Virial coefficients and equation of state of the penetrable sphere model†
Linda
Viererblová
,
Jiří
Kolafa
*,
Stanislav
Labík
and
Anatol
Malijevský
Department of Physical Chemistry, Institute of Chemical Technology, Prague, 166 28 Praha 6, Czech Republic. E-mail: jiri.kolafa@vscht.cz
First published on 7th November 2009
Abstract
We study the penetrable sphere (alias square mound) model in the fluid phase by means of the virial expansion, molecular dynamics simulations, and Ornstein–Zernike integral equation. The virial coefficients up to B8 are expressed as polynomials in the Boltzmann factor with the coefficients calculated by a Monte Carlo integration. New data for pressure and internal energy are obtained by molecular dynamics simulations with attention paid to finite-size errors and properties of the Andersen thermostat. The data and virial coefficients are correlated by a formula for the Helmholtz free energy. We also propose a new closure for the Ornstein–Zernike equation and test several other closures.
1. Introduction
The penetrable sphere model (PSM), called also the square mound potential,1 is an extension of the hard sphere model in which the overlap of spheres is penalized by a finite constant energy. It qualitatively describes the effective interaction of colloidal particles like polymer globules or star polymers in a suitable solvent.2,3 The polymeric chains may interweave, which is entropically penalized. Another popular example of these so called bounded potentials is the Gaussian core model introduced by Stillinger.4The PSM became popular after 1989 due to Marquest and Witten,5 who suggested it as a prototype for the interaction between micelles in a solvent. Since then, the PSM has been studied by a variety of methods. Virial coefficients up to the fifth were calculated by Katsura and Aby.1 Structural and thermodynamical properties have been studied using integral equation theories based on the Ornstein–Zernike equation with Percus–Yevick and hypernetted-chain closures.6,7 More sophisticated closures tailored to the PSM were also suggested.8–10
Likos et al. studied the phase diagram using a combination of cell and density functional theory (DFT) for the solid phase together with integral equations and simulations for the fluid phase.7 Besides freezing transition, they also described the second-order clustering transitions in a solid. DFT based on the bridge density functional and the fundamental-measure theory have been also used to investigate the structural properties of one-component and two-component penetrable spheres in a spherical pore.11
Khampour and Hashim recently proposed an equation of state for the penetrable spheres fluid based on the properties of an equivalent hard sphere fluid as defined by the perturbation theory of simple liquids.12
Many other original approaches have been applied to the penetrable sphere model.13,14 Recent contributions include the density expansion of the radial distribution function,6 low- and high-temperature limits by integral equation theories15 and kinetic properties.16
In section 2 we first review the necessary theory to calculate virial coefficients, to perform molecular dynamical simulations and estimate finite-size errors, and to calculate the structure from the Ornstein–Zernike equation. Results are commented on in section 3. Extensive tables of results are available as ESI†.
2. Theory
2.1 The penetrable sphere model
The penetrable sphere model (PSM) is defined by the pair potential | (1) |
We note that constant ε must not be negative. A model with ε < 0 (attractive penetrable spheres) does not have a thermodynamic limit in the usual sense – all particles condense to one blob whose size depends on the number of particles N and absolute temperature T.
2.2 Virial expansion
 | (2) |
Values of other thermodynamic quantities can be also obtained from the virial expansion.17 By integrating the compressibility factor we get the residual (with respect to the ideal gas, also called excess) Helmholtz energy (free energy) per particle Ares,
 | (3) |
 | (4) |
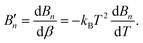 | (5) |
 | (6) |
 | (7) |
 | (8) |
Throughout the paper we will use the reduced quantities: reduced number density ρ* = ρσ3, reduced temperature T* = kBT/ε, reduced energy U* = U/ε, and reduced time t* = (m/ε)1/2σ. To simplify notation, we will omit the superscripts *. Loosely, we assume σ = 1, ε/kB = 1 and particle mass m = 1.
