Natural and artificial peptide motifs: their origins and the application of motif-programming
Kiyotaka Shiba*
Division of Protein Engineering, Cancer Institute, Japanese Foundation for Cancer Research, Tokyo 135-8550, Japan. E-mail: kshiba@jfcr.or.jp; Fax: +81-3-3570-0461; Tel: +81-3-3570-0489
First published on 2nd September 2009
Abstract
In this tutorial review, I discuss the nature and application of peptide motifs. Motifs are usually identified through analysis of the sequence of natural proteins and are linked to particular biological functions, though the association between a motif and its function is only speculative in some cases. In other cases, however, the transplantability and functional independence of motifs have been experimentally proven, providing us with the opportunity to use those motifs as programming units for biotechnological application. In addition to natural motifs, peptide aptamers created using in vitro evolution systems can also serve as motif units. The associated functions of these artificial motifs are related to their binding ability. Numerous binders against both natural biomolecules and inorganic materials have been created from peptide phage systems. By programming these natural and artificial motifs, artificial proteins with the potential to contribute to medical diagnosis and treatment, nanotechnology, and various areas of basic science have been created. In addition, the transplantability and functional independence of motifs provide insight into the nature of protein evolution.
 Kiyotaka Shiba | Kiyotaka Shiba received his BS and PhD degrees in molecular biology from the Faculty of Science, Kyoto University, where he studied the mechanism of protein secretion using bacterial genetics. He then worked as an assistant professor in the Department of Biophysics and Biochemistry at the University of Tokyo. He subsequently took a position as a postdoctoral research fellow at the Massachusetts Institute of Technology until he joined the Cancer Institute in 1991. He is currently chief of the Division of Protein Engineering, Cancer Institute, Japanese Foundation for Cancer Research in Tokyo. His research interests include the evolution of genes, protein engineering and bionanotechnology. |
Introduction
Motif-programming is a method for constructing artificial proteins by embedding peptide motif(s) within artificial protein sequences. Recent advances in genome science have revealed a variety of naturally occurring motifs. In addition, evolutionary molecular engineering has enabled us to create artificial motifs that interact with selected target molecules assigned by the researchers. Motif-programming is method to rationally utilize the accumulating knowledge on motifs, and is expected to contribute to both applied and basic sciences. Here, I first discuss natural and artificial motifs, and then introduce some examples of motif programming.Motifs as recurring and motivating elements
The term “motif” is used in wide variety of contexts, including music, painting, literature, mathematics, biology and the game of chess, among many others. Despite subtle variation in its definition, the term commonly denotes a recurring element discerned within a larger structure. The term has its origin in the Latin word motus, from such words as motivate, move, emotion and promote have evolved. This history of the word implies that a recurring element has the ability to move something, or to induce an activity (Fig. 1). Indeed, motifs in works of art often move our heart, and motifs in biological systems are, as shown below, the motivating forces for a variety of biological activities.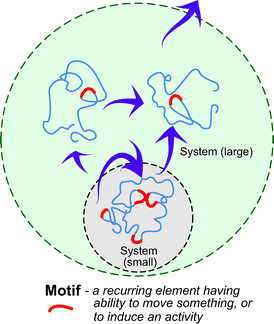 | ||
| Fig. 1 What are motifs? Motifs are elements that recur in a given system, and have the ability to induce activity in part or all of a system. Depending on the definition of the system, a motif can be viewed to act outwardly from the system. | ||
When a motif, or recurring element, is used in a biological context, the definition of “element” differs depending on how we define the larger structure. For researchers who study networks of biological molecules, motifs represent simple patterns of connection within those networks.1 By contrast, if the researchers’ interest is the tertiary structures of proteins, they regard motifs as characteristic alignments of secondary structures (which are also referred to as supersecondary structures).2 When their interest is the primary structure of proteins, motifs are defined as recurring arrays of amino acid residues found within the protein sequences. In this review, I will focus on these sequence motifs found in the primary structures of proteins. I will first introduce how these motifs have been defined or created, and I will then present an overview of recent advances in motif-related research in the biotechnology field.
How peptide motifs are defined from protein sequences
As proteins evolve, there is inherent diversification of their amino acid sequences. This sequence diversification occurs within the constraint that the amino acid alternations cannot diminish the proteins’ functionality. As a consequence, some amino acid residues are rather conservative with respect to their identities, owing to their involvement in the functioning of their proteins, whereas others are not. The conserved residues are most often identified when two or more proteins from different species are compared (Fig. 2A). When proteins diversify concurrently with speciation, they are said to have an “orthologous” relationship, and we refer to the conserved residues as motifs.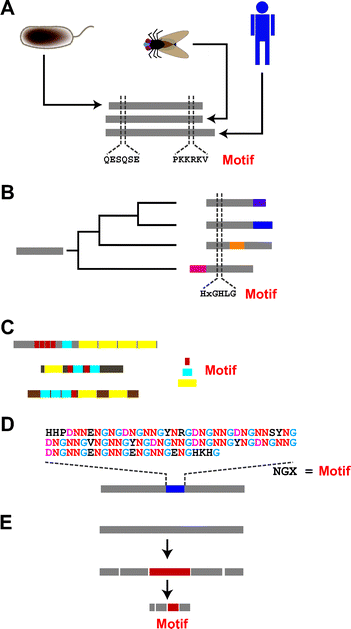 | ||
| Fig. 2 Several ways to identify motifs within protein sequences. (A) From comparison of orthologous proteins. When the same proteins from different species are compared, we may notice some regions are better conserved than others. The conserved segments are often defined as motifs. Gray bars represent protein sequences. Amino acids are shown by one-letter codes. (B) From comparison of paralogous proteins. Proteins can evolve through gene duplication; such proteins are defined as being paralogous. Comparison of paralogous proteins can enable identification of motifs. Colored bars indicate segments that were acquired after gene duplication. (C) From analysis of “shuffled” proteins. Some proteins, especially proteins involved in signal transduction, appear to have evolved through the shuffling of genetic fragments. A variety of motifs have been proposed based on analysis of these proteins. Red, blue and yellow bars represent different repeating motifs. (D) From proteins having repetitive sequences. Repeats of short amino acid sequences are often observed within parts or even the entire structure of a protein. These repeating units are often defined as motifs. The blue bar shows a repetitive region. (E) From non-sequence analysis. In a limited number of cases, series of biochemical, biological and/or biophysical analyses have enabled investigators to track down small peptide segments (shown by a red bar) within the protein structure. In these cases ((D) and (E)), comparison of two or more proteins is not necessary. | ||
In addition, many proteins have built up kin groups over time through gene duplication.3 Proteins that appear to have evolved through gene duplication are said to have a “paralogous” relationship. Comparison of the sequences of paralogous proteins has also enabled the identification of motifs (Fig. 2B). An early example is the identification of the HIGH motif from the comparison of methionyl-, isoleucyl-, tyrosyl- and glutaminyl-tRNA synthetases,4 which are believed to have emerged through gene duplication before the separation of the Archaea, Eucarya and Bacteria domains.
