Dealloying to nanoporous Au/Pt alloys and their structure sensitive electrocatalytic properties†
Caixia
Xu
abd,
Rongyue
Wang
b,
Mingwei
Chen
c,
Yan
Zhang
b and
Yi
Ding
*ab
aKey Laboratory for Liquid-Solid Structural Evolution and Processing of Materials, Ministry of Education, Shandong University, Jinan 250061, China. E-mail: yding@sdu.edu.cn; Fax: +86-531-88366280; Tel: +86-531-88366513
bSchool of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China
cWPI Advanced Institute for Materials Research, Tohoku University, Sendai 980–8577, Japan
dSchool of Chemistry and Chemical Engineering, University of Jinan, Jinan 250022, China
First published on 10th November 2009
Abstract
A simple and general dealloying method is employed to fabricate nanoporous Au/Pt alloys with pre-determined alloy compositions. Structural characterization by electron microscopes demonstrates that selective etching of Cu from Au/Pt/Cu alloy precursors results in the formation of three-dimensional bicontinuous porous network structures with uniform pores and ligaments less than 10 nm. X-Ray photoelectron spectroscopy and X-ray diffraction demonstrate that nanoporous Au/Pt alloys have a single-phase cubic structure with relatively uniform compositions across the samples. These high surface area alloy nanostructures show much enhanced specific activity and distinct surface reactivity toward the electrooxidation of some small organic molecules, such as methanol and formic acid, as the Au content varies within the structure, thus holding great potential for use in clean energy and environmental applications.
1. Introduction
Nanostructured gold has attracted great interest due to its unique catalytic activities towards many important reactions, such as CO oxidation,1–3 water gas shift,4 and selective oxidation of hydrocarbons,5,6 and so on. Meanwhile, platinum is one of the most useful metals involved in the fields of fuel cell technology, chemical synthesis, and environmental protection, with extraordinary catalytic activities. While it has been well documented that alloying Pt with a second metal can often improve the catalytic activity of Pt as well as reduce the cost, it is only recently that people began to realize that the combination of Au with Pt represents a novel class of materials with interesting properties. For instance, Lou et al.7 observed enhanced methanol electrooxidation activity on Au/Pt alloy nanoparticles, and Zhou et al.8 recently found that dendritic Au/Pt nanoaggregates exhibited a remarkable CO-tolerance in hydrogen activation, as compared with pure Pt catalyst. Therefore, there are tremendous efforts for the development of new approaches to functional Au/Pt bimetallic nanostructures, which is of both scientific and technological importance.9At present, great efforts have been devoted to fabricate various Au/Pt bimetallic nanostructures, mostly in the form of alloy nanoparticles.10 These nanoparticles are usually prepared by simultaneous reduction of the respective metal salt precursors, with the assistance of surfactants and reducing agents, at relatively high temperatures. For instance, Xu et al. reported the synthesis of Au/Pt alloy nanoparticles by co-reduction of a N,N-dimethylformamide (DMF) coordinated Au3+–Pt4+ complex using DMF as the reducing agent.11 Zhou et al. prepared Au/Pt alloy nanoparticles (NPs) by rapid reduction of Au3+ and Pt4+ precursors with butyllithium in oleylamine at more than 200 °C.8 During these reactions, different reduction rates, nucleation, and agglomeration behaviors between Au and Pt interplay and contribute to the structure formation. Therefore, core-shell-type nanostructures are the most common products due to phase separation of Au and Pt.12,13 In addition, these methods are practically less feasible for pre-deciding the structure and composition of the resulting nanoalloys.12,14,15 Moreover, the massive use of organic agents may pose additional economic issues and environmental problems, thus hindering their industrial application. Consequently, it is highly desirable to search for an alternative, environmentally-benign strategy that can efficiently fabricate high surface area Au/Pt bimetallic nanostructures with tunable chemical compositions in high throughput.16
Recently, a simple alloy corrosion (also called dealloying) method has attracted increasing interest for creating functional nanoporous metals with tailored porosity.17 In particular, nanoporous gold (NPG) made by dealloying homogeneous Au/Ag alloys18,19 has received tremendous attention because it exhibits a series of intriguing properties in catalysis,20 sensing,21 and surface plasmon resonance.22 It should be emphasized that nearly one century ago, Raney employed a similar strategy to leach Al from Ni/Al alloys in alkaline solutions to produce high surface area metal dusts, which were very effective in catalytic hydrogenation reactions.23 Considering the great success of RANEY® metals and NPG, it is quite unexpected that so far dealloying has not been recognized as an effective method to prepare useful alloy nanocatalysts.24 Possibly due to the fact that corrosion has been traditionally regarded as a destroying process, research efforts in this field have for many years been focusing on analyzing and modeling this process from a thermodynamic or electrochemical viewpoint with an eye on its avoidance.25
In the current manuscript, we extend the dealloying method to multi-component alloy systems with an emphasis on the fabrication and characterization of an important class of catalytic nanostructures, nanoporous Au/Pt alloys (Au/Pt NPAs). By selectively leaching Cu from ternary Au/Pt/Cu source alloys, a series of highly porous Au/Pt alloy nanostructures were prepared with relatively uniform Au/Pt compositions, well controlled length scales and morphologies. These Au/Pt NPAs display distinct catalytic activities for electrooxidation of small organic molecules as the Au content varies. These results not only confirm the success in generating new nanostructures by expanding the known dealloying method, but also provide significant insights into the interplay between Au and Pt in important energy-related catalytic systems.
