A bifunctional strategy towards experimentally (synthetically) attainable molecules with planar tetracoordinate carbons
Yan-Bo
Wu
ab,
Jin-Liang
Jiang
a,
Hua
Li
a,
Zhongfang
Chen
c and
Zhi-Xiang
Wang
*a
aCollege of Chemistry and Chemical Engineering, Graduate University of Chinese Academy of Sciences, Beijing, 100049, People’s Republic of China. E-mail: zxwang@gucas.ac.cn; Fax: +86 10 8825 6693; Tel: +86 10 8825 6674
bInstitute of Molecular Science, the Key Laboratory of Chemical Biology and Molecular Engineering of Education Ministry, Shanxi University, Taiuan, Shanxi, 030006, People’s Republic of China
cDepartment of Chemistry, Institute of Functional Nanomaterials, University of Puerto Rico, San Juan, PR 00931, Puerto Rico
First published on 7th November 2009
Abstract
Bifunctional strategy, including efficiently utilizing valence electrons and offering steric protection, has been proposed to advance C2Al4 global minimum with double planar tetracoordinate carbons (ptC) to a new family of ptC molecules which could be promising for synthetic realization.
Stabilizing the extremely unfavorable planar tetracoordinate carbon (ptC) has always been a challenge.1,2 The first prediction of the ptC molecule (1,1-dilithiocyclopropane)3 encouraged strategic developments, and more computational and experimental realizations.4–9 In the past decade, the great efforts from computational chemists have advanced the ptC chemistry to the general planar hypercoordinate carbons (phC) where the coordination numbers of the flat carbons are more than three (hence, including ptC). Numerous computed phC molecules have emerged.4–17 In contrast, experimentally (synthetically, in particular) accessible phC molecules are rare. Only several transition metal and lithium-containing phC compounds have been synthesized.4 The recently reported CAl42− and its analogues were only observed in the gas phase.18–20 In these computed phC molecules, the electron deficient elements (e.g. B, Al, Li, and transition metals) were frequently used as electron acceptors and the ligands were mostly exposed externally. Exposing such electron deficient atoms results in their high reactivity, and the lack of “anchors” to attach protecting groups in these species further limits the operations of conventional synthetic strategies. In many cases the computed phC molecules are not the global minima, which is a concern; this also presents difficulties for experimental realizations even in the gas phase. For example, the global minimum, CAl42− and its analogues18–20 have been observed, but the efforts to detect the hypercoordinated CB7− and CB62− (not global minima) led to the more favorable species in which the carbons prefer locating on the edges or vertexes.21,22 Therefore, the more challenging task faced by computational chemists is to design phC species attainable experimentally or synthetically in particular (i.e. not just be observable in the gas phase). This will help develop the potential applications of such exotic carbon bonding patterns. Here we propose a bifunctional strategy to advance our newly uncovered global minimum species (D2h C2Al4) with double planar tetracoordinate carbons (dptCs) to those which are promising for synthetic realizations.
Using Gaussian 03,23 most of the reported structures were optimized at the B3LYP/6-311++G** level, while the largest molecule at the B3LYP/6-31G* level. Subsequent frequency analyses were performed to verify the optimized structures to be minima and the unscaled harmonic frequency values were used to evaluate their relative thermodynamic stabilities. The smallest species, C2Al4H8 (1, Fig. 1) was reoptimized at the B3LYP/aug-cc-pVTZ and MP2/aug-cc-pVTZ levels to calibrate suitability of the basis sets and the B3LYP method. The electronic structures were analyzed using the NBO.5.0 program24 at the B3LYP level. The Born–Oppenheimer molecular dynamic (BOMD)25–27 simulations were run at B3LYP/6-31G* level.
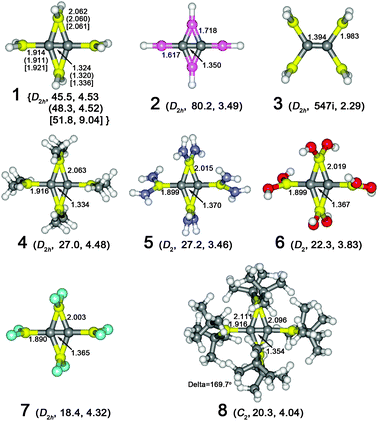 | ||
| Fig. 1 Optimized structures of 1–7 at B3LYP/6-311++G** and 8 at B3LYP/6-31G*. The key bond lengths are given in Å. The numbers after the point groups are the smallest frequencies in cm−1 and the HOMO–LUMO gaps in eV. The values in the parentheses, and brackets given in 1, are at B3LYP/aug-cc-pVTZ and MP2/aug-cc-pVTZ, respectively. Color code, yellow: Al, cyan: F, red: O, blue: N, grey: C, pink: Be, and white: H. | ||
Continuing our interest in phC chemistry, we recently got an intriguing finding, C2Al4 is the simplest neutral global minimum.28 Comparing the electronic structure of C2Al4 with those of CAl42− and CAl5+ led to a succinct equivalence relationship: the central CC moiety in C2Al4 is equivalent to the carbon centers in CAl42− and CAl5+. C2Al4 also contrasts to ethylene by possessing two different molecular orbitals (MO’s). The two MO’s of ethylene, related to the C–H and C–C σ bonds are replaced by the peripheral Al4 and four-center-involved (CAlBRCAlBR) bonding MO’s in C2Al4. Hereafter, AlBR and AlTE represent the bridging and terminal Al atoms, respectively. Although C2Al4 is the global minimum, we speculated that such species can only be detected as transient species in the gas phase and is probably difficult to attain by conventional synthesis due to the reasons mentioned above.
C2Al4 is a global minima, but it does not have optimal electronic structure: the eight more valence electrons than ethylene occupy four non-bonding MO’s and are not effectively utilized for chemical bonding. When designing boraplanes and their analogues, Wang and Schleyer proposed to direct the two non-bonding electrons on ptC into a peripheral B4 bonding orbital in boraplanes (or C–B bonds in their analogs) to utilize the otherwise “wasted” electrons and thus to achieve perfect ptC arrangements.29,30 Following this idea, we expect that directing the eight non-bonding electrons in C2Al4 into bonding MO’s will benefit its stability and thus improve its chemical accessibility. Moreover, applying the equivalence relationship to our31 recently designed CBe4H42− also led to a D2h C2Be4H4 minimum (2). Hence it is anticipated that further replacement of BeH groups in 2 with the valence isoelectronic AlH2 will result in a true D2h minimum with perfect dptCs, 1. This expectation was confirmed by computations at the B3LYP/6-311++G**, B3LYP/aug-cc-pVTZ and MP2/aug-cc-pVTZ levels. The key geometric parameters at these three levels are compared in Fig. 1, which indicates that the B3LYP/6-311++G** is an appropriate level in describing this system. Thus, computational results at this level (B3LYP/6-311++G**) are discussed hereafter, unless otherwise specified.
The four occupied non-bonding MO’s (column 2 in Fig. 2) of the isolated C2Al4 correlate to the eight occupied MO’s (columns 3 and 4) of 1. Although one can observe somewhat non-bonding characters in the eight MO’s of 1, it is apparent that the electrons are much more efficiently utilized for bonding. This is strongly supported by the heat formation of C2Al4 + H2 to 1, −75.4 kcal mol−1 (ΔH). The free energy changes (ΔG) are given in Table 1 for comparison. The characteristic occupied orbitals of C2Al4 (the first three MO’s in column 1) have their counterparts in column 5. The bonding interactions between C and AlTE in the LUMO of C2Al4 indicate its great potential to accept electrons. In contrast, the LUMO of 1 is basically a non-bonding orbital. Therefore 1 is less reactive than C2Al4. This is further quantified by the much larger HOMO–LUMO gap of 1, (4.53 eV) than that of C2Al4 (2.84 eV).
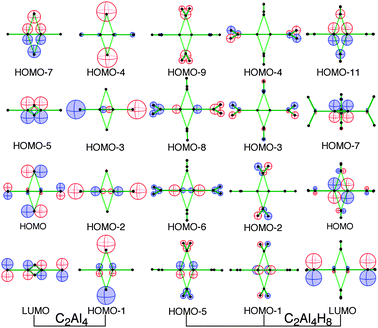 | ||
| Fig. 2 Correlations of the major MO’s of C2Al4 MO’s to those of 1. | ||
Fully hydrogenating C2Al4 to 1 not only utilizes the non-bonding electrons in C2Al4 for Al–H bonds, but also enhances the interactions between the central CC moiety and the peripheral Al4 ring. Compared with C2Al4, the C–C bond (RCC = 1.324 Å) in 1 is elongated by 0.008 Å, but the C–AlTE/C–AlBR bonds are substantially shortened by 0.085/0.100 Å. The more compact C2Al4 core in 1 indicates the more efficient utilization of the valence electrons than in C2Al4. This is also unveiled by NBO electronic structure analyses. The Wiberg bond indices (WBI’s) of C–AlTE/C–AlBR bonds in C2Al4 are 0.53/0.52 (Table 2), while the values in 1 are increased to 0.65/0.67. The ionic interactions between the central CC moiety and the Al4 ring are also strengthened. Due to the relatively large electronegativity of H compared to Al, hydrogen atoms bear negative charges, −0.33e (H–AlTE) and −0.32e (H–AlBR), resulting in much larger positive charges on AlTE/AlBR (1.22e/1.14e) than the 0.68e/0.66e in C2Al4. Although the central carbon atoms in 1 bear less negative charges, −1.07e vs. −1.34e in C2Al4, the overall ionic interactions in 1 are increased on the basis of the Coulomb attraction consideration. Therefore the total interactions between Al and C atoms are enhanced, consistent with the more compact C2Al4 core in 1.
| Q C | Q Al | WBIC | WBIAl | WBICC–Al | WBIAl–Als | |
|---|---|---|---|---|---|---|
| AlTE/AlBR | AlTE/AlBR | AlTE/AlBR | AlTE/AlBR | |||
| C2Al4 | −1.34 | 0.68/0.66 | 3.13 | 0.73/0.83 | 0.52/0.53 | 0.22/0.30 |
| 1 | −1.07 | 1.22/1.14 | 3.41 | 2.52/2.61 | 0.65/0.67 | 0.18/0.21 |
| 4 | −1.09 | 1.65/1.58 | 3.38 | 2.18/2.26 | 0.62/0.67 | 0.14/0.16 |
| 5 | −1.20 | 1.86/1.83 | 3.24 | 1.97/2.00 | 0.57/0.65 | 0.08/0.09 |
| 6 | −1.26 | 2.01/1.95 | 3.20 | 1.74/1.83 | 0.55/0.63 | 0.08/0.08 |
| 7 | −1.27 | 2.11/2.04 | 3.15 | 1.60/1.71 | 0.54/0.62 | 0.06/0.08 |
The electronic and geometric analyses only indicate the optimal electronic structure of 1. The thermodynamic stability is important for the experimental realization. Eqn (1)–(3) (Table 1), give good estimations on its thermodynamic stability. The full hydrogenation of C2Al4 to 1 (eqn (1)) is exothermic by 75.4 kcal mol−1 (ΔH), and the free energy of eqn (1) is −41.0 kcal mol−1. Note that C2Al4 was confirmed to be a global minimum.28 More realistic estimations are given by eqn (2) and (3). Relative to the experimentally32,33 available C2H4 + Al4H4 and C2H2 + Al4H6, 1 is 46.3 and 31.7 kcal mol−1 (ΔH) more stable, respectively. In comparison with the MP2/aug-cc-pVTZ results, the B3LYP underestimates the favorability of 1 by several kcal mol−1 (Table 1). The exothermicity indicates the good thermodynamic stability of 1. The ethylene-like isomer (3, as shown in Fig. 1) of 1 has six imaginary frequencies and is 41.6 kcal mol−1 less stable than 1.
The above discussion demonstrates the good electron structure and thermodynamic stability of 1 in contrast to various experimentally realized species. We further examined its thermal stability by two sets of BOMD (Born–Oppenheimer molecular dynamics) simulations at 323 and 373 K, respectively, starting at 1. As reflected by structural evaluation described by the RMSD (root-mean-square deviation relative to 1) vs. simulation time (Fig. 3), 1 was well maintained in the 35 ps simulation. The dynamic simulations imply that there are no nearby low-lying isomers which can be visited by crossing low energy barriers, though they can not tell whether 1 is a global minimum. Therefore it can be safely concluded that 1 has adequate thermal and thermodynamic stability for experimental realizations.
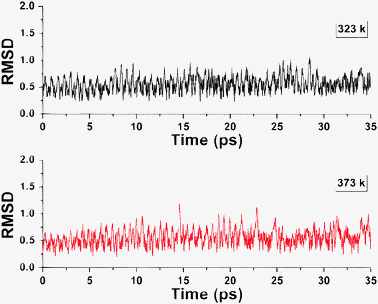 | ||
| Fig. 3 RMSD vs. time in the BOMD simulations of 1 at 323 and 373 K. | ||
Substituting Al’s in C2Al4 by AlH2 groups is a bifunctional approach. On the one hand, the additional eight Al–H bonds can utilize the eight otherwise “wasted” electrons of C2Al4, but on the other hand, the AlH2 groups provide “anchors” to attach protecting groups in the attempts to synthesize such molecules. The use of protecting groups is a common strategy to prepare reactive species such as bottleable carbene.34,35
1 can be served as a prototype for a series of dptC molecules. Elaborating 1 with proper substituents gives new dptC molecules. Replacing all the hydrogen atoms in 1 with CH3, NH2, OH and F groups lead to minima, 4(D2h), 5(D2), 6(D2), and 7(D2h), respectively. Note that the modes corresponding to the smallest vibrational frequencies (values are shown in Fig. 1) are not related to the distortion motions of the planar C2Al4 cores. The C2Al4 cores in all the derivatives maintain structures with perfect dptCs (i.e. C2Al4 core lie in the same plane). Similar to 1, the total WBI’s of CC–AlTE/CC–AlBR (the WBI between CC moiety and AlTE or AlBR) and NBO charges on C and AlTE/AlBR atoms (Table 2) reflect the enhanced covalent and ionic bonding interactions in these derivatives, with respect to the isolated C2Al4. The WBI’s and NBO charges also indicate that, as the electronegativity of the central atoms of the substituents increase (C to N, to O, and to F), the covalent bonding interactions decrease and the ionic interactions increase (see Table 2). The overall interactions between the CC moiety and the Al4 ring, as indicated by the shortened C–AlTE/C–AlBR bonds, increase as the central atom of the substituent moves from C to F in the periodical table.
One can also attach bulky protecting groups to stabilize these species or hinder their possible high reactivity. Molecule 8 in Fig. 1 is the B3LYP/6-31G* optimized structure with the t-Bu groups. As expected, the C2Al4 core in 8 expands slightly due to the crowded substituents (the C–AlTE/C–AlBR bond lengths, 1.960/2.111(2.096) Å vs. 1.913 and 2.063 Å in 4). However, despite the large steric effects, the planarity of C2Al4 is reasonably preserved (the distortion angle ∠AlBRCCAlBR, δ = 169.7°), indicating the rigid C2Al4 cores. The t-Bu groups should bring large enough steric effects to protect the C2Al4 core. Similar strategy should be applicable to 5 and 6. We also found auxiliary supports for promising synthetic realization of such molecules. The C2Cu4 core in the synthesized tetranuclear acetylidocopper(I)36 has a structure similar to the C2Al4 cores.
In conclusion, we advanced our recently computed C2Al4 global minimum with dptCs to the new species which are promising for experimental access. In comparison with the isolated C2Al4, the new molecules have two advantages. Firstly, the eight non-bonding electrons in C2Al4 are more efficiently used for chemical bonding, which results in good thermodynamic stabilities. Secondly, the new molecules have “anchors” to attach protecting groups to hinder their possible high reactivity in the attempts to synthesize such molecules. We invite experimental realizations to move the phC chemistry forward.
The authors thank Prof. G. Merino for constructive discussion. Support in China by Chinese Academy of Sciences, NSFC (Grant No. 20973197 and 20773160) and China Postdoctoral Science Foundation (Grant No. 20090450608) and in US by NSF Grant CHE-0716718, the US Environmental Protection Agency (EPA grant No. RD-83385601), and the Institute for Functional Nanomaterials (NSF Grant 0701525) is acknowledged.
References
- H. J. Monkhorst, Chem. Commun. (London), 1968, 1111 RSC.
- R. Hoffmann, R. W. Alder and C. F. Wilcox, Jr., J. Am. Chem. Soc., 1970, 92, 4992 CrossRef CAS.
- J. B. Collins, J. D. Dill, E. D. Jemmis, Y. Apeloig, P. v. R. Schleyer, R. Seeger and J. A. Pople, J. Am. Chem. Soc., 1976, 98, 5419 CrossRef CAS.
- R. Keese, Chem. Rev., 2006, 106, 4787 CrossRef CAS.
- G. Merino, M. A. Mendez-Rojas, A. Vela and T. Heine, J. Comput. Chem., 2007, 28, 362 CrossRef CAS.
- W. Siebert and A. Gunale, Chem. Soc. Rev., 1999, 28, 367 RSC.
- K. Sorger and P. v. R. Schleyer, THEOCHEM, 1995, 338, 317 CrossRef.
- D. Rottger and G. Erker, Angew. Chem., Int. Ed. Engl., 1997, 36, 812 CrossRef CAS.
- L. Radom and D. R. Rasmussen, Pure Appl. Chem., 1998, 70, 1977 CrossRef CAS.
- Z. X. Wang and P. v. R. Schleyer, Science, 2001, 292, 2465 CrossRef CAS.
- K. Exner and P. v. R. Schleyer, Science, 2000, 290, 1937 CrossRef CAS.
- S. D. Li, C. Q. Miao and G. M. Ren, Eur. J. Inorg. Chem., 2004, 11, 2232 CrossRef.
- Y. Pei and X. C. Zeng, J. Am. Chem. Soc., 2008, 130, 2580 CrossRef CAS.
- Y. Pei, W. An, K. Ito, P. v. R. Schleyer and X. C. Zeng, J. Am. Chem. Soc., 2008, 130, 10394 CrossRef CAS.
- R. M. Minyaev, T. N. Gribanova, A. G. Starikov and V. I. Minkin, Dokl. Chem., 2002, 382, 41 CrossRef CAS.
- K. Ito, Z. F. Chen, C. Corminboeuf, C. S. Wannere, X. H. Zhang, Q. S. Li and P. v. R. Schleyer, J. Am. Chem. Soc., 2007, 129, 1510 CrossRef CAS.
- S. Erhardt, G. Frenking, Z. F. Chen and P. v. R. Schleyer, Angew. Chem., Int. Ed., 2005, 44, 1078 CrossRef CAS.
- L. S. Wang, A. I. Boldyrev, X. Li and J. Simons, J. Am. Chem. Soc., 2000, 122, 7681 CrossRef CAS.
- X. Li, L. S. Wang, A. I. Boldyrev and J. Simons, J. Am. Chem. Soc., 1999, 121, 6033 CrossRef CAS.
- X. Li, H. F. Zhang, L. S. Wang, G. D. Geske and A. I. Boldyrev, Angew. Chem., Int. Ed., 2000, 39, 3630 CrossRef CAS.
- L. M. Wang, W. Huang, B. B. Averkiev, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2007, 46, 4550 CrossRef CAS.
- B. B. Averkiev, D. Y. Zubarev, L. M. Wang, W. Huang, L. S. Wang and A. I. Boldyrev, J. Am. Chem. Soc., 2008, 130, 9248 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 revision E.01, Pittsburgh, PA, 2003 Search PubMed.
- F. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, New York, 2003 Search PubMed.
- K. Bolton, W. L. Hase and G. H. Peshlherbe, Modern Methods for Multidimensional Dynamics Computation in Chemistry, ed. D. L. Thompson, World Scientific, 1998 Search PubMed.
- W. Chen, W. L. Hase and H. B. Schlegel, Chem. Phys. Lett., 1994, 228, 436 CrossRef CAS.
- X. S. Li, J. M. Millam and H. B. Schlegel, J. Chem. Phys., 2000, 113, 10062 CrossRef CAS.
- Y. B. Wu, H. G. Lu, S. D. Li and Z. X. Wang, J. Phys. Chem. A, 2009, 113, 3395 CrossRef CAS.
- Z. X. Wang and P. v. R. Schleyer, J. Am. Chem. Soc., 2001, 123, 994 CrossRef CAS.
- Z. X. Wang and P. v. R. Schleyer, J. Am. Chem. Soc., 2002, 124, 11979 CrossRef CAS.
- Z. X. Wang, C. G. Zhang, Z. F. Chen and P. v. R. Schleyer, Inorg. Chem., 2008, 47, 1332 CrossRef CAS.
- A. Grubisic, X. Li, S. T. Stokes, J. Cordes, G. F. Gantefor, K. H. Bowen, B. Kiran, P. Jena, R. Burgert and H. Schnockel, J. Am. Chem. Soc., 2007, 129, 5969 CrossRef CAS.
- X. Li, A. Grubisic, S. T. Stokes, J. Cordes, G. F. Gantefor, K. H. Bowen, B. Kiran, M. Willis, P. Jena, R. Burgert and H. Schnockel, Science, 2007, 315, 356 CrossRef CAS.
- D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef.
- A. J. Arduengo, Acc. Chem. Res., 1999, 32, 913 CrossRef.
- V. W. W. Yam, W. K. M. Fung and K. K. Cheung, Angew. Chem., Int. Ed. Engl., 1996, 35, 1100 CrossRef CAS.
| This journal is © the Owner Societies 2010 |