During their evolutionary course, proteins also appear to alter their structure in a far less gradual manner by suddenly replacing entire sections of their primary sequences with different sequences, or they may acquire additional new sequences within their preexisting structures. This may occur through the random shuffling of genomic fragments in some sort of recombination mechanism, and through subsequent selection process. Motifs are also identified in these altered or added sequences. One example is motif-N, which has apparently spread into some aminoacyl-tRNA synthetases in the Eucarya lineage (Fig. 2C).5 The acquisition of motif-N was totally independent of the authorized evolutional paths of aminoacyl-tRNA synthetases. Random mutations and selection processes resulted in the current structures, in which the genetic element encoding the motif appears to have roamed around genomic space in search of places where the motif was needed.
Genomic shuffling (exon-shuffling) became especially prominent after the emergence of Chordata. Numerous signaling proteins, which characterize multicellular organisms, are thought to have evolved through genomic (exon) shuffling.6 From such signaling proteins, a long list of motifs have been identified—e.g., SH2, SH3, BH1–BH4, among many others. These motifs within signaling pathways likely played pivotal roles in the evolution the convoluted networks of signaling pathways that characterize multicellular organisms.7
The apparent suddenness of some protein evolution is indicative of the dynamic nature of genomic structure. Indeed, a decade of extensive genomic sequencing has revealed this dynamic nature.8 Particularly noteworthy is the fact that genomic sequences tend to give birth to periodic structures, ranging from those on a scale of one to several nucleotides to those on a subchromosomal scale.9 In some cases, moreover, the repetitive structures within genomic sequences appear to have served as the driving force behind the emergence of new proteins,10 and some existing proteins continue to exhibit this repetitiveness within all or part of their structure (Fig. 1D).11 Motifs have also been identified within these repetitive structures, and in those cases sequence comparison among homologues is not necessary; motifs can be identified from a single repetitive sequence as long as it contains repeats of the motif.
When a motif is very short and is found in number of proteins that have no obvious sequence similarity, except the motif, it is very difficult to determine whether the motifs have the same evolutional origin (divergently evolved) or emerged independently (convergently evolved). If the motifs are three amino acids or so in length, it would be reasonable to suggest that they could have convergent origins. On the other hand, if the motifs are composed of more than several amino acids, they were more likely recruited through genomic shuffling, starting from common ancestor. However, there is always a gray area between divergent and convergent evolution. One must keep in mind that evolutionary analysis inevitably depend on certain models, and all proposed evolutionary history is just the one that has been inferred from those models.
Finally, identification of some motifs does not rely on evolutionary analysis (Fig. 1E). Biochemists or geneticists often dissect proteins or genes into smaller pieces and then try to attribute the functions of the parental proteins (or genes) to these smaller units. When a dissected piece exerts a discernable function, it is considered a motif. Examples include peptides dissected from dentin matrix protein-1 (DMP1), which is a non-collagenous extracellular matrix (ECM) protein present in the matrix of both bone and dentin and believed to be involved in hydroxylapatite (HAP) formation. A minimalist approach to this protein revealed that two peptides, pA (ESQES) and pB (QESQSEQ DS), enhance HAP formation under certain sets of conditions.12
Natural vs. artificial motifs
The term “aptamer” refers to a biomolecule having specific binding activity. The word was coined by Ellington and Szostak when they used their in vitro evolution system to create RNA molecules with the ability to bind to certain dye molecules.13 In that work, they first prepared a large pool of random RNA sequences and then selected clones that specifically bound to the immobilized dye molecules. A similar “selection-from-random” strategy is used to create peptide aptamers; in this case random sequences of DNA are translated into random sequences of amino acids. A peptide phage system is often used to select peptide aptamers.14A phage is a virus that propagates on a bacterial host, though it has its own genome within its capsid architecture. In a peptide phage system, short, near-random sequences of DNA are inserted into the phage genome so that the inserted foreign sequences produce peptide-capsid protein fusion. Several combinations of phages and capsid proteins have been tested for use in peptide phage systems, but the one most commonly used is the M13 (fd) phage and its gp3 (pIII) protein.15 M13 is a filamentous phage with a body that is approximately 1 μm long and 6–7 nm in diameter, and several copies of gp3 protein are located at one end of the phage’s body. As a result, a foreign peptide that is fused to gp3 is displayed at the tip of M13 (Fig. 3A). Typically, peptides comprised of 7–12 amino acids are fused at the N-terminal of gp3. Although as a whole the phages display peptide sequences randomly, there is a one-to-one relation between each peptide that links to the phage and the coding sequence inserted into the phage genome. This makes it possible to deduce the peptide sequence from the DNA sequence in the relevant region of M13’s genome, from which the displayed peptides is transcribed and translated. We call this phage population having molecular diversity in the displayed-peptides “peptide phage library”.