In addition, it is often advantageous to use the packing fraction,
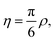 | (9) |
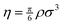 ) instead of density. The packing fraction based virial coefficients are then
) instead of density. The packing fraction based virial coefficients are then | (10) |
The first four virial coefficients of the PSM are known analytically,18
![[B with combining tilde]](https://www.rsc.org/images/entities/i_char_0042_0303.gif) 2 = −4F, 3 = −10F3, 2 = −4F, 3 = −10F3, | (11) |
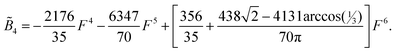 | (12) |
The higher virial coefficients are not known analytically and have to be calculated numerically. The minimum number of f-bonds in a Mayer diagram with n points is n, while the maximum is  . The n-th virial coefficient is then a polynomial in F,
. The n-th virial coefficient is then a polynomial in F,
 | (13) |
Our algorithm is an extension of our previous work on hard spheres and disks.19 Here we briefly review basic ideas.
The first step is a transformation of the Mayer diagrams to the Ree–Hoover (RH) diagrams.20 Pairs of black points not connected by a bond in the Mayer diagrams are assigned factor 1 in the corresponding integral in eqn (7). This factor can be expanded to 1 = e − f. The set of Mayer diagrams is thus replaced by a set of RH diagrams composed of e- and f-bonds (and no unconnected pair). The value of the integral corresponding to a RH diagram is multiplied by the so-called RH weight. It appears that the number of the RH diagrams (with nonzero weight) is actually smaller than of the Mayer diagrams. We performed the topological analysis up to B9,19 and stored the results including the spanning tree analysis (see below) in the form of files protected by checksums.
The numerical integration to obtain Bn is based on random sampling a certain configuration of n spheres represented by a spanning diagram (connected subgraph); e.g., for n = 4 this was a chain, 1–2–3–4, where a bond (dash) denotes an overlap. We generated the configurations of chains by reptation21 and the configurations of more complex spanning diagrams by Monte Carlo simulation.19 More overlaps (e.g., 1–3 and 2–4) may occur in the random configuration. These overlaps can be described by a diagram with an overlap denoted by a bond. In a computer code this diagram is represented by a binary number where each pair of spheres (not connected in the spanning diagram) is represented by one binary digit adopting 1 if the spheres overlap and 0 otherwise. This number then serves as an index to an array (histogram) which accumulates counts corresponding to the configurations.
For hard molecules a configuration can contribute to the value of a RH diagram only if the RH diagram matches the diagram of the configuration (f-bond = overlap, e-bond = no overlap) because for every overlap e = 0 while for a non-overlap f = 0. This is actually the main advantage of the RH representation for hard particles. A computer code based on the Mayer diagrams would have to include summing many histogram bins. Thus Bn is calculated from a sum over the histogram bins, weighted by the RH weights and divided by the so-called unlabeling factors,19 and multiplied by the value of the spanning diagram.
There are two points to consider. First, molecules in a computer program must be numbered (labeled), as in the spanning chain above. A labeled spanning diagram is therefore laid over all possible labelings (numberings) of a RH diagram; this has been done in advance as mentioned above. Several different configurations may thus contribute to the value of one RH diagram; the number of these configurations is the above mentioned unlabeling factor. Second, for n > 5 one spanning diagram is not sufficient to sample all RH diagrams, and several spanning diagrams must be combined. The technology is explained in detail in ref. 19.
Counting the configurations for the PSM is not as simple as for hard bodies because overlaps are only partial, see Table 1. For hard spheres only the selected configurations (shown in a box) have to be counted to calculate Bn because of the one-to-one correspondence between RH diagrams and configurations. For penetrable spheres all configurations with more bonds (overlaps) also have to be counted because the Boltzmann factor eij in the RH integral is no longer zero but E = F + 1. All these additional configurations can be obtained from an RH diagram by generating all subsets of its e-bonds and replacing those e-bonds (alias non-overlaps) by f-bonds.
| Spanning diagrama | RH diagramb | Configurationc | |||
|---|---|---|---|---|---|
|
|
|
|
|
||
| a A line denotes an f-bond if the diagram value is calculated or an overlap if a configuration is generated; no line has no meaning. b A line denotes an f-bond, no line an e-bond. c A line denotes an overlap of spheres, no line missing overlap. | |||||

|

|

|
0 | 0 | 0 |

|

|

|
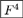
|
0 | |




Our algorithm then proceeded in two separate passes. In the first pass, look-up tables of all needed configurations were calculated and then the Monte Carlo integration was performed. The first pass thus produced tables of counts (probabilities) with which the needed configurations appeared in the sample. To speed up the calculations, these computer programs were generated automatically for each spanning tree with a static code for testing all remaining overlaps. In the second pass all the accumulated contributions were counted and results from all spanning diagrams summed. Because of the large number of diagrams, the maximum virial we managed to calculate was B8.