2. Experimental section
High purity (>99.99%) Pt, Au, and Cu were purchased from Changshu Noble Metal Company. Analytical reagent (AR) grade H2SO4, CH3OH and HCOOH were obtained from Sinopharm Chemical Reagent Co. Ltd and were used as received without further purification. The E-TEK Pt/C and PtRu/C catalysts were purchased from Aldrich with 20 and 28 wt% on carbon powder, respectively. For PtRu/C, the atomic ratio of Pt to Ru was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
Pt/Cu, Au/Cu, and Au/Pt/Cu alloy foils with different Pt/Au compositions were made by refining the corresponding high purity Pt, Au, and Cu (99.99%) at high temperatures under the protection of high purity argon in an arc furnace, and followed by melt-spinning. The relative composition of Pt and Au can be well controlled by adjusting the initial feed ratio. Dealloying of Cu was carried out under potential control on a CHI 1130A potentiostat in a conventional three-electrode electrochemical cell using a Pt foil as the counter electrode and saturated calomel electrode (SCE) as the reference electrode. As the working electrode, Au/Pt/Cu alloy foils were etched in 1 M H2SO4 solution at a desired potential (1.2–1.4 V vs. RHE) for 3–10 h at 0–60 °C. The electrolyte was renewed constantly to avoid complications from the Cu2+ ions generated during etching. The prepared Au/Pt NPAs were ground into powders and dried in a desiccator at room temperature in air. The catalyst suspensions were made by mechanically mixing 4.0 mg catalyst powder, 4.0 mg carbon powder, 200 μl isopropanol, and 200 μl Nafion solution (0.5 wt%). The commercial Pt/C and PtRu/C catalysts were prepared in a similar way except that further addition of carbon powder was not needed. All mixtures were sonicated for 30 min to form a uniform suspension. Catalyst ink was placed on a polished 4 mm diameter glassy carbon electrode as the working electrode for electrochemical testing.
Powder X-ray diffraction data were collected on a Bruker D8 ADVANCE X-ray diffractometer using Cu Kα radiation (λ = 1.5418 Å). The measurements were done in reflection geometry and the diffraction (Bragg) angles, 2θ, were scanned at a rate of 0.02 s−1. The structure and chemical composition of all samples were characterized on a JEOL JSM-6700F field emission scanning electron microscope (SEM), equipped with an Oxford INCA X-sight energy dispersive X-ray spectrometer (EDS). Transmission electron microscopy (TEM) images were obtained through a 300 kV field-emission Philips CM300FEG and a 200 kV JEM-2100 high-resolution transmission electron microscope. Au/Pt catalysts were sonicated and suspended in ethanol solution and were drop cast onto carbon-coated copper grids, followed by solvent evaporation in air at room temperature. The surface structures of these samples were analyzed with a VGESCALAB X-ray photoelectron spectrometer (XPS), using monochromatized Mg Kα X-ray as the excitation source, and choosing C 1s (284.60 eV) as the reference line.
All electrochemical measurements were performed on a CHI 760C potentiostat in a standard three-electrode cell with Pt foil serving as a counter electrode, and mercury sulfate electrode (MSE) or reverse hydrogen electrode (RHE) selected as the reference electrode depending on the experimental requirements. All potential values in this report were converted to the RHE scale for clarity. Prior to the measurements, the solutions were deoxygenated by bubbling with high-purity N2 for at least 30 min. All current densities were normalized by the electrochemical active surface areas of these Pt-based electrodes. The Pt surface area of the PtRu electrode catalyst is estimated from the CO stripping charge,26 while those of the rest of the catalysts were calculated by integrating the H desorption charge on platinum.27
3. Results and discussion
Three kinds of Au/Pt/Cu ternary alloys and two kinds of binary alloys (Au/Cu, Pt/Cu) were refined in an arc furnace, and then melt-spinned to make thin foils under a nitrogen-protected atmosphere. The composition of these ternary source alloys were analyzed by EDS (Fig. S1†) and summarized in Table 1, which proves that their compositions are in good agreement with the original feed compositions of Au/Pt ratios around 4:1, 1:1, 1:4, and the overall Cu content at ∼80 at%. For simplicity, they are denoted as Au16Pt4Cu80, Au10Pt10Cu80, and Au4Pt16Cu80.