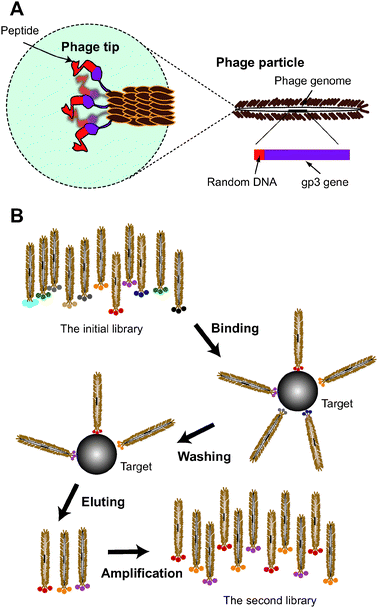 | ||
| Fig. 3 A peptide phage system. (A) Schematic representation of a filamentous phage. M13 phage has a body approximately 1 μm long in length and 6–7 nm in diameter, within which the single-stranded phage genome is packed. DNA sequences encoding peptides can be inserted into a specific site within the genome so that the translated peptides are displayed at one tip of the phage body as the fusion protein with gp3.15 Note that each phage displays five copies of a unique peptide sequence that is encoded by the inserted DNA contained in the phage particle. (B) Scheme of phage panning. The panning procedure is comprised of binding, washing, eluting and amplification steps.15 During the binding step, phages displaying various peptides are incubated with the target molecules. If the peptides have affinity for the target, the phages displaying those peptides are moored to the target molecule. The moored and unbound phages are separated in the washing step. The interaction between the peptides and the target is non-covalent. Usually, the bound phages are eluted from the target using solutions having lower and lower pH. The recovered phages are re-amplified on E. coli to prepare the secondary library. In this library, the fraction of phages that have affinity for the target is increased. After repeating this panning procedure, the library is mostly composed of phages that display the binder peptides. | ||
When we want to create a peptide sequence that specifically binds to an assigned target, we first incubate the target with a library of peptide phages. If the phage displays a peptide that associates with the target molecule, the phage is moored to the target via the displayed peptide. For this purpose, target molecules are usually immobilized on the surface of a plastic tube or magnetic beads, which makes it easy to remove unbound phages by washing the target-phage complex. The bound phages are recovered by elution with solutions having lower pH, or by some other method. In many cases, the recovered phages are re-amplified on Escherichia coli K strain (laboratory bacteria) to prepare a phage sublibrary. This sublibrary is no longer random (naïve) and contains a large fraction of phages that have affinity for the assigned target. Together, the binding, washing, eluting and amplification steps are referred to as a “panning” procedure (Fig. 3B).15 By repeating the panning procedure, more and more of the biased library is occupied by phages able to bind to the target. In some cases, selective pressures such as shortening the binding time or increasing the concentration of a competitor are applied to the panning procedure to promote selection of strong binders.
The peptide phage system was a groundbreaking invention in molecular biology. Its earliest use focused on identification of epitopes for antibodies.16 An epitope is the portion of a protein (or other molecule) recognized by an antibody (or other immune system component, including B and T cells). When phage panning was carried out against a monoclonal antibody, the obtained peptide aptamers often shared sequence similarities with parts of the antigen protein, and these sequences corresponded to the epitope recognized by the antibody. Thus, phage panning enabled identification yet-to-be-identified epitopes or antigens for given antibodies. This system has been used to identify ligands for orphan receptors—i.e., receptors whose endogenous ligand has not yet been identified. Notably, the created peptide aptamers do not necessarily share sequence similarities with their counterparts in natural proteins—e.g., some peptide aptamers against certain antibodies do not resemble the natural epitope sequence, though they bind the same part of the antibody (paratope). In these cases, the created peptides are believed to mimic the 3D shape of the epitope.17 Thus, an in vitro evolution system can provide solutions to a given question (function) that nature has not yet realized.
Panning procedures are most often performed in plastic tubes. Since the middle of the 1990s, however, panning has also been carried out in vivo. In that case, a phage library is injected into the tail or other vein of a mouse, and phage clones that localize at specific sites, including tumors, are collected, re-amplified and re-injected, in what constitutes an in vivo panning cycle.18 The purpose of in vivo panning is to create artificial peptides that can direct foreign molecules to specific organs or lesion sites, which would be a highly desirable for the diagnosis and treatment of illnesses.19 In fact, medically oriented in vivo panning has reached a point where human subjects have been used for panning a phage library.20
In 2000, Belcher’s group extended the targets of a peptide phage system from biomolecules to inorganic molecules. They isolated 12-mer peptides that specifically bound to the surfaces of semiconductors such as GaAs and InP.21 About the same time, the field of nanotechnology emerged, and material-binding peptides began attracting much attention as powerful tools for nanofabrication. Indeed, Belcher’s findings were followed by numerous reports on the isolation of material-binding peptides whose targets include gold, silver, cadmium sulfide, carbon nanohorns, carbon nanotubes, and many others (see review22 for the references). These material-binding peptides are used not only for directing foreign nanomaterials to specific positions, but also for facilitating mineralization of inorganic materials.23,24
Peptide aptamers can be regarded as artificial motifs because they are inherently associated with binding functions and, like natural motifs, they are composed of arrays selected from the 20 amino acids. The only difference between natural and artificial motifs is whether their sequences were extracted from natural proteins or were created through in vitro evolution. Indeed, similar motifs have been identified in both natural proteins and in vitro evolution systems. For instance, the RGD (that was named after one letter code for Arg-Gly-Asp) motif is a well-known adhesion tri-peptide found in ECM (= extracellular matrix; connective tissue in animals) proteins such as fibronectin.25 When peptide aptamers were selected against platelet cells, the RGD tripeptide was identified among the evolved aptamer sequences,26 which is indicative of the equality between natural and artificial motifs.