Alternatively, we could have started from the Mayer diagrams instead of the RH diagrams. In this case the second line of Table 1 would contain contributions F6, F5, F4, which would simplify the second pass. However, the number of the RH diagrams is smaller than the number of the Mayer diagrams. It is not clear which approach is more efficient.
We used a four-tap register-shift random number generator22 extensively tested in our previous work23 for the MC integration. The MC calculations were performed in blocks of 4 × 109 spanning configurations. About 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 blocks were generated in total. Details on merging the results over several spanning diagrams and on estimating the errors are given in our previous work.19
000 blocks were generated in total. Details on merging the results over several spanning diagrams and on estimating the errors are given in our previous work.19
2.3 Molecular dynamics
Point (i) is controlled by events. All possible events (like a possible particle collision, measurement, change of velocity by a thermostat, stop simulation, etc.) are organized in time slices so that the forthcoming event can be easily chosen. The events in time slices are implemented as an unsorted linked-list. More technical details can be found in our previous work.23
As regards point (ii), three types of collisions may occur for a pair of colliding penetrable spheres:16
1. The particles initially do not overlap and their radial kinetic energy (calculated from the radial components of both velocities with respect to the center of mass) is less than the potential barrier ε. The particles then reflect as hard spheres; their velocity components perpendicular to their tie-line are preserved while the parallel components revert sign.
2. The particles initially do not overlap and their radial kinetic energy is greater than the potential barrier ε. The particles then “refract in”; their velocity components perpendicular to their tie-line are again preserved while the parallel components can be easily calculated from the conservation of momentum and energy.
3. The particles initially overlap. They then “refract out”.
To calculate collisions of particles in point (iii), a cutoff sphere is drawn around each particle. Only particles within this cutoff sphere are checked for a possible collision. A variant of the linked-cell list method24,25 is used to scan all neighbors lying within the cutoff. In addition, the event “particle leaves its cutoff sphere” is scheduled and if it happens, all collisions are recalculated. The longer cutoff, the more precalculated collisions are wasted because the particles have collided with another particle; the shorter cutoff, the more frequent recalculation of all collisions. Therefore there is an optimum cutoff value, so are an optimum cell size and time slice.
Particles are placed in a cubic box of volume V and subject to the periodic boundary conditions. The initial configuration is a lattice with possible overlaps or vacancies, the initial velocities are drawn from the Maxwell–Boltzmann distribution. Production runs start after a period of carefully monitored equilibration.
A constant temperature is maintained by the Andersen thermostat.24,25 A random particle is chosen at regular time intervals tA/N, where tA is the thermostat time constant, and new velocities are assigned according to the Maxwell–Boltzmann distribution; then, collisions are recalculated. We used a high-quality four-tap register-shift random number generator22 so that the reported artifacts26 can be excluded.