| Sample | Au/Pt/Cu source alloys | Au/Pt NPAs | ||
|---|---|---|---|---|
| Feed composition (at%) | Bulk composition (by EDS, at%) | Bulk compisition (by EDS, at%) | Near surface composition (by XPS, at%) | |
| Au20Pt80 | Au4Pt16Cu80 | Au3.7Pt14Cu82.3 | Au21.6Pt76Cu2.4 | Au10Pt75Cu15 |
| Au50Pt50 | Au10Pt10Cu80 | Au11Pt10Cu79 | Au51Pt48Cu1 | Au30Pt58Cu12 |
| Au80Pt20 | Au16Pt4Cu80 | Au15.8Pt4.2Cu80 | Au72Pt25.7Cu2.3 | Au77Pt17Cu6 |
Fig. 1 shows the XRD patterns of the corresponding Au/Pt/Cu source alloys. For the Au4Pt16Cu80 alloy, the diffraction peaks at 42.2, 49.2, and 72.0 can be assigned to the (111), (200), and (220) diffractions of the face-centered cubic (fcc) structure. For the Au10Pt10Cu80 and Au16Pt4Cu80 alloys, the diffraction peaks for the (111), (200), and (220) planes shift slightly to lower angles, indicative of a lattice constant increase with an increase of Au content. It should be noted that in the latter two samples, there appears five additional diffraction peaks at 24.0, 34.1, 55.3, 61.1, and 76.9. Being quite low in intensity, they can be ascribed to the reflections of the (100), (110), (210), (211), and (300) planes, respectively, for an ordered AuCu3-type intermetallic structure.28 Because the (111), (200), and (220) diffractions are located at almost the same sites for these two types of structures, by comparing the obtained data with the standard XRD patterns (especially the relative intensity among peaks) for both ordered and disordered AuCu3 structures, it can be seen that these ternary source alloys are possibly mixed phases of these two structures, in particular for Au10Pt10Cu80 and Au16Pt4Cu80. The slight difference in ordering is related to the phase behaviors between source elements, which can be tuned by changing the preparation conditions. However, we will show below that the slight non-uniformity of the phase structure of alloy precursors does not affect the formation of uniform nanoporous alloys, because within the structures, Au, Pt, and Cu atoms are distributed uniformly at the sub-nanometer scale.
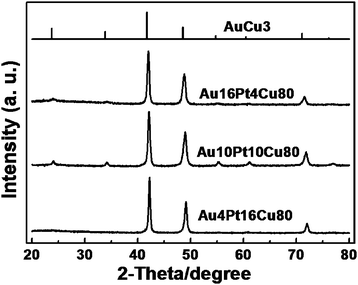 | ||
| Fig. 1 XRD patterns of Au/Pt/Cu source alloys. The standard pattern of intermetallic AuCu3 (JCPDS 65-3249) is attached at the top for comparison. | ||
Because Pt and Au are significantly more noble than Cu,27 either chemical or electrochemical dealloying methods can be employed to dissolve Cu from the source ternary alloys.29,30Fig. 2a illustrates a typical morphology of the resulting sample after dissolving Cu from the Au10Pt10Cu80 alloy, which is characterized by an open porous structure with a narrow pore and ligament size distribution of ∼5 nm, very similar to that of NPG.17 TEM provides more details for this structure, and the clear contrast between the dark skeletons and inner bright regions further indicates the formation of three-dimensional (3D) bicontinuous nanoporous structures (Fig. 2b). Fig. 2c provides a typical high resolution transmission electron microscopy (HRTEM) image for this structure, where the lattice spacing was calculated to be ∼0.233 nm, corresponding to the (111) crystal plane of the AuPt alloy structure. It is interesting to find that the lattice fringes extend continuously from one ligament to the other along (111) planes, indicating a single-crystalline structure across several layers of pores and ligaments within a grain typically orders of microns in size. While it is well known that NPGs made by dealloying Ag/Au alloys often adopt a single-crystalline porous grain structure,19 this interesting structural feature also exists in the present nanoporous Au/Pt system. The selected-area electron diffraction (SAED) image in the inset of Fig. 2b also reveals a single-crystal pattern recorded along the [110] zone axis direction. As show in Fig. 2d–i, dealloying of Au4Pt16Cu80 and Au16Pt4Cu80 alloys also resulted in similar 3D bicontinuous porous networks with ligament sizes averaging 4 and 9 nm, respectively. Accordingly, selected- area electron diffractions (shown as insets) resolve well defined diffraction spots that can be indexed to cubic crystallographic structures. A careful inspection shows that under the same dealloying conditions, nanoporous alloys with higher Pt contents generally give smaller feature sizes. And this trend can be extended to the dealloying of the binary Pt/Cu alloy (Pt20Cu80), where the resulting nanoporous Pt shows a beautiful 3D porous structure with feature sizes of around 3 nm (Fig. S2†), which is in good agreement with the literature value reported by Corcoran et al.31
 | ||
| Fig. 2 SEM, TEM, and HRTEM images of nanoporous alloys by dealloying (a, b, c) Au10Pt10Cu80, (d, e, f) Au4Pt16Cu80, and (g, h, i) Au16Pt4Cu80 alloys, respectively. | ||