Motifs as building blocks
A number of motifs have been identified from bio-informational analyses (Fig. 4A),27,28 but their associated functions have not always been proven in biochemical or genetical experiments. Nonetheless, for some of motifs, the relationships between the annotated functions and the peptide sequences have been appropriately confirmed at different levels. The most rudimentary confirmations are derived from mutational analyses. If substitution of an amino acid within a putative motif impairs the function of the protein in which it resides, we know that the motif plays a critical role in that function (Fig. 4B). A stronger linkage is established when the proposed function can be transferred to a foreign molecule by transferring a motif-containing peptide (Fig. 4C). Examples of this type of motif include various “tag” motifs such as HA tag, Flag tag, Myc-tag etc. And in rare instances, an isolated (or synthetic) peptide can exert its associated function without being embedded within the structure of a larger molecule (Fig. 4D). The potential transplantability and functional independence of motifs suggests that they can be treated as building blocks, with which to rationally program artificial molecules so that they exhibit a desired combination of functions. Below, I will introduce some examples of such “motif-programming.”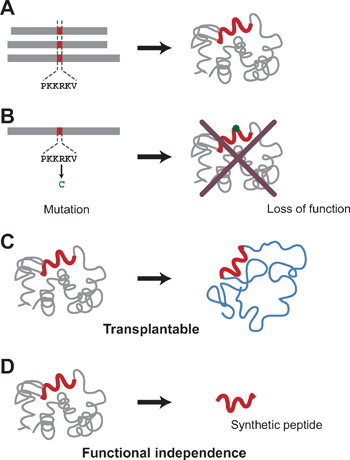 | ||
| Fig. 4 Levels of assessment of motif-function relationships. (A) Putative motifs. The association of motifs with their functions is only speculative and not confirmed by genetic or biochemical analysis. At this level, we do not know whether or not the motifs can serve as programming units. (B) Motifs proven by genetic analysis. When mutations within a motif sequence impair the associated function exerted by the entire protein, the linkage between the motif and function is strengthened. However, we still do not know whether the motif can endow other molecules with the associated function. (C) Transplantable motifs. When the transplantability of a motif is high, its associated function can be transferred to a target molecule by conjugating the peptide to the target. (D) Functionally independent motifs. In some cases, a synthetic peptide motif can exert its associated function by itself, without conjugation to a larger molecule. The functional independence of the motif is considered to be high in those cases. | ||
Motif-programming experiments
Motif-programming is our approach to construction of artificial proteins by rationally embedding functional peptide motifs within artificial protein sequences.29,30 In this section, I will introduce several examples of motif-programming by focusing on results from our group.Programming natural motifs
BH motifs (BH1–BH4) (Table 1) are shared among the Bcl-2 family proteins, which are involved in mitochondria-dependent programmed cell death and have been divided into three classes based on the motifs they contain.31 There are the anti-apoptotic multidomain members (Bcl-xL, Bcl-2, MCL-1, A1, Bcl-W), the pro-apoptotic multidomain members (BAX, BAK), and the pro-apoptotic BH3-only members (Bim, Bid, Puma, Noxa).31 These proteins are believed to form complex signaling networks in which the BH peptide contributes by providing protein-protein interaction sites. It has already been shown that a 16-amino acid peptide that includes the 9-amino acid BH3 motif can induce apoptosis when injected into mammalian cells.32 Thus the functional independence of BH3 is well established, and the associated function of BH3 is induction of cell death.| Name | Sequencea | Associated function | Ref. |
|---|---|---|---|
| a Shown by one-letter code of amino acid. | |||
| BH3 | LRRFGDKLN | Signal transduction of cell death (apoptosis) | 31 |
| PTD | YGRKKRRQRRR | Translocation of proteins across cell membrane | 33 |
| BH1 | ELFRDGVN | Signal transduction of cell death (apoptosis) | 31 |
| BH2 | ENGGWDTF | Signal transduction of cell death (apoptosis) | 31 |
| BH4 | RELVVDFL | Signal transduction of cell death (apoptosis) | 31 |
| minTBP-1 | RKLPDA | Peptide aptamer against Ti | 42 |
| RGD | RGD | Cell attachment | 25 |
| NGX | NGD or NGN | Shell formation | 47 |
| Motif-A in DMP1 | ESQES | Bone formation | 12 |
| Motif-B in DMP1 | QESQSEQDS | Bone formation | 12 |
PTD (Table 1) is an 11-amino acid peptide motif located in the tat protein from HIV (the virus that leads to AIDS).33 This motif was not identified through sequence analysis, but from inquiry into the function of tat protein. It was found that the motif has the ability to translocate across the plasma membrane and by fusing or attaching the motif to a foreign molecule, it can be used to introduce proteins, nucleic acids and other molecules into cells. Thus the transplantability of PTD motif is well established, and the associated function of PTD is cell invasion.
Our expectation was that by combining the BH3 and PTD motifs, we could create a bifunctional protein that when added to culture medium would automatically penetrate into cells and induce apoptosis.34 To that end, we prepared a library of artificial proteins that contained the two motifs in various numbers and orders (Fig. 5A). To prepare that library, we employed a system we developed called MolCraft (Fig. 6).29,35
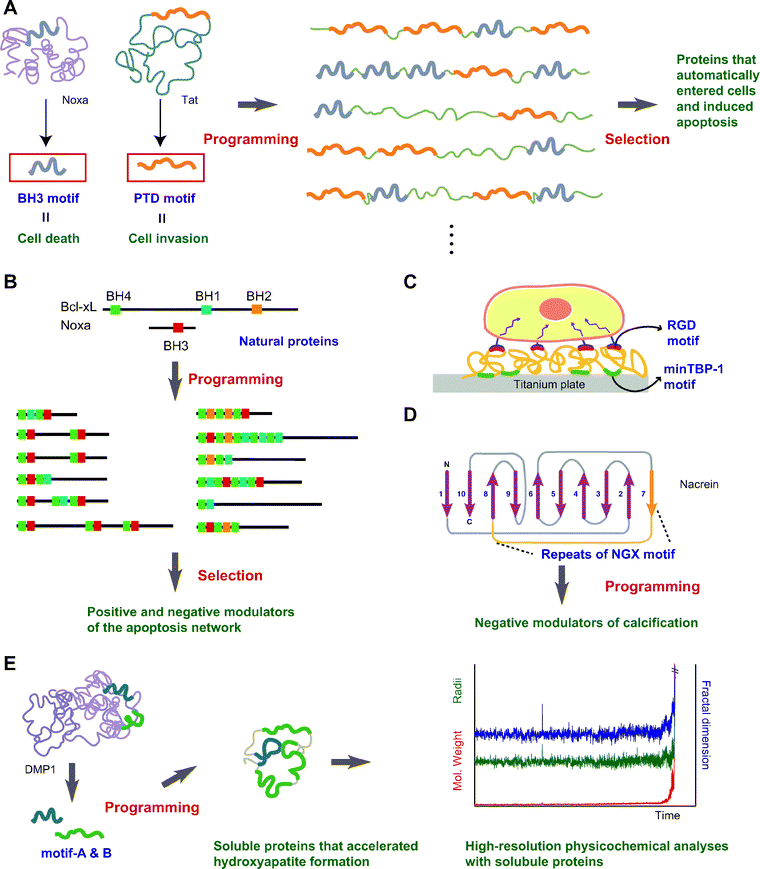 | ||
| Fig. 5 Examples of motif-programming experiments. (A) Programming motifs mediating cell death and cell invasion.34,40 BH3 and PTD are natural motifs whose functions are induction of cell death and cell invasion, respectively. By embedding these motifs in artificial proteins, we created bifunctional proteins that automatically entered cells and induced apoptosis. (B) Shuffling BH1, BH2, BH3 and BH4 motifs to create artificial signaling molecules.7,30 Natural components of apoptotic signaling networks share combinations of BH1-4 motifs. From among the artificial proteins created by embedding these motifs, we selected positive and negative modulators of the apoptosis network. (C) Artificial ECM.44 RGD is a well-known natural motif that has cell-binding ability. By combining RGD with minTBP-1, an artificial motif mediating Ti binding, we constructed artificial ECM proteins that endow a Ti surface with the ability to bind cells. (D) Confirmation of the putative function of a motif.50 The motif NGX was identified in pearl oyster and is thought to be involved in shell formation. To confirm the putative involvement of this motif in biomineralization, artificial proteins were prepared through motif-programming, after which we selected those that inhibited calcification. (E) More detailed analysis of motifs.48 Motifs from DMP1 have been shown to enhance calcium phosphate formation when immobilized on a glass substrate. However, such immobilization is an obstacle to detailed analysis of the motifs. To enable high-resolution physicochemical analysis, soluble proteins with the capacity to accelerate calcium phosphate formation were created through motif-programming, after which time-resolved static light scattering analysis was carried out. | ||
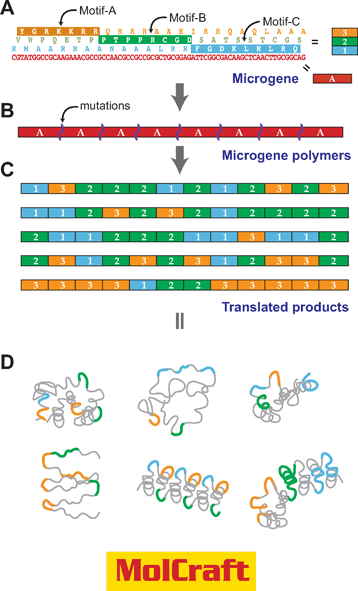 | ||
| Fig. 6 Motif-programming using MolCraft.29,35 With MolCraft, motifs are initially embedded in different reading frames of a single short DNA sequence, which we call a microgene. The designer microgene is then polymerized to generate tandem repeats of the microgene unit. In this process, we use special conditions that allow the random insertion or deletion of mutations at junctions of microgene units. Because of these mutations, the reading frame of the polymers shifts randomly at the junctions. As a result, the translated products make up a library of combinatorial polymers of the three reading frames (motifs) encoded by a single microgene. | ||
The exon theory of genes, proposed by W. Gilbert in 1987,36 suggested the possibility that new combinations of exons (microgenes) could give birth to novel genes. Mimicking the exon-shuffling in vitro to create novel proteins have been attempted by recombination using sexal PCR 37 and by polymerization of microgenes.38,39 These efforts are expected to establish a new system for in vitro protein creation. MolCraft is a simplified hierarchical protein evolution system in which a single gene block is used to make larger genes. In MolCraft, a microgene is initially designed so that it encodes motifs in its different translational frames (Fig. 6A) and then tandemly polymerized with insertion or deletion mutations at the junctions between microgene units (Fig. 6B). Because of the junctional perturbations,35 proteins translated from a single microgene polymer are molecularly diverse, originating from the combinatorics of three reading frames, and are thus combinatorial polymers of motifs (Fig. 6C). Notably, repetitiousness retained in the overall structure of proteins contributes to the formation of ordered structures, and enhances the chances of reconstituting biological activity rationally encrypted in the microgene unit (Fig. 6D).29
In the motif programming of BH3 and PTD, we first designed the microgene that codes BH3 and PTD motifs in different reading frames. Although the microgene unit is just tandemly polymerized in the MolCraft system, because of random insertion and deletion mutations at junctions of the microgene, we were able to prepare a combinatorial library of BH3 and PTD motifs by randomly shifting between reading frames within the polymers. The resultant long reading frames were cloned into an expression vector, and then translated in E. coli cells. Their functions of the artificial proteins were then investigated after purification. Among 21 proteins tested in the (BH3 + PTD) programming experiments, one clone showed a notable ability to penetrate through cell membranes and induce apoptosis in several cancer cell lines with a GI50 of approximately 5 μM.34,40 In addition, transfection of the artificial gene led to effective expression of the artificial protein within cells, which in turn caused apoptosis at a level similar to that seen in cells transfected with naturally occurring pro-apoptotic Noxa or Bax.40 Thus both protein transduction and DNA transfection showed that the programmed protein was able to induce apoptosis in human cells. These results substantiate the concept of motif-programming and suggest that motif-programmed proteins have the potential to serve as novel agents for the treatment of maladies.
Among BH motifs, BH3 and BH4 motifs are associated with pro-apoptotic and anti-apoptotic phenotypes, respectively.31 Indeed, pro-apoptotic activity could be programmed by embedding the BH3 motif in artificial proteins, as described above, and a synthetic peptide derived from BH4 reportedly prevented apoptosis otherwise induced by an anticancer drug.41 However, considering the fact that apoptotic signaling is governed via a convoluted network of BH-bearing proteins, the execution of pro- and anti-apoptotic activities by BH motifs should be a dynamic process, so that simple allocation of functions to each BH motif is likely an oversimplification. With that in mind, we next created artificial proteins composed of combinations of BH1, BH2, BH3 and BH4 motifs (Fig. 5B).7 For this purpose, we first had to develop a second generation of the MolCraft system, MolCraft II,30 because the original MolCraft cannot make combinatorial polymers of more than three motifs. In the MolCraft II, more than two microgenes were constructed and polymerized from the stochastic assembly of sense- and antisense oligonucleotides in a single pot reaction, which enabled the shuffling of the BH1-4 motifs. From the prepared libraries, we randomly selected 41 clones and found that 28 yielded stable proteins when transfected into mammalian cells. We then assessed the pro-apoptotic activity of the 28 clones by scoring the viability of cells after transfection, and found that one clone containing two BH3, two BH1 and four BH4 motifs induced apoptosis, which was consistent with the aforementioned 〈BH3 + PTD〉 programming experiment. To determine whether the library also contained anti-apoptotic proteins, we next scored the capacity of the 28 clones to relieve the growth inhibition caused by the natural pro-apoptotic peptide Bim, and found that two of the proteins reproducibly increased cell viability. Taken together, these results demonstrate that functional proteins exerting opposing effects can emerge from a single pool prepared from common motifs and suggest that motifs contribute to the plastic evolvability of protein networks.
Combinations of natural and artificial motifs
TBP-1 is a 12-amino acid artificial motif that was isolated as a peptide aptamer against titanium (Ti) using a peptide phage system.42 Mutational analysis revealed that the first six residues, RKLPDA (minTBP-1, Table 1), is sufficient for binding, and of those arginine (R1), proline (P4) and aspartate (D5) residues are essential. Proline is known to introduce a kink into the main chain of peptides, and the side chains of arginine and asparate, respectively, act as a Lewis base and Lewis acid at neutral pH. Because of the kink introduced by P4, R1 and D5 are expected to be oriented in the same direction, so that the base (R1) and the acid (D5) are situated in close proximity to one another. Based on this analysis and the fact that the surface of Ti in water is covered with an oxide film displaying both acidic (–O−) and basic (–OH2+) hydroxyl groups, we proposed that minTBP-1 binds reversibly and electrostatically to Ti—i.e., R1 and D5, respectively, interact with –O− and –OH2+ on the surface of the oxide film covering the Ti.42When we detached TBP-1 from the phage body and evaluated the synthetic TBP-1 peptide, we found that bound comparatively weakly to Ti (the estimated dissociation constant for the peptide binding to Ti was approximately 13 mM).43 On the other hand, when the core of TBP-1, which is comprised of the six amino acids of minTBP-1, was transferred to ferritin, a naturally occurring, spherical, cage protein, the modified ferritin was endowed with the ability to selectively bind Ti. The estimated dissociation constant of the modified ferritin for binding to Ti was approximately 3.8 nM, which is comparable to the strong interaction between an antigen and antibody. Ferritin is composed of 24 subunits and in the modified ferritin each subunit displayed minTBP-1 at its N-terminal end. This multivalent presentation of the motif may be responsible for the strong binding of the modified ferritin. Thus, although the functional independence of the artificial TBP-1 motif is limited, it has applicable transplantability.
As mentioned above, the RGD motif (Table 1) is a well-known cell adhesion peptide found in ECM proteins such as fibronectin and vitronectin.25 In addition, this motif is often found within the sequences of peptide aptamers isolated as organ or cell binders. The motif is recognized by a set of integrin family proteins (αvβ1, αvβ3, etc.), and the interaction of RGD and integrin is known to trigger a cascade of overlapping reactions, including cell attachment, cell spreading and cytoskeletal reorganization. The functional independence and transplantability of the motif are already been well established. For instance, Arap et al. isolated a peptide aptamer containing the RGD through in vivo panning against tumor blood vessels, and by making a peptide–drug conjugate, they succeeded in using the peptide to actively target an anticancer drug to the tumor in model animals.19
With the aim of developing methods for the functionalization of dental Ti implants, we created artificial proteins that contain both minTBP-1 and RGD, which were expected to endow the surface of Ti implants with artificial ECM-like activity (Fig. 5C).44 The artificial proteins created were confirmed to rapidly associate with the Ti surface and to dissociate relatively slowly, which is consistent with the properties observed with the aforementioned minTBP-1-ornamented ferritin molecules. The proteins were also confirmed to have a capacity for cell attachment and to mediate the development of a well-organized network of F-actin stress fibers and focal adhesions on Ti-plates. Intriguingly, some of the artificial proteins showed an even greater ability to mediate ECM-induced intracellular signaling than fibronectin. And while both the artificial proteins and fibronectin contained the RGD motif, their respective modes of interaction with Ti were totally different. In buffer, the artificial proteins rapidly bound to and detached from Ti, which may reflect the inherent properties of the embedded minTBP-1 motif. By contrast, the dissociation of fibronectin from Ti was very slow, indicating that hydrophobic interactions dominate the association. Given the observation that the signaling ability of some artificial proteins is superior to that of fibronectin, we believe the reversible immobilization is well suited for development of protein-based biomaterials, as most biological activities proceed in a reversible manner. We also observed a similar reversible interaction in a recent experiment in which a cytokine was immobilized on a Ti surface using minTBP-1-programmed artificial proteins.45
Determining the function of motifs
Biomineralization is a process in which organisms form crystals (including amorphous ones) of inorganic material and use them as parts of their structures. The formation of seashells from calcium carbonate or formation of bone from calcium phosphate are typical biomineralization phenomena; other examples include the utilization of silica by sea urchins and magnetite grains by certain bacteria. It is well known that a variety of proteins are involved in biomineralization, and these mineralization-related proteins often contain repeats of short amino acid sequences. In other words, they are repositories of motifs. However, the mechanisms by which these proteins or the motifs regulate crystal growth are not yet fully understood. Clarification of these mechanisms will not only deepen our understanding of biological crystallization, but will also provide guidance for the application of biomineralization in bionanotechnology.46 Therefore, the synthesis of artificial proteins from the mineralization-related motifs should provide insight into how biomolecules regulate biomineralization. Of course, the research aimed at directly translating the biomineralization process into “bottom-up” nanofabrication that has recently come into the spotlight is one of the most relevant areas of research in nanobiotechnology.The NGX (Asn-Gly-Asp or Asn) motif (Table 1) was identified in nacrein, a major soluble organic matrix protein from the nacreous layer of pearls cultured in Pinctada fucata.47 Nacrein is a member of the carbonic anhydrase family of enzymes and has an idiosyncratic insertion domain comprised of about 130 amino acids arranged in simple repeats of NGX. It has been proposed that nacrein somehow modulates the calcification process, although no direct in vivo evidence has yet been reported. With the aim of gaining insight into the role of the NGX motif in calcification, we created a set of artificial proteins containing repeats of the Asn-Gly-Asx sequence (Fig. 5D). Among the created proteins, we found that some clones impeded the formation of CaCO3in vitro, indicating the ability of the NGX motif to modulate calcification. Notably, Turbo marmoratus, which belongs to a separate class of Pinctada, uses an NG repeat instead of the NGX motif present in the idiosyncratic domain of nacrein. Thus, two related organisms appear to have separately evolved appendix domains by respectively reiterating the Asn-Gly-Asx and Gly-Asn motifs. The resemblance of these motifs may imply that there is more than one solution for certain biomineralization activities.
Recently, we also applied our synthetic approach to a motif found in DMP1 (dentin matrix protein 1) (Table 1), which, as mentioned earlier, is an acidic protein expressed in bone and dentin (Fig. 5E). Previous work demonstrated that when immobilized on a glass substrate, peptides (motifs-A and B) corresponding to parts of DMP1 enhance HAP (hydrooxylapatitite) formation.12 Because immobilization of motifs represents an impediment to high-resolution physicochemical analysis, we initially used an artificial protein library prepared from DMP1-programmed proteins to search for clones that accelerate HAP formation without immobilization. After identifying several soluble proteins that accelerated HAP formation in vitro, we used them in time-resolved static light scattering analysis. Under the experimental conditions, amorphous calcium phosphate particles with radii of approximately 300 nm were formed during the initial phase before precipitation. In the presence of the motif-programmed proteins, the particles increased their fractal dimension and molecular weight without increasing their gyration radii during a short period prior to precipitation.48 This suggests that in the presence of appropriate motifs, amorphous calcium phosphate particles are directly transformed to HAP through rearrangement of their internal structural units.
Rational and irrational aspects of motif programming
The concept underlying motif programming is quite simple. If one wants to create molecules having functions A, B and C, one can just link three motifs respectively associated with those three functions. In a limited number of cases, this simple motif arithmetic does work. We have shown, for instance, that a peptide containing the BH3 motif could be fused to a protein transduction motif (poly Arg) to create a bifunctional peptide that penetrates into cells and induced apoptosis.49 In this experiment, however, the 9-amino acid BH3 motif was accompanied by flanking non-motif sequences (total 20 amino acids). Indeed, when the core BH3 motif was simply fused to the PTD motif, the conjugated peptide did not induce apoptosis, though it did enter cells.34 Similarly, simple conjugations of peptide motifs in our other experiments usually failed to reconstitute functional peptides.48,50 This difficulty in motif programming likely stems from the weakness of the transplantability and functional independence of the motifs. Apparently, motifs rely in large part on the context of their parental proteins for their functionality. Franking sequences may influence the structure of the motifs, which might be prerequisite for the functionality. Once they are extracted from their parent and put into a new context, motifs do not necessarily recapitulate their original functions. Because we do not know the appropriate context for expression of a given motif’s function, our combinatorial approach—i.e., selection of clones having expected functions from a library containing the motifs in various numbers and orders—helps us with motif-programming. In other words, motif-programming has both a rational and an irrational aspect. Based on the linkages between peptide sequences and their functions, we can rationally design artificial multifunctional proteins. But because we do not know the best arrangement of these motifs within polypeptides, we have to depend on an irrational selection-from-random strategy. Additionally, during motif programming experiments, we sometimes encounter unexpected functions that could not have been predicted from the functions associated with the motifs.44,51–53 Motif sequences may thus possess a capacity to evolve new functions within the context of a biological system.Summary
Motifs are minimal peptide units that exert biological functions. They can be extracted from natural proteins or artificially created using in vitro evolution systems. Based on the linkage between motifs and their functions, we can rationally program artificial proteins with a desired functionality. However, the linkage between a motif’s sequence and its function is often weak. Consequently, in many cases simple arithmetic addition does not work for motif-programming. The manifestation of associated functions is influenced substantially by the motif’s context. In practical terms, this capriciousness in motif-programming can be avoided by taking a combinatorial approach in which clones having the desired functions are selected from sequence pools prepared through the combinatorial linkage of motifs. These motif-programming experiments also deepen our understanding of the role of motifs in the evolution of proteins.References
- R. Milo, S. Shen-Orr, S. Itzkovitz, N. Kashtan, D. Chklovskii and U. Alon, Science, 2002, 298, 824–827 CrossRef CAS.
- C.-I. Brändén and J. Tooze, Introduction to Protein Structure, Garland Publishing, Inc., New York and London, 1991 Search PubMed.
- S. Ohno, Evolution by Gene Duplication, Springer, Heidelberg, 1970 Search PubMed.
- T. A. Webster, H. Tsai, M. Kula, G. A. Mackie and P. Schimmel, Science, 1984, 226, 1315–1317 CrossRef CAS.
- K. Shiba, J. Mol. Evol., 2002, 55, 727–733 CrossRef CAS.
- M. Long, Curr. Opin. Genet. Dev., 2001, 11, 673–680 CrossRef CAS.
- H. Saito, S. Kashida, T. Inoue and K. Shiba, Nucleic Acids Res., 2007, 35, 6357–6366 CrossRef CAS.
- E. S. Lander, L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar and M. Doyle, et al., Nature, 2001, 409, 860–921 CrossRef CAS.
- E. M. Marcotte, M. Pellegrini, T. O. Yeates and D. Eisenberg, J. Mol. Biol., 1999, 293, 151–160 CrossRef CAS.
- S. Ohno, J. Mol. Evol., 1984, 20, 313–321 CrossRef CAS.
- H. Tseng and H. Green, Cell, 1988, 54, 491–496 CrossRef CAS.
- G. He, T. Dahl, A. Veis and A. George, Nat. Mater., 2003, 2, 552–558 CrossRef CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS.