The compressibility factor was calculated from the virial of force,24
 | (14) |
Another way how to get the compressibility factor is via the radial distribution function (RDF), g(r),
 | (15) |
| y(r) = eβu(r)g(r). | (16) |
 | (17) |
2.4 Ornstein–Zernike equation
Structural and thermodynamic properties of fluids can be predicted by integral equation theories.17 The most common approach is based on the Ornstein–Zernike (OZ) equation, | (18) |
To solve the OZ equation, another relation between h and c is needed. This so-called closure has a general form
| g(r) = exp[−βu(r) + γ(r) + B(r)], | (19) |
| γ(r) = h(r) − c(r) | (20) |
The simplest approximation is the hypernetted chain (HNC) closure, where the bridge function is assumed to be zero,
| B(r) = 0. | (21) |
| B(r) = ln[γ(r) + 1] − γ(r). | (22) |
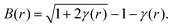 | (23) |
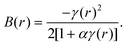 | (24) |
 | (25) |
 | (26) |
| α = 1.0185eT − 0.2685ρ. | (27) |
| B(r) = F5BHS(r). | (28) |
3. Results and discussion
3.1 Virial coefficients
The virial coefficients for the ρ expansion are shown in Fig. 1; we remind the reader that we assume σ = 1. The curves start at T = 0 (hard sphere limit) and at very low temperatures are indistinguishable from the hard sphere values. On the other hand, at high temperatures (high probability of overlap) they tend to zero. Coefficients B2 and B3 are positive (they are positive for all positive potentials), the higher coefficients are negative at some temperature regions. The shape of the curves suggests that the convergence of the virial expansion will be bad for temperatures in the range about [0.1,0.5]. | ||
| Fig. 1 Virial coefficients for the ρ expansion as a function of reduced temperature T. | ||
The B2 and B3 coefficients are similar in shape as for the Gaussian core model;31 higher coefficients, which in principle could be negative, are not available.
Full tables of the calculated coefficients Bn,i of polynomials (13) are accessible as ESI along with a computer program (in C) to calculate the virial coefficients Bn, truncated virial expansions, and finite size corrections†. These tables also include error estimates of Bn,i. For B4 to B7 tables of functions Bn(F) for −1 ≤ F ≤ 1 are also given along with the error estimates. Since the errors of Bn,i are highly correlated, it is not possible to calculate error estimates of Bn(F) from the errors of Bn,i. Combining the errors of Bn,i as uncorrelated gives a very pessimistic estimate of the error of Bn(T). For instance, for F = −1 (hard sphere) the direct calculation gives B7 = 53.3401 ± 0.0019 while the “uncorrelated error” is ±7.7 because the sum is composed of large alternating contributions.
In Table 2 we compare the calculated virial coefficients with selected known data. These include the virial coefficients of the hard sphere fluid (the T = 0 limit) up to B8 published by several authors. The comparison with the analytical results for B4 also successfully passed. Some deviations of B5,i from the old data1 are commensurate with the precision then available.
| Quantity | This work | Literature | Ref. |
|---|---|---|---|
|
a There is a typo in ref. 32, Table 2.2. The correct weighted average of values from ref. 19 and 33 is 8(0) = 68.5394(88).
|
|||
|
3(0) |
9.999990(6) | 10 | 32 |
|
4(0) |
18.36480(3) | 18.36477 | |
|
5(0) |
28.22441(8) | 28.22445(10) | |
|
6(0) |
39.81541(34) | 39.81545(34) | |
|
7(0) |
53.3401(19) | 53.3418(15) | |
|
8(0) |
68.563 | 68.539(9)a | |
|
4,4
|
−62.17149(5) | −62.17143 | 6 |
|
4,5
|
−90.67153(9) | −90.67143 | |
|
4,6
|
−10.135247(14) | −10.135232 | |
|
5,5
|
−389.9907(2) | −389.99 | 1 |
|
5,6
|
−1503.1437(11) | −1503.14 | |
|
5,7
|
−1555.6541(13) | −1555.22 | |
|
5,8
|
−486.2638(5) | −485.74 | |
|
5,9
|
−78.0563(1) | −77.81 | |
|
5,10
|
−6.0692(0) | −6.04 |
| t A | Z vir | Z RDF | U res | T |
|---|---|---|---|---|
| N = 100 | ||||
| 0.5 | 1.90257(6) | 1.89683(16) | 0.25715(3) | 0.60006(3) |
| 1 | 1.90232(5) | 1.89670(15) | 0.25717(3) | 0.60000(3) |
| 2 | 1.90218(6) | 1.89708(15) | 0.25715(3) | 0.59994(4) |
| 3 | 1.90192(6) | 1.89718(14) | 0.25715(3) | 0.60001(5) |
| 5 | 1.90177(7) | 1.89674(16) | 0.25714(4) | 0.59998(6) |
| 8 | 1.90151(8) | 1.89667(14) | 0.25722(4) | 0.60008(7) |
| N = 200 | ||||
| 0.5 | 1.90175(4) | 1.89903(13) | 0.25794(2) | 0.60003(3) |
| 8 | 1.90125(7) | 1.89892(13) | 0.25793(4) | 0.59999(6) |
|
N = 20000 |
||||
| 3 | 1.90096(3) | 1.90089(7) | 0.25870(2) | 0.59999(2) |
3.2 Molecular simulations
We performed most simulations with N = 20000 particles. Even though this number of particles is large, it is necessary to take into account possible finite-size errors.