This coarsening phenomenon in Au/Pt NPAs with an increase in Au content can be ascribed to the different surface diffusivity between Pt and Au along alloy/electrolyte interfaces.32 Because Pt has a much smaller diffusivity,33 during the dealloying process, it effectively restrains the structure from coarsening. To have an idea of how the presence of Pt may affect the pore formation and evolution of the resulting alloy structures, we have also examined the dealloying process of the Au20Cu80 alloy under the same etching conditions. As shown in Fig. S3,† the process also generated a similar 3D nanoporous structure, with however, a much coarsened feature size of around 25 nm. These observations are also consistent with Erlebacher’s recent results that doping a little Pt in an Au/Ag alloy could markedly reduce and stabilize the feature sizes of the resulting porous gold structures.34 Like NPG, the structures of Au/Pt NPAs are tunable in a controlled way from a few nanometers to microns simply by tailoring the processing conditions, such as dealloying and post-annealing parameters. Fig. 3 illustrated an Au20Pt80 NPA sample that was made by dealloying the Au10Pt10Cu80 ternary alloy in concentrated HNO3 for 48 h, and the resulting structure shows quite uniform spongy morphology with average ligament size of around 6 nm. Upon thermal annealing at 500 °C for 15 min, the structure sees a dramatic coarsening to a self-similar porous framework with a structure unit of around 50 nm. By controlling the annealing temperature or time, nanoporous Au/Pt alloys with a characteristic dimension of up to several microns can also be routinely obtained. In sharp contrast to other bimetallic nanostructures, the size-controllable and tunable character endows Au/Pt NPAs with rich opportunities for surface functionalization and structure regulation, which are highly beneficial for catalysis and sensing applications.
 | ||
| Fig. 3 SEM image of (a) the sample after dealloying the Au10Pt10Cu80 alloy in concentrated HNO3 for 48 h, and (b) the Au20Pt80 NPA after annealing at 500 °C for 15 min. | ||
It remains an interesting question if this dealloying process is just a surface phenomenon, considering that the alloy foils are quite thick (around 50 μm), and the resulting nanopores are very small. Imaging of the fractured surfaces shows that the bicontinuous porous structures occur through the entire thickness of the foils. Considering that Au and Pt in bulk have a big miscibility gap over a wide composition and temperature range,35,36 one may expect non-uniform composition variations across the entire structure. The compositional analysis with EDS for these three samples gives rather consistent compositional information over different areas and the nominal compositions of Au21.6Pt76Cu2.4, Au51Pt48Cu1, and Au72Pt25.7Cu2.3 (Table 1, and see Fig. S4†) indicate that most of the Cu has been leached out from the source alloys during dealloying.
Considering that XPS is more sensitive to the surface structures and properties, Fig. 4 presents the Pt 4f and Au 4f core level spectra for the Au/Pt NPAs. The binding energies of the Pt 4f7/2 region for Au20Pt80, Au50Pt50, and Au80Pt20 NPAs peak at 70.9, 70.8, and 70.6 eV, respectively, revealing a gradual shift to a lower binding energy with an increase in Au content, as compared with 71.2 eV for pure Pt.37 The 4f7/2 lines of Au for the Au/Pt NPAs are at 83.8, 83.8, and 83.9 eV, which also exhibit a slight downshift, relative to that of pure Au (84.0 eV), with an increase of Pt content. The shift of the binding energy for Pt and Au in Au/Pt NPAs could be ascribed to the charge transfer between them due to the formation of the Au/Pt alloy,38,39 which will be further supported by the XRD characterization discussed below. The near surface compositions of Au/Pt NPAs were obtained by analyzing the area under each core level peak based on XPS spectra. The metal composition within the detection depth of XPS was determined to be Au10Pt75Cu15, Au30Pt58Cu12, and Au77Pt17Cu6, respectively (see Table 1). Compared with the EDS results presented above, the Au20Pt80 and Au50Pt50 samples have seen a certain degree of Pt and Cu enrichment in the near surface region. The preferential segregation of active components, such as Cu, to the surface is understandable because the formation of more stable oxide provides the driving force for such kinds of surface segregation, which is frequently seen in other alloy nanostructures.40,41 Interestingly, for the Au80Pt20 alloy sample, dealloying seems to have induced a slight segregation of Au and Cu to the surface layer. Ruban42 and co-workers have systematically studied the surface segregation behaviors in transition metal alloys. And for the Au/Pt bimetallic system, they suggested a preferential formation of a core/shell structure with segregated Au on the surface, especially when the Pt content is low. However, due to the complexity of our structures, the exact mechanism remains unclear at the present stage.
