co)polymers with original properties and would clearly increase the number of applications for polyesters.
Notes and references
- (a) S. Mecking, Angew. Chem., Int. Ed., 2004, 43, 1078 CrossRef CAS; (b) M. Okada, Prog. Polym. Sci., 2002, 27, 87 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef.
- Recently polyesters have also been produced by copolymerization of epoxides and anhydrides. See for instance: R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041 Search PubMed and references therein.
- A. Duda and S. Penczek, in Polymers from Renewable Resources: Biopolyesters and Biocatalysis, ed. C. Scholz and R. A. Gross, ACS Symposium Series, Washington, DC, 2000, vol. 764, p. 160 Search PubMed.
- While not well established, there is reason to be concerned over the toxicity of β-substituted β-propiolactones. In particular, the possible hazards associated with β-butyrolactone are disclosed in IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: β-butyrolactone, 1976, 11, 225.
- The ROP of ε-caprolactone was also extensively investigated but cannot be included in this paper on stereocontrolled ROP of cyclic esters. L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762 Search PubMed.
- (a) H. R. Kricheldorf, M. Berl and N. Scharnagl, Macromolecules, 1988, 21, 286 CrossRef CAS; (b) P. Dubois, R. Jérôme and P. Teyssié, Makromol. Chem., Macromol. Symp., 1991, 42/43, 103 Search PubMed; (c) P. Dubois, C. Jacobs, R. Jérôme and P. Teyssié, Macromolecules, 1991, 24, 2266 CrossRef CAS.
- C. Wang, H. Li and X. Zhao, Biomaterials, 2004, 25, 5797 CrossRef CAS.
- R. E. Drumright, P. R. Gruber and E. Henton, Adv. Mater., 2000, 12, 1841 CrossRef CAS.
- W. Dittrich and R. C. Schulz, Angew. Makromol. Chem., 1971, 15, 109 CrossRef CAS.
- J. Baran, A. Duda, A. Kowalski, R. Szymanski and S. Penczek, Macromol. Rapid Commun., 1997, 18, 325 CrossRef CAS.
- J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 4072 CrossRef CAS.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510 CrossRef CAS.
- (a) T. M. Ovitt and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4686 CrossRef CAS; (b) C. P. Radano, G. L. Baker and M. R. Smith, J. Am. Chem. Soc., 2000, 122, 1552 CrossRef CAS; (c) T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291 CrossRef CAS.
- Nomura’s and Gibson’s alkyl initiators have to be mixed in situ with benzyl alcohol to be catalytically active: (a) N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938 CrossRef CAS; (b) P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688 CrossRef CAS.
- A. F. Douglas, B. O. Patrick and P. Mehrkhodavandi, Angew. Chem., Int. Ed., 2008, 47, 2290 CrossRef CAS.
- P. L. Arnold, J.-C. Buffet, R. P. Blaudeck, S. Sujecki, A. J. Blake and C. Wilson, Angew. Chem., Int. Ed., 2008, 47, 6033 CrossRef CAS.
- A. Otero, J. Fernández-Baeza, A. Lara-Sánchez, C. Alonso-Moreno, I. Márquez-Segovia, L. F. Sánchez-Barba and A. M. Rodríguez, Angew. Chem., Int. Ed., 2009, 48, 2176 CrossRef CAS.
- (a) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229 CrossRef CAS and references therein; (b) L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239 CrossRef CAS.
- M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717 CrossRef CAS.
- A. Finne, Reema and A. C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3074 CrossRef CAS and references therein.
- A. J. Chmura, C. J. Chuck, M. G. Davidson, M. D. Jones, M. D. Lunn, S. D. Bull and M. F. Mahon, Angew. Chem., Int. Ed., 2007, 46, 2280 CrossRef CAS.
- A. Pietrangelo, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2009, 2736 RSC.
- S. McLain, T. Ford and N. Drysdale, Polym. Prepr. (Am. Chem. Soc., Polym. Chem.), 1992, 33, 463 Search PubMed.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316 CrossRef CAS.
- A. Amgoune, C. M. Thomas and J.-F. Carpentier, Pure Appl. Chem., 2007, 79, 2013 CrossRef CAS.
- F. Bonnet, A. R. Cowley and P. Mountford, Inorg. Chem., 2005, 44, 9046 CrossRef CAS.
- I. Palard, A. Soum and S. M. Guillaume, Chem.–Eur. J., 2004, 10, 4054 CrossRef.
- A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem.–Eur. J., 2006, 12, 169 CrossRef CAS and references therein.
- X. Liu, X. Shang, T. Tang, N. Hu, F. Pei, D. Cui, X. Chen and X. Jing, Organometallics, 2007, 26, 2747 CrossRef CAS.
- A. Amgoune, C. M. Thomas and J.-F. Carpentier, Macromol. Rapid Commun., 2007, 28, 693 CrossRef CAS.
- H. Ma, T. P. Spaniol and J. Okuda, Angew. Chem., 2006, 118, 7982 CrossRef and references therein.
- S. Bloembergen, D. A. Holden, T. L. Bluhm, G. K. Hamer and R. H. Marchessault, Macromolecules, 1989, 22, 1656 CrossRef CAS.
- A. Le Borgne and N. Spassky, Polymer, 1989, 30, 2312 CrossRef CAS.
- M. Zintl, F. Molnar, T. Urban, V. Bernhart, P. Preishuber-Pflügl and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 3458 CrossRef CAS.
- (a) Y. Hori and T. Hagiwara, Int. J. Biol. Macromol., 1999, 25, 237 CrossRef CAS; (b) H. R. Kricheldorf and S. Eggerstedt, Macromolecules, 1997, 30, 5693 CrossRef CAS.
- J. E. Kemnitzer, S. P. McCarthy and R. A. Gross, Macromolecules, 1993, 26, 1221 CrossRef CAS.
- (a) J. E. Kemnitzer, S. P. McCarthy and R. A. Gross, Macromolecules, 1993, 26, 6143 CrossRef CAS; (b) Z. Jedlinski, M. Kowalczuk, P. Kurcok, G. Adamus, A. Matuszowicz, W. Sikorska, R. A. Gross, J. Xu and R. W. Lenz, Macromolecules, 1996, 29, 3773 CrossRef CAS; (c) H. R. Kricheldorf, S.-R. Lee and N. Scharnagi, Macromolecules, 1994, 27, 3139 CrossRef CAS.
- The steric bulk of the ortho-phenolate substituents plays a crucial role in achieving high selectivity: A. Amgoune, C. M. Thomas, S. Ilinca, T. Roisnel and J.-F. Carpentier, Angew. Chem., 2006, 118, 2848 (Angew. Chem., Int. Ed., 2006, 45, 2782) Search PubMed.
- N. Ajellal, M. Bouyahyi, A. Amgoune, C. M. Thomas, A. Bondon, I. Pillin, Y. Grohens and J.-F. Carpentier, Macromolecules, 2009, 42, 987 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |