Resolving ligand hyperfine couplings of type 1 and 2 Cu(II) in ascorbate oxidase by high field pulse EPR correlation spectroscopy†
Alexey
Potapov
a,
Israel
Pecht
b and
Daniella
Goldfarb
*a
aDepartments of Chemical Physics, Weizmann Institute of Science, Rehovot Israel 76100. E-mail: daniella.goldfarb@weizmann.ac.il; Fax: +972-8-9344123; Tel: +972-8-9342016
bDepartments of Immunology, Weizmann Institute of Science, Rehovot Israel 76100
First published on 7th November 2009
Abstract
Ascorbate oxidase contains two paramagnetic Cu(II) binding sites, type 1 (T1) and type 2 (T2) and in both sites the Cu(II) is coordinated to histidine residues. We use several pulse EPR techniques at high field (95 GHz) to determine ligand 1H and 14N hyperfine couplings in the two sites and identify the T1 signals by a new triple resonance correlation technique named THYCOS.
Information about the coordination shells of paramagnetic metal ions is usually obtained by high resolution EPR techniques that determine the hyperfine couplings of nuclei in the close vicinity of the metal ion. These techniques, often referred to as hyperfine spectroscopy, include electron spin echo envelope modulation (ESEEM),1 continuous-wave (cw) and pulse electron-nuclear double resonance (ENDOR)2 and electron–electron double resonance (ELDOR) detected NMR (ED-NMR).3 Metalloproteins often have a number of paramagnetic centres with overlapping EPR spectra that should be resolved. Sometimes this can be achieved by specific chemical modifications, which can eliminate one of the centres. In other cases they can be resolved on the basis of different relaxation times by applying a relaxation filter.4 Yet, spectroscopic methods providing additional resolution and correlations between signals are highly desired.
The most popular correlation method in hyperfine spectroscopy is the ESEEM derived technique, hyperfine sublevel correlation spectroscopy (HYSCORE).5 It correlates between nuclear frequencies in the α and β electron-spin manifolds. Another technique is pulse TRIPLE,6 which has also been extended to two dimensions (2D) and correlates ENDOR lines belonging to the same paramagnetic species and the same electron spin manifold.7 With the development of high field EPR the ED-NMR technique has become an attractive method for measuring nuclear frequencies and it performs well for low γ nuclei with broad ENDOR lines.8 Recently ED-NMR has been extended into a correlation method, referred to as THYCOS (triple resonance hyperfine sublevel correlation spectroscopy),9 the sequence of which is shown in Fig. 1.
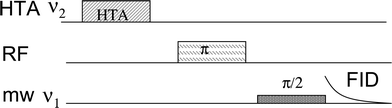 | ||
| Fig. 1 The THYCOS pulse sequence. | ||
THYCOS combines ENDOR and ED-NMR in a manner similar to the TRIPLE. In the THYCOS sequence (Fig. 1) the high turning angle (HTA) microwave (mw) pulse, with frequency ν1, transfers populations across a forbidden EPR transition (ΔMS = ±1, ΔMI = ±1). This reduces the population difference across the corresponding allowed transitions (ΔMS = ±1, ΔMI = 0) that is detected via a free induction decay (FID) produced by a long selective pulse with a frequency of ν2. An echo detection is possible as well. A change in the FID intensity occurs when a radiofrequency (RF) pulse transfers population from any level affected by either of the mw pulses. The FID intensity decreases if the RF excites the same nucleus as the HTA pulse (not interesting) and it recovers if another nucleus is involved. Therefore the THYCOS experiment links lines in the ED-NMR spectrum with lines in the ENDOR spectrum of nuclei belonging to the same site and opposite electron spin manifolds. The feasibility of this new technique has been so far demonstrated only on model systems, a single crystal of Cu2+-doped L-histidine and a frozen solution of the Cu2+-histidine complex.
The purpose of the present work is to resolve the 1H and 14N hyperfine couplings of type 1 (T1) and type 2 (T2) Cu(II) sites in ascorbate oxidase (AO) by state of the art high field (W-band, 95 GHz) pulse ENDOR and the correlation techniques, THYCOS and HYSCORE.
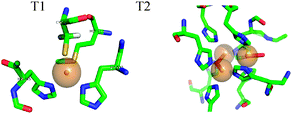
AO, a member of the multi-copper oxidases family, catalyzes the oxidation of ascorbate with the concomitant four electron reduction of oxygen.10 It contains four copper ions, two of which are paramagnetic in the oxidized state. T1 is involved in electron uptake from the substrate. Here the Cu(II) is coordinated to a methionine weak axial ligand and two histidines and one cysteine in the equatorial plane (See Fig. 2). T2 is part of the tri-nuclear centre, where the O2 molecule is reduced. The other two Cu(II) atoms in this centre are anti-ferromagnetically coupled and EPR silent. The T2 Cu(II) has a normal tetragonal environment and it is coordinated to histidine and water ligands (see Fig. 2).11 T1 Cu(II) has an intense blue colour, an unusually small 63,65Cu A‖ hyperfine coupling, and a large spin density on the cysteine sulfur. These unique properties were attributed mainly to the covalent cysteine thiolate ligation.12 Most interestingly, the Cu(II) hyperfine coupling of the C112D/M121L azurin mutant lacking the cysteine ligand in the T1 site, has been reported recently to have a character between T1 and T2 in the absence of sulfur ligation.13
Hyperfine interactions of ligand nuclei in the T1 centre yield experimental data that, together with other EPR parameters, provide a basis for the determination of the electronic structure of the T1. While T1 and T2 have significantly different 63,65Cu A‖ values they have close g-values, (gT1 = (2.036,2.058,2.227) and gT2 = (2.053,2.053,2.242).14 Accordingly, at W-band (95 GHz) they overlap throughout almost the whole spectrum. Consequently, hyperfine spectroscopy will generate spectra with contributions from both T1 and T2 and these should be assigned to their specific sites.
AO was a generous gift from Boehringer Manheim FRG. The final protein concentration was 1.3 mM (potassium phosphate buffer pH = 6.0). Fig. 3A shows the W-band echo-detected EPR spectrum of AO and Fig. 3B shows a series of W-band 1H Davies ENDOR spectra recorded at several fields along the EPR powder pattern. The spectrum taken at 3324 mT reveals two doublets corresponding to two protons with hyperfine couplings A1 = 14 MHz and A2 = 35 MHz. The frequencies of these signals vary slightly with the observed magnetic field, showing that the hyperfine coupling is primarily isotropic in character. This large isotropic hyperfine coupling is characteristic of the cysteine β-protons in T1.15,16 Simulations (see Fig. S1, ESI†) of the 1H signals of the β-proton with the larger coupling (Fig. 3B) gave [Axx, Ayy, Azz] = [30,30,36] MHz and Euler angles with respect to the g principal axis system of (α,β,γ) = (0, 50, 0)°. Due to the lack of distinct spectral features for the β-proton with the smaller coupling no simulations were attempted but from the field dependence we obtained Aiso = 13 MHz.
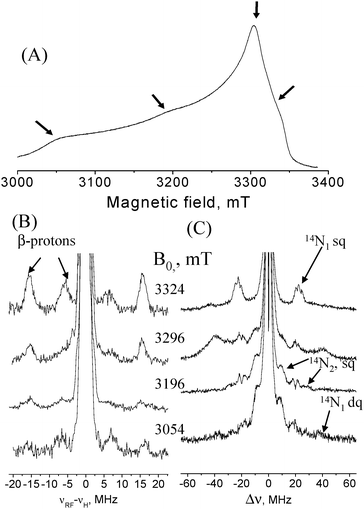 | ||
| Fig. 3 (A) Field-sweep echo-detected EPR spectrum of AO. Arrows mark the fields where ED-NMR and ENDOR spectra were measured. (B) Davies ENDOR spectra recorded at various fields with tinv = 200 ns, trf = 25 μs, t90/t180 = 100/200 ns; (C) ED-NMR spectra recorded at the same fields as (B) with tHTA = 10 μs, tdet = 3 μs. Measurement temperature was 12 K. | ||
ED-NMR spectra, recorded at the same field positions as the ENDOR spectra, are shown in Fig. 3C. At B = 3324 mT a group of three 14N lines appear at Δν = ν1 − ν2 ∼ ±20 MHz and are assigned to the high frequency component of a hyperfine doublet of 14N nuclei (N1) with A ∼ 20 MHz. The fine structure is due to quadrupole splittings and the three peaks may suggest that either two 14N nuclei with slightly different A-values are present or that they arise from some special orientation selection effects of only a single nucleus. 14N lines around this frequency appear in all recorded spectra, suggesting that they are associated with a small hyperfine anisotropy. The low frequency component of the hyperfine doublet is not observed as it overlaps with the central broad line at Δν = 0. To observe it we carried out W-band HYSCORE measurements, where low frequency peaks can be easily detected, in contrast to ED-NMR and ENDOR. A 14N hyperfine coupling of 20 MHz falls within the cancellation condition, |A/2 − νN| ≈ 0, (νN is the 14N Larmor frequency) at W-band, giving rise to pronounced nuclear modulations.
The HYSCORE spectrum recorded at B = 3324 mT is shown in Fig. 4. Cross peaks are observed in both quadrants and appear at (21, 1.7), (21, 3.5), (21, 5.2) and (18, 1.7) MHz. These are assigned to one 14N nucleus with single-quantum (sq) transition frequencies of 18 and 21 MHz in one manifold and 1.7 and 3.5 MHz in the other manifold. The double quantum (dq) transition frequency of the latter is at 5.2 MHz. The high frequency sq set is consistent with the frequencies observed for N1 in the ED-NMR spectrum and confirm their assignment. An additional peak at (23, 1.7) MHz is observed in the (+,+) quadrant again consistent with the three peaks observed in the ED-NMR spectrum. More HYSCORE spectra recorded at different fields are shown in Fig. S2.† Analysis of the frequencies using first order expressions and a series of simulations considering one nucleus (see Fig. S3 in ESI†), aiming at reproducing the peaks’ positions in the HYSCORE spectra, gave an isotropic hyperfine coupling Aiso ∼ 23 MHz, an anisotropic component, T⊥ ∼ −1 MHz and a quadrupole coupling constant, e2Qq/h ∼ 3 MHz. The latter falls within the range of couplings reported for imidazole nitrogens.17
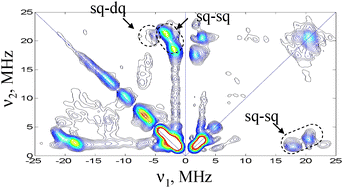 | ||
| Fig. 4 W-band HYSCORE spectrum of AO, taken at 3324 mT, t90 = 12.5 ns, t180 = 25 ns, = 135.5 ns. Measurement temperature was 12 K. Acquisition time ∼1 h. | ||
Signals attributed to another type of 14N with larger hyperfine couplings, A ∼ 40 MHz are resolved in the ED-NMR spectrum at Δν = ±10 and ±30 MHz and appear at all fields measured except 3324 mT. These signals, assigned to N2, were not observed in HYSCORE spectra recorded at similar fields. The broad signals observed at Δν ∼ ±40 MHz at B = 3296 mT are either of 63,65Cu (Larmor frequency is 37 and 39 MHz), which has a small A⊥or due to 14N dq transitions.
The absence of the Δν = ± 10 and ±30 MHz signal in the 3324 mT spectrum, which was recorded close to the gxx of T1, suggests that these signals are most probably due to T2 and that N1 belongs to T1. To substantiate this assignment THYCOS was carried out at B = 3296 mT with Δν = 20 MHz and the RF was scanned over the 1H region. Fig. 5 shows the THYCOS spectrum together with the ENDOR spectrum recorded at the same field. It shows the β-protons signal at 125 MHz while the high frequency partner is not observed. This confirms the assignment of N1 to T1. Moreover, it shows that the 14N high frequency hyperfine component and the low frequency β-protons component belong to different manifolds, namely their hyperfine couplings have the same sign. THYCOS performed with the Δν = 10 MHz value did not reveal any β-protons signals nor other protons.
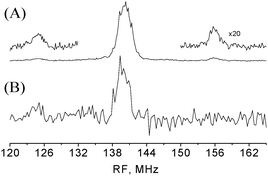 | ||
| Fig. 5 (A) Davies ENDOR with 1H spectra lines belonging to the β-protons are expanded. Experimental conditions are as in Fig. 3b. (B) THYCOS experiment with Δν = 20 MHz. tHTA = 10 μs, and the duration echo detection pulses were 100 and 200 ns, respectively. The spectrum was collected for 12 h. For both spectra B0 = 3296 mT. | ||
The 14N hyperfine couplings are compared with those of T1 in other proteins in Table 1, showing a rather large spread. The AO 14N couplings are very close to those of plastocyanin (poplar),16 which has the same trigonal pyramid geometry as in AO. The similarity to plastocyanin may be significant; indicating that the same geometry results in the same 14N, but it can also be accidental. To the best of our knowledge no obvious relationship between the magnitude of 14N couplings and geometrical factors has been reported so far either from experimental results or from quantum chemical calculations. More results are needed to establish such a correlation.
| A iso(14N), MHz | A iso (1H) MHz | Geometry | |
|---|---|---|---|
| Plastocyanin,15 T1(poplar) | 22 | 27, 16 | Trigonal pyramidal19 |
| Azurin,23 T1 | 25, 18 | 21, 21 | Trigonal bipyramidal24 |
| Stellacyanin15, T1 | 33, 17 | 20, 20 | Distorted tetrahedral25 |
| Plastocyanin(spinach),26 T1 | 23, 17 | Trigonal pyramidal27 | |
| Ascorbate oxidase (this work), T1 | 23 | 32, 13 | Trigonal pyramidal11 |
| Ascorbate oxidase (this work), T2 | 40 | — |
The hyperfine couplings of the T1 β-protons are also compared in Table 1 with those of other T1 Cu(II) reported so far. The Aiso value of the β-protons arises partially from hyperconjugation and it was shown to depend on the dihedral angle Cu–S–C–H, φ, according to:15
| Aiso = ρs(Bsin2φ + C) | (1) |
To summarize, in this work we determined the hyperfine couplings of the directly coordinated 14N ligand in the T1 and T2 Cu(II) sites in AO and found that the two histidines in T1 have very similar couplings. In addition, we have determined the 1H hyperfine couplings of the T1 β-protons and used their signals to assign the 14N signals employing THYCOS. We have demonstrated the feasibility and effectiveness of W-band ED-NMR, HYSCORE and particularly the new THYCOS correlation technique in resolving and assigning signals in overlapping spectra of two paramagnetic sites of a protein.
Acknowledgements
This work was supported by the Binational USA-Israel Science foundation (BSF). DG holds the Erich Klieger Chair in Chemical Physics.References
- (a) W. B. Mims, Rev. Sci. Instrum., 1965, 36, 1472–1479 CrossRef CAS; (b) S. A. Dikanov and Y. D. Tsvetkov, Electron Spin Echo Envelope Modulation (ESEEM) Spectroscopy, CRC Press, Boca Raton, FL, 1992 Search PubMed.
- (a) E. R. Davies, Phys. Lett. A, 1974, 47, 1–2 CrossRef CAS; (b) W. B. Mims, Proc. R. Soc. London, Ser. A, 1965, 283, 452–457 CrossRef CAS.
- P. Schosseler, T. Wacker and A. Schweiger, Chem. Phys. Lett., 1994, 224, 319–324 CrossRef CAS.
- T. Maly and T. Prisner, J. Magn. Reson., 2004, 170, 88–96 CrossRef CAS.
- P. Hőfer, A. Grupp, H. Nebenfűhr and M. Mehring, Chem. Phys. Lett., 1986, 132, 279–282 CrossRef.
- M. Mehring, P. Höfer and A. Grupp, Ber. Buns. Ges. Phys. Chem. Chem. Phys., 1987, 91, 1132–1137 Search PubMed.
- (a) B. Epel and D. Goldfarb, J. Magn. Reson., 2000, 146, 196–203 CrossRef CAS; (b) D. Goldfarb, B. Epel, H. Zimmermann and G. Jeschke, J. Magn. Reson., 2004, 168, 75–87 CrossRef CAS.
- (a) M. Flores, A. G. Agrawal, M. van Gastel, W. Gartner and W. Lubitz, J. Am. Chem. Soc., 2008, 130, 2402–2403 CrossRef CAS; (b) G. Jeschke and H. Spiess, Chem. Phys. Lett., 1998, 293, 9–18 CrossRef CAS.
- A. Potapov, B. Epel and D. Goldfarb, J. Chem. Phys., 2008, 128, 052320 CrossRef.
- E. Adman, Copper Protein Structures, in Metalloproteins: Structural Aspects, 1991, vol. 42 Search PubMed.
- (a) A. Messerschmidt, A. Rossi, R. Ladenstein, R. Huber, M. Bolognesi, G. Gatti, A. Marchesini, R. Petruzzelli and A. Finazzi-Agró, J. Mol. Biol., 1989, 206, 513–529 CrossRef CAS; (b) A. Messerschmidt, R. Ladenstein, R. Huber, M. Bolognesi, L. Avigliano, R. Petruzzelli, A. Rossi and A. Finazzi-Agró, J. Mol. Biol., 1992, 224, 179–205 CrossRef CAS.
- (a) E. I. Solomon, J. W. Hare and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 1389–1393 CrossRef CAS; (b) E. I. Solomon, J. W. Hare, D. M. Dooley, J. H. Dawson, P. J. Stephens and H. B. Gray, J. Am. Chem. Soc., 1980, 102, 168–178 CrossRef CAS.
- K. M. Lancaster, K. Yokoyama, J. H. Richards, J. R. Winkler and H. B. Gray, Inorg. Chem., 2009, 48, 1278–1280 CrossRef CAS.
- A. Marchesini and P. M. H. Kroneck, Eur. J. Biochem., 1979, 101, 65–76 CAS.
- M. M. Werst, C. E. Davoust and B. M. Hoffman, J. Am. Chem. Soc., 1991, 113, 1533–1538 CrossRef.
- J. E. Roberts, J. F. Cline, V. Lum, H. B. Gray, H. Freeman, J. Peisach, B. Reinhammar and B. M. Hoffman, J. Am. Chem. Soc., 1984, 106, 5324–5330 CrossRef CAS.
- J. W. A. Coremans, O. G. Poluektov, E. J. J. Groenen, G. W. Canters, H. Nar and A. Messerschmidt, J. Am. Chem. Soc., 1997, 119, 4726–4731 CrossRef CAS.
- G. Schaftenaar and J. H. Noordik, J. Comput.-Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS.
- J. M. Guss, H. D. Bartunik and H. C. Freeman, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 790–811 CrossRef.
- S. A. Moggach, S. J. Clark and S. Parsons, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, o2739–o2742 CrossRef.
- E. I. Solomon, R. K. Szilagyi, S. Debeer George and L. Basumallick, Chem. Rev., 2004, 104, 419–458 CrossRef CAS.
- S. Shadle, J. Penner-Hahn, H. Schugaryl, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 1993, 115, 767–776 CrossRef CAS.
- M. Fittipaldi, G. C. M. Warmerdam, E. C. de Waal, G. W. Canters, D. Cavazzini, G. L. Rossi, M. Huber and E. J. J. Groenen, ChemPhysChem, 2006, 7, 1286–1293 CrossRef.
- H. Nar, A. Messerschmidt, R. Huber, M. van De Kamp and G. W. Canters, J. Mol. Biol., 1991, 221, 765–72 CrossRef CAS.
- P. J. Hart, A. M. Nersissian, R. G. Herrmann, R. M. Nalbandyan, J. S. Valentine and D. Eisenberg, Protein Sci., 1996, 5, 2175–83 CrossRef CAS.
- I. Bertini, C. O. Fernández, B. G. Karlsson, J. Leckner, C. Luchinat, B. G. Malmström, A. M. Nersissian, R. Pierattelli, E. Shipp, J. S. Valentine and A. J. Vila, J. Am. Chem. Soc., 2000, 122, 3701–3707 CrossRef CAS.
- Y. Xue, M. Okvist, O. Hansson and S. Young, Protein Sci., 1998, 7, 2099–105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HYSCORE spectra recorded at several magnetic fields, simulated HYSCORE and ENDOR spectra. Tables with calculated ρs. See DOI: 10.1039/b919069d |
| This journal is © the Owner Societies 2010 |