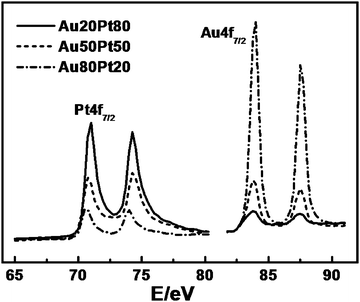 | ||
| Fig. 4 XPS spectra for Au/Pt NPAs in the Pt 4f and Au 4f regions. | ||
XRD was also used to monitor the structure formation of Au/Pt NPAs. From Fig. 5, it can be clearly observed that all samples show only a set of broad diffraction peaks, which can be assigned to the diffraction of (111), (200), (220) planes from an fcc structure, indicating the formation of alloy structures. As expected, the peak positions gradually shift to the pure Au (JCPDS 65-2870) side with an increase of Au content. While the respective ternary source alloys may contain some AuCu3-type intermetallic compounds, it is interesting to note that for Au50Pt50 and Au80Pt20 NPAs, their phase structures have undergone a natural transition from the initial ordered structure to the targeted fcc structure by the removal of Cu atoms. Despite the thermodynamic immiscibility of Pt and Au at low temperatures, here we observed the formation of relatively uniform Au/Pt alloy structures. This is actually not unusual because nanoscale alloying has been quite frequently observed in other systems, even at low temperatures.43 A careful inspection of the XRD pattern for the Au80Pt20 NPA reveals a noticeable tail at the lower-angle side. Taken together with the XPS results above and the theoretical calculations,40 we suggest that there exists an Au-rich surface layer in the Au80Pt20 NPA.
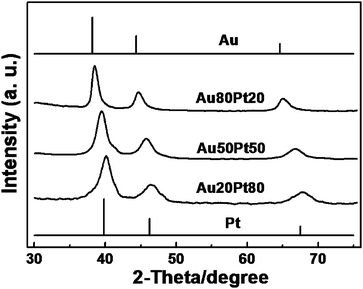 | ||
| Fig. 5 XRD patterns of the resulting Au/Pt NPAs. The standard patterns of pure Pt (JCPDS 65-2868) and Au (JCPDS 65-2870) are attached for comparison. | ||
Au/Pt NPAs represent a new class of high-surface-area alloy nanostructures, where the interconnected interstices and channels extending in all three dimensions allow unblocked transport of medium molecules and electrons, which is particularly desirable to catalysis. It is thus interesting to explore the electrocatalytic performance of Au/Pt NPAs by way of evaluating their potential in important energy-conversion applications, such as anode catalysts in fuel cell technology. Moreover, the exploration of their catalytic activities may provide fundamental insights into the synergistic effect between metal components as the composition varies. Fig. 6 shows the cyclic voltammetry (CV) profiles of Au/Pt NPA catalysts recorded in 0.5 M H2SO4 solution, which are characterized by a typical hydrogen region, electrochemical double-layer, and metal redox behaviors. The potential region between 0.05–0.35 V is associated with hydrogen adsorption/desorption (denoted hereafter as the under-potentially deposited hydrogen, Hupd) on Au/Pt NPAs, along with the adsorption/desorption of anions of the supporting electrolyte. Notably, the signal associated with Hupd on the catalyst surface gradually weakens as the Au content increases. The voltammetric features related to the reduction of Pt and Au oxides can be clearly observed in the backward scan.44 The comparison of the amplitudes of the reduction peaks for Pt and Au oxides roughly reflects the relative bimetallic compositions. Notably, no Cu dissolution was detected on any of the Au/Pt NPAs catalysts, indicating that most of the residual Cu atoms are buried within the ligament structures, which consequently means that more catalytically active precious metal atoms are exposed to the surface, thus favoring the enhancement of catalyst utilization.
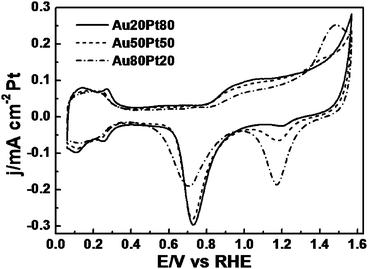 | ||
| Fig. 6 CV profiles of Au/Pt NPAs in 0.5 M H2SO4 solution at room temperature, Scan rate: 50 mV s−1. | ||
Fig. 7a presented the CV profiles of Au/Pt NPAs for the methanol oxidation reaction (MOR) together with that of the commercial PtRu/C catalyst. It can be clearly found that among all catalysts, there are evident differences for the current density and peak potential besides the deviation between forward- and backward-oxidation peaks. As reported in previous literatures, the sharp rising current in the forward scan can be ascribed to the characteristic methanol oxidation on the electrode surface, forming adsorbed carbonaceous intermediates, such as CO and HCO−, which will be oxidized at higher potential due to the formation of Pt–OH and Pt–O species.45 During the backward sweep, the desorption of OHads or reduction of Pt oxides regenerates the active metallic Pt surface which allows further oxidation of methanol molecules at lower potentials. However, the generated CO intermediates can not be removed due to the low electro-potential, which results in the poisoning of the catalyst, forming the second anodic peak at lower potential.46,47
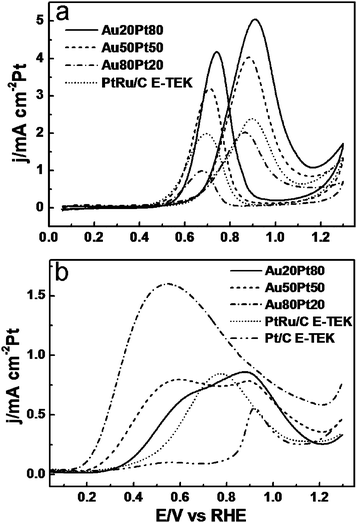 | ||
| Fig. 7 (a) CVs of MOR on Au/Pt NPAs and PtRu/C catalysts in 0.5 M H2SO4 + 1.0 M CH3OH solution. (b) The positive sweeping curves of Au/Pt NPAs, PtRu/C, and Pt/C catalysts in 0.5 M H2SO4 + 0.5 M HCOOH mixed solution, respectively. Scan rate: 50 mV s−1. | ||
During the methanol electrooxidation, the peak potential and current density for the forward anodic oxidation peak can be used to evaluate the catalytic activity of the electrocatalysts. As shown in Fig. 7a, the peak potentials of Au/Pt NPAs for the first (forward) anodic wave are located within a narrow range of 0.87–0.91 V, which is quite close to that of PtRu/C (0.89 V). The slight negative shift of the peak potential as the Au content increases might indicate more favorable reaction kinetics on Au-rich NPAs.48 Interestingly, the specific activity of Au/Pt NPAs takes the opposite trend and the activity radically improves when the Pt content increases. For the Au20Pt80 NPA, the specific activity reaches ∼5 mA cm−2 Pt, which is more than twice that of the PtRu/C catalyst (∼2.4 mA cm−2 Pt). It is well known that methanol electrooxidation occurs preferentially on electrode surfaces with continuous neighboring Pt sites.49 Therefore, when the Au content is higher in the Au/Pt alloy, a number of Pt atoms will exist in the form of monomers (single atoms), dimers, or groups of Pt atoms which do not provide effective dissociative adsorption of methanol molecules.50 Meanwhile, Au itself is much less active to MOR in acidic conditions, so the decline of activity can be correlated to the accumulation of more Au atoms on the surface. To summarize, compared with the commercial PtRu alloy nanocatalyst, Au/Pt NPAs with tailored surface structures exhibit distinct performances for MOR, such as improved reaction kinetics and enhanced oxidation activity. And in general, Au/Pt NPAs with higher Pt contents possess better catalytic activity towards MOR.
In recent years, direct formic acid fuel cell has been gaining increasing attention as a highly efficient and low toxicity power source. The catalytic activities of the prepared Au/Pt NPAs were thus evaluated for formic acid oxidation (FAO) along with that of commercial Pt/C and PtRu/C catalysts, included for comparison. For clarity, only the forward scan voltammetric curves were illustrated and the complete profiles were provided in Fig. S5.† As shown in Fig. 7b, the voltammetric response of Pt/C can be characterized by a dominant anodic peak at ∼0.92 V with a small shoulder at ∼0.56 V. For the PtRu/C catalyst, there appears one broad peak centered at 0.77 V. In comparison, all Au/Pt NPAs exhibit broad and highly specific activity over a wide range of 0.25–1.2 V. Specifically, for the Au20Pt80 sample, the oxidation peak mainly emerges at ∼0.90 V with a strong shoulder peak at ∼0.66 V, while for the Au50Pt50 NPA, it shows a saddle type of behavior with two adjoining oxidation peaks located at 0.63 and 0.92 V with a similar specific current intensity of ∼0.8 mA cm−2. The trend of negative potential shift for FAO as the Au content increases is the most pronounced for Au80Pt20, which shows a broad anodic peak centered at 0.58 V with a remarkable current density of ∼1.6 mA cm−2. The density value is an order of magnitude higher than those of Pt/C and PtRu/C at the same potential, and is nearly 2 and 3 times that of the peak current of PtRu/C and Pt/C (Table 2), respectively.
For the FAO reaction on Pt-based electrodes, a dual-path mechanism is usually adopted to describe the surface reactions, where the low-potential peak at ∼0.5 V is attributed to the direct oxidation of formic acid via a dehydrogenation process, and the high-potential peak at ∼0.9 V can be assigned to the oxidation of adsorbed CO species generated from a dehydration process.26,51 Previous studies have shown that this FAO reaction is highly sensitive to the actual surface structure of the Pt electrode, which is usually denoted as an ensemble effect.52 This ensemble effect was recently confirmed by density functional calculations and that for the FAO reaction on a Pt-based electrode, three neighboring Pt atoms are required in order for the dehydration path to occur, while the dehydrogenation path proceeds preferentially on isolated Pt atoms.53 This can explain well the observed results here because as the Au content increases, the surface Pt atoms can be effectively isolated by the neighboring Au atoms, which makes the dehydrogenation path dominant. Similar results have also been observed for Pt deposits on Au(111)26 and bimetallic Au/Pt nanoparticles.11
As to the distinct electrocatalytic performances on Au/Pt NPAs, the alloying of Au and Pt obviously leads to the modulation of the electronic structure of surface active sites, which should be the main reason for the markedly enhanced catalytic activities as compared to the commercial nanoparticle catalysts. In addition, Au/Pt NPAs, as a novel class of nanostructured electrode materials, have particular structural advantages which can be summarized as: (a) made by dealloying in aqueous solutions, NPAs have extremely clean surfaces readily available for surface electrode reactions; (b) possessing a bicontinuous porous network structure extending in all three dimensions, NPAs allow infinite opportunities for transport of electrons and target molecules, thus greatly facilitating the reaction kinetics; (c) being support free and particle free, NPAs completely eliminate the support corrosion problem and particle aggregation/sintering problem. Finally, the interaction between constituent metal elements may lead to special surface ensembles with unique structural properties, which can exhibit strong synergistic effects with much enhanced catalytic activities under appropriate conditions.
4. Conclusions
In summary, we demonstrated that dealloying can be extended to prepare nanoporous Au/Pt alloys with uniform size, controllable composition, and homogeneous structure. These novel Au/Pt nanostructures exhibit surface sensitive reactivity towards the electrooxidation of small organic molecules, which can be ascribed to their special structure characteristics which provide good electronic conductivity, and mass transport along the entire length scale. In view of functional optimization for practical applications, with this concept, we can readily obtain desired activities and properties by adjusting the initial feed ratio of source alloys and tailoring the dealloying process. It should be emphasized that this dealloying method is unusually simple and highly repeatable with nearly no loss of precious metals during fabrication. Involving only bench top processing in aqueous solutions, dealloying can be deemed as a “green” method to synthesise high-surface-area alloy nanostructures in high throughput. Considering that there exists nearly infinite amounts of multi-component alloys, the dealloying method is expected to be very versatile and powerful for the fabrication of other functional nanostructures, which should hold great promise for potential applications in energy- and environment-related science and technology.Acknowledgements
This work was supported by the National Science Foundation of China (20928003, 20906045, 90923011), the National 863 (2006AA03Z222) and 973 (2007CB936602) Program Projects of China, the Natural Science Foundation of Shandong Province (Y2007B33), and the Key Project of Chinese Ministry of Education (108078). Y.D. is a Tai-Shan Scholar supported by the SEM-NCET Program and the Shandong Natural Science Fund for Distinguished Young Scholars.References
- H. Yin, C. Wang, H. Zhu, S. H. Overbury, S. Sun and S. Dai, Chem. Commun., 2008, 4357 RSC.
- M. Haruta, N. Yamada, T. Kobayahsi and S. Iijima, J. Catal., 1989, 115, 301 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647 CrossRef CAS.
- H. Sakurai, A. Ueda, T. Kobayashi and M. Haruta, Chem. Commun., 1997, 271 RSC.
- Y. Murakami and K. Konishi, J. Am. Chem. Soc., 2007, 129, 14401 CrossRef CAS.
- M. D. Hughes, Y. J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. E. King, H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132 CrossRef CAS.
- Y. B. Lou, M. M. Maye, L. Han, J. Luo and C. J. Zhong, Chem. Commun., 2001, 473 RSC.
- S. H. Zhou, K. McIlwrath, G. S. Jackson and B. Eichhorn, J. Am. Chem. Soc., 2006, 128, 1780 CrossRef.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241 CrossRef CAS.
- M. Schrinner, S. Proch, Y. Mei, R. Kempe, N. Miyajima and M. Ballauff, Adv. Mater., 2008, 20, 1928 CrossRef CAS.
- J. B. Xu, T. S. Zhao, Z. X. Liang and L. D. Zhu, Chem. Mater., 2008, 20, 1688 CrossRef CAS.
- M. L. Wu, D. H. Chen and T. C. Huang, Chem. Mater., 2001, 13, 599 CrossRef CAS.
- P. Hernández-Fernández, S. Rojas, P. Ocón, J. L. Gómez de la Fuente, J. S. Fabián, J. Sanza, M. A. Peňa, F. J. García-García, P. Terreros and J. L. G. Fierro, J. Phys. Chem. C, 2007, 111, 2913 CrossRef CAS.
- T. S. Ahmadi, Z. L. Wang, T. C. Green, A. Henglein and M. A. El-Sayed, Science, 1996, 272, 1924 CrossRef CAS.
- H. Lang, S. Maldonado, K. J. Stevenson and B. D. Chandler, J. Am. Chem. Soc., 2004, 126, 12949 CrossRef CAS.
- D. Zhao and B. Xu, Angew. Chem., Int. Ed., 2006, 45, 4955 CrossRef CAS.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Nature, 2001, 410, 450 CrossRef CAS.
- Y. Ding and J. Erlebacher, J. Am. Chem. Soc., 2003, 125, 7772 CrossRef CAS.
- Y. Ding, Y. J. Kim and J. Erlebacher, Adv. Mater., 2004, 16, 1897 CrossRef CAS.
- C. X. Xu, J. X. Su, X. H. Xu, P. P. Liu, H. J. Zhao, F. Tian and Y. Ding, J. Am. Chem. Soc., 2007, 129, 42 CrossRef CAS.
- Z. Liu and P. C. Searson, J. Phys. Chem. B, 2006, 110, 4318 CrossRef CAS.
- J. Biener, G. W. Nyce, A. M. Hodge, M. M. Biener, A. V. Hamza and S. A. Maier, Adv. Mater., 2008, 20, 1211 CrossRef CAS.
- B. W. Hoffer and J. A. Moulijn, Appl. Catal., A, 2009, 352, 193 CrossRef CAS.
- S. Koh and P. Strasser, J. Am. Chem. Soc., 2007, 129, 12624 CrossRef CAS.
- R. C. Newman and K. Sieradzki, MRS Bull., 1999, 24, 12.
- J. Kim, C. Jung, C. K. Rhee and T. H. Lim, Langmuir, 2007, 23, 10831 CrossRef CAS.
- K. Kuśmierczyk, M. Lukaszewski, Z. Rogulski, H. Siwek, J. Kotowski and A. Czerwiński, Polish J. Chem., 2002, 76, 607 Search PubMed.
- H. Abe, F. Matsumoto, L. R. Alden, S. C. Warren, H. D. Abruña and F. J. DiSalvo, J. Am. Chem. Soc., 2008, 130, 5452 CrossRef CAS.
- P. Strasser, S. Koha and J. Greeley, Phys. Chem. Chem. Phys., 2008, 10, 3670 RSC.
- S. Koh, N. Hahn, C. Yu and P. Strasser, J. Electrochem. Soc., 2008, 155, B1281 CrossRef CAS.
- D. V. Pugh, A. Dursun and S. G. Corcoran, J. Mater. Res., 2003, 18, 216 CAS.
- J. Erlebacher, J. Electrochem. Soc., 2004, 151, C614 CrossRef CAS.
- D. V. Pugh, A. Dursun and S. G. Corcoran, J. Electrochem. Soc., 2005, 152, B455 CrossRef.
- J. Snyder, P. Asanithi, A. B. Dalton and J. Erlebacher, Adv. Mater., 2008, 20, 4883 CrossRef CAS.
- V. Ponec and G. C. Bond, Catalysis by Metals and Alloys, Elsevier, Amsterdam, 1995 Search PubMed.
- M. M. Mariscal, S. A. Dassie and E. P. M. Leiva, J. Chem. Phys., 2005, 123, 184505 CrossRef.
- Handbook of X-ray Photoelectron Spectroscopy, ed. J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Physical Electronics, Inc., Eden Prairie, USA, 1995, p. 181 Search PubMed.
- Z. Bastl and S. Pick, Surf. Sci., 2004, 566–568, 832 CrossRef CAS.
- X. Ge, X. Yan, R. Wang, F. Tian and Y. Ding, J. Phys. Chem. C, 2009, 113, 7379 CrossRef CAS.
- V. Zielasek, B. Jurgens, C. Schulz, J. Biener, M. M. Biener, A. V. Hamza and M. Bäumer, Angew. Chem., Int. Ed., 2006, 45, 8241 CrossRef CAS.
- F. Tao, M. E. Grass, Y. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932 CrossRef CAS.
- A. V. Ruban, H. L. Skriver and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15990 CrossRef.
- J. Luo, M. M. Maye, V. Petkov, N. N. Kariuki, L. Wang, P. Njoki, D. Mott, Y. Lin and C. J. Zhong, Chem. Mater., 2005, 17, 3086 CrossRef CAS.
- H. Möller and P. C. Pistorius, J. Electroanal. Chem., 2004, 570, 243 CrossRef CAS.
- T. J. Schmidt, H. A. Gasteiger and B. J. Behm, Electrochem. Commun., 1999, 1, 1 CrossRef CAS.
- N. M. Markovic and P. N. Ross, Surf. Sci. Rep., 2002, 45, 117 CrossRef CAS.
- H. A. Gasteiger, N. Markovic, P. N. Ross and E. J. Cairns, J. Electrochem. Soc., 1994, 141, 1795 CAS.
- S. Sriramulu, T. D. Jarvi and E. M. Stuve, Electrochim. Acta, 1998, 44, 1127–1134 CrossRef CAS.
- T. Iwasita, Electrochim. Acta, 2002, 47, 3663 CrossRef CAS.
- B. Du and Y. Tong, J. Phys. Chem. B, 2005, 109, 17775 CrossRef CAS.
- H. Okamoto, W. Kon and Y. Mukouyama, J. Phys. Chem. B, 2005, 109, 15659 CrossRef CAS.
- S. Park, Y. Xie and M. J. Weaver, Langmuir, 2002, 18, 5792 CrossRef CAS.
- M. Neurock, M. Janik and A. Wieckowski, Faraday Discuss., 2009, 140, 363 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: EDS compositional analysis of Au/Pt/Cu source alloys, SEM images of dealloyed Pt20Cu80 and Au20Cu80 samples, EDS compositional analysis of the resulting Au/Pt NPAs, CVs of Au/Pt NPAs and Pt/C catalysts in HCOOH mixed solutions. See DOI: 10.1039/b917788d |
| This journal is © the Owner Societies 2010 |