In Fig. 2 we analyze the N-dependence of the compressibility factor for T = 0.6 and ρ = 0.4. It is seen that the molecular dynamics results for Z based on the RDF, (15), match the Monte Carlo results. In addition, these results match the theoretical 1/N dependence (17) as calculated from the equation of state (see section 3). Since the RDF is calculated entirely from configurations, we have thus verified that the Andersen thermostat provides the canonical distribution of configurations. Similar results have been obtained for the residual internal energy (see the ESI†).
 | ||
| Fig. 2 The compressibility factor in dependence on 1/N for T = 0.6 and ρ = 0.4. Both routes, virial (14) and via the RDF (15), are shown for MD while for standard canonical (NVT) and grand canonical (μVT) MC only RDF-based results are available. The theoretical NVT slope is given by eqn (17). | ||
We also ran two grand canonical (μVT) Monte Carlo simulations to verify that there is no ensemble error of the order 1/N in this ensemble, as assumed by formula (17). The chemical potential was set so that the averaged number of particles was 100 and 200, respectively. The results are not distinguishable within the error bar from the thermodynamic limit.
The compressibility factor calculated from the virial of force shows a different N-dependence, although the thermodynamic limit 1/N = 0 is the same. To elucidate this phenomenon further, we ran several simulations for the same system and N = 100 and 200, but for various thermostat time constants tA, see Table 3. The results show that the RDF-based compressibility factor, energy, and averaged temperature do not depend on the velocity update time. The same applies to the averaged kinetic energy (expressed via the kinetic temperature) which matches the thermostat temperature T = 0.6. However, the virial of force shows a pronounced dependence on the thermostat time constant.
Consequently, the Andersen thermostat does not provide the correct canonical correlations between coordinates and momenta. Rather, it can be viewed as a set of short microcanonical samples with different energies. The error is of the order of 1/N.
A further examination shows that for N = 20000 the statistical errors are comparable to the finite-size errors only at the lowest density ρ = 0.1. For larger densities the systematic finite-size errors are several times smaller than the statistical errors. We therefore did not include any finite-size correction in the data.
Fig. 3 and 4 show the compressibility factor and residual internal energy, respectively, as functions of density. Full tables of the results are provided as ESI†.
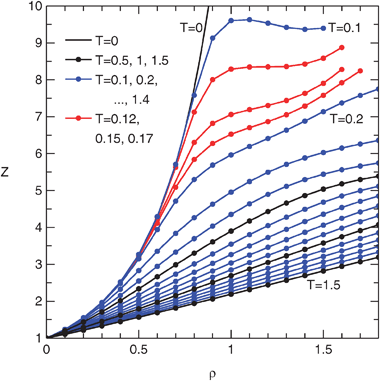 | ||
| Fig. 3 Compressibility factor isotherms of the penetrable sphere fluid from MD simulations. | ||
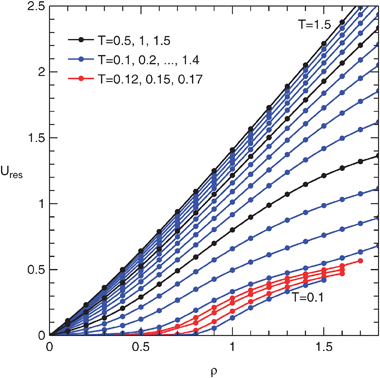 | ||
| Fig. 4 Residual internal energy of the penetrable sphere fluid from MD simulations. | ||
It is seen that the behavior of the fluid is unusual at low temperatures. The particles are almost hard, and therefore at low densities the fluid is similar to the hard sphere fluid. The energy is negligible, i.e., only rarely a pair of overlapping spheres is found. As the density increases, the chemical potential grows as well, eventually overcoming the repulsion. A typical configuration consists of pairs (dumbbells) of particles; the compressibility factor drops and the energy approaches 1/2 per particle. At even higher densities triples etc. form. It should be noted that the fluid is in a metastable state, but we have not observed spontaneous freezing.
At slightly higher temperatures the fluid behavior is not so singular and various clusters of particles can be detected. In Fig. 5 we show the dependence of a clustering probability on density for T = 0.17. The clustering probability of cluster C is defined as the probability that a randomly chosen particle is a part of this cluster, or equivalently by p(C) = 〈|C|N(C)〉/N, where |C| is the cluster size (number of spheres connected by an overlap) and N(C) the number of such clusters found in a configuration; thus  . It is seen that the number of monomers decreases with density while the number of higher oligomers increases; relatively stable dimers are a subject of both trends and their number passes through a maximum. Trimers exhibit the same tendency towards clustering: linear trimers prevail at low density while cyclic ones thrive as density increases. At higher temperatures and sufficiently high density a percolation occurs, i.e., most particles are connected in one large cluster. The graphical abstract shows a small (N = 512) sample configuration for T = 0.17 and ρ = 1 with color-coded clusters.
. It is seen that the number of monomers decreases with density while the number of higher oligomers increases; relatively stable dimers are a subject of both trends and their number passes through a maximum. Trimers exhibit the same tendency towards clustering: linear trimers prevail at low density while cyclic ones thrive as density increases. At higher temperatures and sufficiently high density a percolation occurs, i.e., most particles are connected in one large cluster. The graphical abstract shows a small (N = 512) sample configuration for T = 0.17 and ρ = 1 with color-coded clusters.
 | ||
| Fig. 5 Clustering probabilities p(C) as a function of density for T = 0.17. | ||
3.3 Equation of state
The simplest equation of state is the virial equation. Solid black lines in Fig. 6 show the differences of this equation truncated at B8 from the simulation data. Since at low densities the virial equation matches closely the data, the data are consistent with the virial coefficients. | ||
| Fig. 6 Deviations of compressibility factors calculated by various EOSs from the MD data. Black: virial expansion, color: eqn (29) with various maximum densities ρmax (green: 0.6, red: 0.7, blue: 0.8). The curves are shifted by multiples of 0.5. | ||
We have developed an equation of state based on a combination of the effective hard sphere approach by Khanpour and Hasim12 and the popular Carnahan–Starling-like expansion in η/(1 − η).
To do this, we write the compressibility factor in the form
 | (29) |
 | (30) |
Different forms of Q(T) were tested, for example a polynomial in T. Finally we fitted the simulation data for each temperature separately (which is not exactly the same because the T-dependence propagates to Ures, cf.eqn (4)) to obtain the optimal values of Q(T) and then suggested the appropriate form
| Q = be−a/T − 1, | (31) |
The data on the compressibility factor and potential energy were fitted simultaneously by the least-square method in the whole temperature range and various density ranges ρ ≤ ρmax. The quality of the fit deteriorates with increasing density and also with smaller number of terms considered in eqn (29). The optimized parameters are a = 0.6483, b = 6.8352 for ρmax = 0.8, a = 0.6273, b = 7.1452 for ρmax = 0.7, and a = 0.6228, b = 7.7922 for ρmax = 0.6.
Fig. 6 shows the differences of various EOSs from the simulation data. It is seen that for large temperatures (and also for T = 0) the proposed eqn (29) is considerably better than the virial expansion. It is not, however, true for small temperatures, the worst case being T = 0.1.
The equation nevertheless enabled us to calculate a reliable estimate of the finite-size correction term shown in Fig. 2.
3.4 The Ornstein–Zernike equation
The OZ equation was solved by technology explained in ref. 34. Only densities up to ρ = 1 were considered. No solution was found with the HNC closure in the region of small but nonzero temperatures and high densities (T∈[0.1,0.17] and ρ ≥ 0.8, T = 0.2 and ρ ≥ 0.9, and T = 0.3 and ρ = 1). In addition, the CG closure does not converge at T = 0 for ρ ≥ 0.9 and the MS closure for ρ ≥ 1 (which was interpreted as an indication of freezing18).In order to compare performance of closures at a number of state points we used the mean quadratic relative error of the radial distribution function at interval [0.5,2] defined by
 | (32) |
Seven closures are compared in Fig. 7. The proposed HSB closure works best at low densities, ρ ≤ 0.2; at lower temperatures its performance extends to slightly higher densities and at T = 0 it is exact because it uses the hard sphere bridge function. This is exactly what one would expect from its construction based on the ρ-expansion. A practical impact of this closure is limited because at low densities all closures work well enough for most purposes. At higher densities a simple scaling the hard sphere bridge function (28) is not appropriate because more diagrams become important, which should be scaled by higher powers of F. This closure is in fact the worst one at high densities and temperatures.
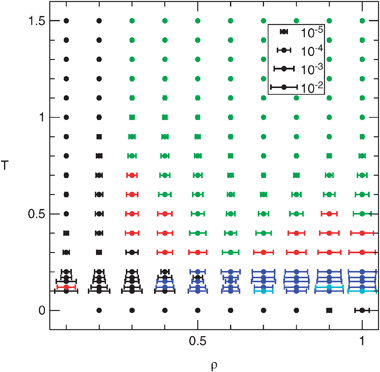 | ||
| Fig. 7 The best closure as a function of density and temperature. Black: HSB, blue: ZHOU, red: CG, green: HNC, cyan: VM; PY and MS are never the best. See section 2.4 for the acronyms. The value of the mean quadratic relative deviation of the radial distribution function (32) is indicated as a horizontal error bar in logarithmic scale. | ||
Closures ZHOU and CG, proposed for the PSM, are the best at lower temperatures and moderate and high densities; ZHOU is better at lower temperatures, CG at the higher. However, for states where the clustering effect is significant, that is for low temperatures together with high densities, no closure gives fully satisfactory results.
As already noted,15 the simplest HNC closure is the best option at higher temperatures except for the lowest densities. Thus for ρ > 0.2 the bridge function of the penetrable sphere fluid is better approximated by zero than by the rescaled hard sphere value.
Fig. 8 shows one selected state point at the low temperature and high density region. Here both CG and ZHOU give similar qualitatively correct results. HNC is of similar accuracy, but with the deviation of the opposite sign. Other closures deteriorate in the order VM, MS, PY, HSB.
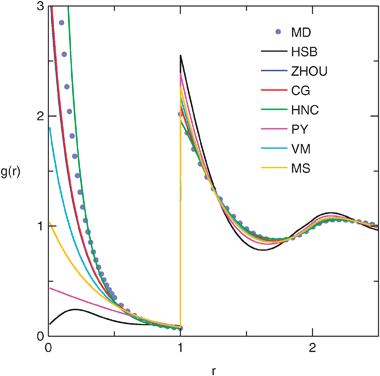 | ||
| Fig. 8 Radial distribution function for T = 0.3 and ρ = 0.8 from the OZ equation compared to selected MD data. Closure acronyms are defined in section 2.4. | ||
4. Concluding remarks
The aim of this work was to generate accurate up-to-date thermodynamic and structural data of the penetrable sphere model in the fluid range. We checked successfully consistency of data obtained from different sources (molecular dynamics, virial coefficients, Monte Carlo).In order to avoid possible systematic errors, a special attention was paid to finite size effects. In particular, we examined properties of the Andersen thermostat with respect to the convergence to the thermodynamic limit.
Finally, the structure of the penetrable sphere fluid was studied by the Ornstein–Zernike equation with four classical general closures, two closures tailored for the model, and one new closure. While the new closure is good at low densities only, the simplest HNC closure is surprisingly the best one in a wide range of conditions.
Acknowledgements
This work was supported by the Ministry of Education, Youth and Sports of the Czech Republic under the projects 6046137307 and LC512 (Center for Biomolecules and Complex Molecular Systems).References
- S. Katsura and Y. Abe, J. Chem. Phys., 1963, 39, 2068 CrossRef CAS.
- C. N. Likos, H. Lowen, M. Watzlawek, B. Abbas, O. Jucknischke, J. Allgaier and D. Richter, Phys. Rev. Lett., 1998, 80, 4450 CrossRef CAS.
- A. Jusufi, M. Watzlawek and H. Löwen, Macromolecules, 1999, 32, 4470 CrossRef CAS.
- F. H. Stillinger, J. Chem. Phys., 1976, 65, 3968 CrossRef CAS.
- S. Marquest and T. A. Witten, J. Phys. (France), 1989, 50, 2068 Search PubMed.
- A. Santos and A. Malijevský, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 75, 021201 CrossRef.
- C. N. Likos, M. Watzlawek and H. Löwen, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 58, 3135 CrossRef CAS.
- M. J. Fernaud, E. Lomba and L. L. Lee, J. Chem. Phys., 2000, 112, 810 CrossRef CAS.
- N. Choundhury and S. K. Ghosh, J. Chem. Phys., 2003, 119, 4827 CrossRef.
- S. Zhou, Commun. Theo. Phys., 2006, 46, 323 Search PubMed.
- S.-C. Kim and S.-Y. Suh, J. Chem. Phys., 2002, 117, 9880 CrossRef CAS.
- M. Khanpour and R. Hashim, J. Chem. Phys., 2008, 129, 164508 CrossRef.
- L. Acedo and A. Santos, Phys. Lett. A, 2004, 323, 427 CrossRef CAS.
- M. Schmidt, J. Phys.: Condens. Matter, 1999, 11, 10163 CrossRef CAS.
- A. Malijevský, S. B. Yuste and A. Santos, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 76, 021504 CrossRef.
- A. Santos, e-print cond-mat/0501068.
- J. P. Hansen and I. R. McDonald, Theory of Simple Liquids, Academic Press, Oxford, 1986 Search PubMed.
- G. A. Martynov and G. N. Sarkisov, Mol. Phys., 1983, 49, 1495 CAS.
- S. Labík, J. Kolafa and A. Malijevský, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 71, 021105 CrossRef.
- F. H. Ree and W. G. Hoover, J. Chem. Phys., 1964, 40, 939 CrossRef; F. H. Ree and W. G. Hoover, J. Chem. Phys., 1967, 46, 4181 CrossRef CAS.
- E. Leontidis, J. J. DePablo, M. Laso and U. W. Suter, Adv. Polym. Sci., 1994, 116, 283 CAS.
- R. M. Ziff, Comput. Phys., 1998, 12, 385 CrossRef.
- J. Kolafa, S. Labík and A. Malijevský, Phys. Chem. Chem. Phys., 2004, 6, 2335 RSC.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, Clarendon Press, Oxford, 1986 Search PubMed.
- D. Frenkel and B. Smit, Understanding Molecular Simulation, Academic Press, San Diego, 2002 Search PubMed.
- D. S. Cerutti, R. Duke, P. L. Freddolino, H. Fan and T. P. Lybrand, J. Chem. Theory Comput., 2008, 4, 1669 CrossRef CAS.
- L. Verlet, Mol. Phys., 1980, 41, 183.
- S. Labík, A. Malijevský and W. R. Smith, Mol. Phys., 1991, 73, 87 CAS.
- A. Malijevský and S. Labík, Mol. Phys., 1987, 60, 663 CrossRef CAS.
- S. Labík and A. Malijevský, Mol. Phys., 1989, 67, 431 CrossRef CAS.
- A. A. Louis, P. G. Bolhuis and J. P. Hansen, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 7961 CrossRef CAS.
- A. Malijevský and J. Kolafa, J. Lect. Notes Phys., 2008, 753, 27 Search PubMed.
- N. Clisby and B. M. McCoy, J. Stat. Phys., 2006, 122, 15 CrossRef.
- J. Kolafa, S. Labík and A. Malijevský, Mol. Phys., 2002, 100, 2629 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of results. See DOI: 10.1039/b917204a |
| This journal is © the Owner Societies 2010 |