- J. K. Scott and G. P. Smith, Science, 1990, 249, 386–390 CrossRef CAS.
- C. F. Barbas III, D. R. Burton, J. K. Scott and G. J. Silverman, Phage Display: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 2001 Search PubMed.
- C. W. Stephen and D. P. Lane, J. Mol. Biol., 1992, 225, 577–583 CrossRef CAS.
- Y. C. J. Chen, K. Delbrook, C. Dealwis, L. Mimms, I. K. Mushahwar and W. Mandecki, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1997–2001 CrossRef CAS.
- R. Pasqualini and E. Ruoslahti, Nature, 1996, 380, 364–366 CrossRef CAS.
- W. Arap, R. Pasqualini and E. Ruoslahti, Science, 1998, 279, 377–380 CrossRef CAS.
- W. Arap, M. G. Kolonin, M. Trepel, J. Lahdenranta, M. Cardo-Vila, R. J. Giordano, P. J. Mintz, P. U. Ardelt and V. J. Yao, et al., Nat. Med., 2002, 8, 121–127 CrossRef CAS.
- S. R. Whaley, D. S. English, E. L. Hu, P. F. Barbara and A. M. Belcher, Nature, 2000, 405, 665–668 CrossRef CAS.
- C. Tamerler and M. Sarikaya, Acta Biomater., 2007, 3, 289–299 CrossRef.
- K. T. Nam, D. W. Kim, P. J. Yoo, C. Y. Chiang, N. Meethong, P. T. Hammond, Y. M. Chiang and A. M. Belcher, Science, 2006, 312, 885–888 CrossRef CAS.
- K. Sano, H. Sasaki and K. Shiba, J. Am. Chem. Soc., 2006, 128, 1717–1722 CrossRef CAS.
- E. Ruoslahti and M. D. Pierschbacher, Science, 1987, 238, 491–497 CrossRef CAS.
- S. Fong, L. V. Doyle, J. J. Devlin and M. V. Doyle, Drug Dev. Res., 1994, 33, 64–70 CrossRef CAS.
- V. Neduva, R. Linding, I. Su-Angrand, A. Stark, F. de Masi, T. J. Gibson, J. Lewis, L. Serrano and R. B. Russell, PLoS Biol., 2005, 3, e405 CrossRef.
- R. J. Edwards, N. Moran, M. Devocelle, A. Kiernan, G. Meade, W. Signac, M. Foy, S. D. Park and E. Dunne, et al., Nat. Chem. Biol., 2007, 3, 108–112 CrossRef CAS.
- K. Shiba, J. Mol. Catal. B: Enzym., 2004, 28, 145–153 CrossRef CAS.
- H. Saito, T. Minamisawa and K. Shiba, Nucleic Acids Res., 2007, 35, e38 CrossRef.
- J. T. Opferman and S. J. Korsmeyer, Nat. Immunol., 2003, 4, 410–415 CrossRef CAS.
- C. Moreau, P. F. Cartron, A. Hunt, K. Meflah, D. R. Green, G. Evan, F. M. Vallette and P. Juin, J. Biol. Chem., 2003, 278, 19426–19435 CrossRef CAS.
- A. Vocero-Akbani, M. A. Chellaiah, K. A. Hruska and S. F. Dowdy, Methods Enzymol., 2001, 332, 36–49 CAS.
- H. Saito, T. Honma, T. Minamisawa, K. Yamazaki, T. Noda, T. Yamori and K. Shiba, Chem. Biol., 2004, 11, 765–773 CrossRef CAS.
- K. Shiba, T. Takahashi and T. Noda, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 3805–3810 CrossRef CAS.
- W. Gilbert, Cold Spring Harbor Symposia on Quantitative Biology, 1987, 52, 901–905 CAS.
- W. P. C. Stemmer, Nature, 1994, 370, 389–391 CrossRef CAS.
- K. Nord, J. Nilsson, B. Nilsson, M. Uhlén and P. A. Nygren, Protein Eng., Des. Sel., 1995, 8, 601–608 CrossRef CAS.
- K. Shiba, T. Hatada and T. Noda, Protein Eng., 1996, 9, 813–814.
- H. Saito, T. Minamisawa, T. Yamori and K. Shiba, Cancer Science, 2008, 99, 398–406 Search PubMed.
- S. Shimizu, A. Konishi, T. Kodama and Y. Tsujimoto, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3100–3105 CrossRef CAS.
- K. Sano and K. Shiba, J. Am. Chem. Soc., 2003, 125, 14234–14235 CrossRef CAS.
- K. Sano, H. Sasaki and K. Shiba, Langmuir, 2005, 21, 3090–3095 CrossRef CAS.
- K. Kokubun, K. Kashiwagi, M. Yoshinari, T. Inoue and K. Shiba, Biomacromolecules, 2008, 9, 3098–3105 CrossRef CAS.
- K. Kashiwagi, T. Tsuji and K. Shiba, Biomaterials, 2009, 30, 1166–1175 CrossRef CAS.
- J. Aizenberg, D. A. Muller, J. L. Grazul and D. R. Hamann, Science, 2003, 299, 1205–1208 CrossRef CAS.
- H. Miyamoto, T. Miyashita, M. Okushima, S. Nakano, T. Morita and A. Matsushiro, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9657–9660 CrossRef CAS.
- T. Tsuji, K. Onuma, A. Yamamoto, M. Iijima and K. Shiba, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16866–16870 CrossRef CAS.
- A. Letai, M. C. Bassik, L. D. Walensky, M. D. Sorcinelli, S. Weiler and S. J. Korsmeyer, Cancer Cell, 2002, 2, 183–192 CrossRef CAS.
- K. Shiba and T. Minamisawa, Biomacromolecules, 2007, 8, 2659–2664 CrossRef CAS.
- K. Shiba, Y. Takahashi and T. Noda, J. Mol. Biol., 2002, 320, 833–840 CrossRef CAS.
- K. Shiba, T. Honma, T. Minamisawa, K. Nishiguchi and T. Noda, EMBO Rep., 2003, 4, 148–153 CrossRef CAS.
- K. Shiba, T. Shirai, T. Honma and T. Noda, Protein Eng., Des. Sel., 2003, 16, 57–63 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |