Supramolecular nanoarchitectures for light energy conversion
Taku Hasobe*ab
aSchool of Materials Science, Japan Advanced Institute of Science and Technology (JAIST), Nomi, Ishikawa, 923-1292, Japan. E-mail: t-hasobe@jaist.ac.jp
bPRESTO, Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama, 332-0012, Japan
First published on 7th September 2009
Abstract
Recent developments in synthetic and supramolecular techniques have made it possible to control precisely, organize and arrange molecules at the nanometre level. Such synthetic and supramolecular strategies enable us to construct photofunctional molecular architectures for light energy conversion, such as photovoltaics. In photovoltaic cells, processes such as light-harvesting, charge separation for carrier generation, and carrier transport are generally required. Therefore, the construction of supramolecular assemblies based on these three processes is interesting and promising for the future development of photovoltaics. In this perspective, the focus is on the recent developments of supramolecular systems for light energy conversion, which are mainly composed of porphyrin dyes and nanocarbon materials, such as fullerenes and carbon nanotubes. The specific topics are as follows: (i) preparation, photodynamics, and photoelectrochemistry of self-assembled porphyrin nanoparticles prepared by simple blend, (ii) highly organized supramolecular nanoassemblies of porphyrins and fullerenes using gold nanoparticles, dendritic and polypeptide structures, (iii) the supramolecular formation and photoelectrochemical property of carbon nanotubes, and (iv) supramolecular photofunctional nanorods of porphyrins.
 Taku Hasobe | Taku Hasobe is a lecturer in the School of Materials Science, Japan Advanced Institute of Science and Technology (JAIST). He received his PhD degree from the Department of Materials and Life Science, Graduate School of Engineering, Osaka University (supervisor: Prof. S. Fukuzumi) in 2004. Then, he joined the Radiation Laboratory, University of Notre Dame as a postdoctoral research fellow under the supervision of Prof. P. V. Kamat. He was also awarded a research fellowship of the Japan Society for the Promotion of Science (JSPS) for Young Scientists from 2002 to 2005. He joined JAIST as an assistant professor in 2005 and started the current faculty position in 2006. Now, he is also a concurrent researcher of the PRESTO project, Japan Science and Technology Agency (JST). He was presented with the PCCP Prize 2009 for Outstanding Achievement of Young Scientists in Physical Chemistry and Chemical Physics by the Royal Society of Chemistry. His research interest is in the construction of supramolecular systems for solar energy conversion. |
1. Introduction
The recent rapid consumption of fossil fuels has created serious environmental problems, such as the greenhouse effect and air pollution.1–4 In order to protect and maintain the quality of human life and the global environment, clean and renewable energy resources such as those obtained by using photovoltaics are required.5 Attention has been drawn to the development of photovoltaic systems composed of organic molecules as a photoactive layer.5–16 Therefore, novel strategies and approaches for the fabrication of organic photovoltaics are essential for the future development of next generation devices.17–19Photovoltaic cells convert sunlight directly into electricity by the photovoltaic effect. In the multilayer films, energy conversion is generally performed by the following three processes: (i) light-harvesting and exciton diffusion, (ii) charge separation, and (iii) carrier transport.20 In such organic blends, the photons are absorbed by either the donor or the acceptor, or by both. Fig. 1A provides examples of solar irradiance spectra. The sun produces a broad spectrum of light output from the ultraviolet to the infrared region, whereas the absorption region of organic molecules (monomer) is generally very limited. Therefore, the cooperative aggregation of organic molecules contributes effectively to the broadening of the absorption region (this is the 1st reason for the requirement of a molecular assembly for photovoltaics). Next, if the donor moieties absorb most of incident light, electron–hole pairs are generated on the donor domain, following promotion of a π-electron from the HOMO level to the LUMO level (Fig. 1B). To dissociate into charge carriers, the excitons have to migrate toward a donor/acceptor interface (exciton diffusion). However, the exciton diffusion range is generally limited in the case of organic molecules.20 Consequently, the close organization between donor and acceptor moieties for efficient charge separation is essential to the utilization of the excitation energy (2nd reason). Finally, the created charges have to escape from their mutual Coulomb attraction before migration toward the electrodes takes place, typically via the hopping mechanism.20,21 In this process, the mobilities of the molecular aggregated structures largely dominate the carrier transport properties. As reported previously,22–24 mobility is largely dependent on the grain structures (i.e., grain-size/boundary). This means that the size and morphological control of molecular assemblies is also a critical factor in the efficient transport of charge carriers (3rd reason).
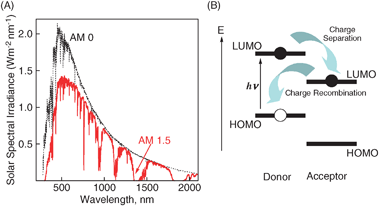 | ||
| Fig. 1 (A) Solar irradiance spectra. (B) Illustration of exciton dissociation (charge separation) and charge recombination in a donor–acceptor pair (one-electron picture for clarity). | ||
Recent research on supramolecular assemblies have made rapid progress. Supramolecular assemblies are defined as the organization of chemical structures through, or mediated by, noncovalent bonds, such as π–π interactions,25–27 hydrogen bonds,28–30 van der Waals interactions,31,32 and coordination bonds.33–35 These bonds play a complementary role toward covalent synthesis since nanometre or larger dimensional organization is not possible by the use of covalent bonds alone. Such organized molecular architectures with well-defined shapes and structures are applicable to those for photovoltaic systems. However, so far, little attention has been given to utilize such supramolecular systems in photovoltaic cells.
Promising candidates for light energy conversion are porphyrins, which are known as chromophores and electron donors in a number of important biological systems, such as the photosynthetic reaction centers.36 Therefore, porphyrins are also suitable for efficient electron transfer because of their small reorganization energies.37,38 In addition, the rich and extensive absorption features of porphyrinoid systems guarantee increased absorption cross-sections and an efficient use of the solar spectrum.39,40 On the other hand, porphyrins are also promising building blocks for superstructures. So far, various supramolecular nanoassemblies composed of porphyrins, such as particles,41 tubes42,43 and rods,44–47 have been widely reported.
Recent developments in nanoscale carbon materials, such as fullerenes and carbon nanotubes, provide us with new ways to design and construct energy conversion architectures. Although the construction of molecular architectures based on π-electronic conjugation in cylindrical or spherical structures has already been examined, new approaches for the fabrication of photovoltaics are desired. Fullerene derivatives, such as [6,6]-phenyl-C61-butyric acid methyl ester (PCBM),48,49 which act as electron acceptors have been extensively used in organic photovoltaics.50 The electron-transfer process to C60 is thereby highly favored because of the minimal changes in the structure and solvation reorganization following its reduction.51–53 Based on these basic properties, bulk heterojunction organic solar cells, which possess an active layer of a conjugated donor polymer and an acceptor fullerene, have been extensively studied.7,9,10,48,54,55
Moreover, a combination of porphyrins and fullerenes seems ideal for enhancing the light-harvesting efficiency of chromophores and the resulting charge separation by photoinduced electron transfer throughout the solar spectrum.40,56,57 Porphyrins and fullerenes are also known to form supramolecular complexes, which contain close contacts between one of the electron-rich 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 bonds of the guest fullerene and the geometric center of the host porphyrin. The porphyrin–fullerene interaction energies are reported to be in the range of −16 to −18 kcal mol−1.58 Such a strong interaction between porphyrins and fullerenes is likely to be a good driving force for the formation of supramolecular complexes.59,60
:6 bonds of the guest fullerene and the geometric center of the host porphyrin. The porphyrin–fullerene interaction energies are reported to be in the range of −16 to −18 kcal mol−1.58 Such a strong interaction between porphyrins and fullerenes is likely to be a good driving force for the formation of supramolecular complexes.59,60
The usefulness of carbon nanotubes in optical, electronic and catalytic applications has prompted researchers to synthesize various carbon nanostructures. Carbon nanotubes (CNT) possess both semiconducting and metallic characteristics.61,62 Of particular interest are the optical and electronic properties of semiconducting CNT.63–66 These initial studies show the ability of carbon nanostructures in promoting charge separation via bandgap excitation. Moreover, surface modification or the covalent linkage of molecules often facilitates the solubilization of CNT. However, designing supramolecular assemblies remains a challenge. The utilization of molecular organizations, such as J- and H-aggregates of porphyrins, are useful methods to form ordered assemblies.67–69
This perspective is thereby intended to focus on the recent advancement in supramolecular assemblies for light energy conversion, such as photovoltaics. The formation of supramolecular assemblies of donor and acceptor moieties plays an important role in attaining efficient energy conversion properties. The structural and photophysical properties, as well as quantitative discussions of light energy conversion properties, are presented here.
2. Self-assembled porphyrin nanoparticles
2.1 Size-control of porphyrin nanoparticles and their quenching properties of the excited states
Although the preparation of porphyrin nanoparticles has been reported previously,41,70 so far little attention has been drawn to their photophysical properties. A systematic examination of the structural and photophysical properties provides us with fundamental information for future optical and electronic applications.Stabilizers, such as ethylene glycol (EG), are useful for the formation of stable colloidal systems by the host–guest solvent method. Four EG derivatives, triethylene glycol (TriEG), tetraethylene glycol (TetraEG), hexaethylene glycol (HexaEG) and heptaethylene glycol (HeptaEG), with different chain lengths are shown in Fig. 2. Porphyrin nanoassemblies are prepared by adding an excess volume of a poor solvent (H2O) to a H2P(CO2H)4 solution in a good solvent (THF) mixed with a small volume of EG surfactants, while vigorously stirring. In all systems, the final concentrations of H2P(CO2H)4 are fixed as 0.05 mM in H2O–THF (15:1, v/v) containing 1.5% volume of the EG derivative. A reference system without EG is also made under the same condition (H2O:THF = 15:1, v/v).
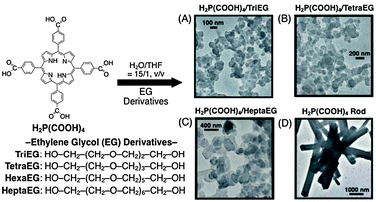 | ||
| Fig. 2 Size-control of porphyrin nanoparticles using EG derivatives. (A) H2P(CO2H)4/TriEG, (B) H2P(CO2H)4/TetraEG, (C) H2P(CO2H)4/HeptaEG and (D) pristine H2P(CO2H)4 rods.71 | ||
Transmission electron micrographs (TEM) are employed to examine the sizes and shapes of these porphyrin nanoassemblies, as shown in Fig. 2. In the case of H2P(CO2H)4–EG composite nanoassemblies (Fig. 2A–C), we can see a lot of nanoparticles, which is in sharp contrast to the much larger size of the pristine H2P(CO2H)4 rods without EG (Fig. 2D).71 This indicates that H2P(CO2H)4 moieties are effectively organized by EG derivatives to form nanoparticle assemblies. Additionally, with increasing chain lengths of EG surfactants, the average diameters of nanoparticles also increase from ca. 100 to 400 nm. This trend is in good agreement with that measured by dynamic light scattering (DLS). These indicate that porphyrin nanoparticles are formed in a mixed H2O–THF solution.
X-Ray diffraction (XRD) measurements further demonstrate the following two important structural aspects. One is that all H2P(CO2H)4–EG nanoparticles have similar internal crystal alignments. The other is that H2P(CO2H)4 assemblies surrounded by EG surfactants possess enhanced crystallinities and organized internal structures, as compared to the pristine H2P(CO2H)4 rods.71 Additionally, the porphyrin nanoparticles are stable in solution, with no precipitation for several days.
To examine the excited species of H2P(CO2H)4 nanoparticles, transient absorption spectra were measured following a 532 nm laser light pulse irradiation of the H2P(CO2H)4 moiety. Upon the excitation of H2P(CO2H)4–TriEG nanoparticles in H2O–THF (15:1, v/v), transient absorption spectra are observed, as shown in Fig. 3, where the broad bands appear in the 460 and 880 nm regions. The observed broad bands in the 460 and 880 regions are ascribed to the triplet state of porphyrin [3H2P*(CO2H)4]. The inset shows the decay, which obeys first-order kinetics with a rate constant range of 1.5 × 107 s−1 to 3.9 × 107 s−1 (Table 1). The triplet lifetimes of the transient species are calculated to range from 67 to 26 ns (Table 1), which is much shorter than that of the corresponding monomer, H2P(CO2H)4 (40 μs). The efficient triplet quenching of the H2P(CO2H)4 nanoparticle is due to the aggregation effects of H2P(CO2H)4 moieties, such as triplet–triplet (T–T) annihilation (vide infra). Moreover, the lifetime of the triplet state decreases with decreasing particle sizes (Table 1). This trend is also very similar to that of fluorescence lifetime (i.e., excited singlet state).36
![Nanosecond transient absorption spectra of H2P(CO2H)4–TriEG nanoassemblies (H2O–THF, 15 : 1, v/v) observed by 532 nm laser light (ca. 3 mJ per pulse) irradiation at 0.1 μs (●) and 1.0 μs (○) in an Ar-saturated solution. Inset: time profile at 460 nm. [H2P(CO2H)4] = 0.05 mM in H2O–THF (15 : 1, v/v).71](/image/article/2010/CP/b910564f/b910564f-f3.gif) | ||
| Fig. 3 Nanosecond transient absorption spectra of H2P(CO2H)4–TriEG nanoassemblies (H2O–THF, 15:1, v/v) observed by 532 nm laser light (ca. 3 mJ per pulse) irradiation at 0.1 μs (●) and 1.0 μs (○) in an Ar-saturated solution. Inset: time profile at 460 nm. [H2P(CO2H)4] = 0.05 mM in H2O–THF (15:1, v/v).71 | ||
| Sample | λmax/nm | kq/s−1 | τ0T/ns | ϕTqa | γT/mol−1 dm3 s−1b |
|---|---|---|---|---|---|
| a Calculated from the equation: ΦTq = [0.96 −ΦSq(ave)], assuming ΦS of the 1H2P*(CO2H)4 monomer is 0.96.b Calculated from eqn (4).71 | |||||
| H2P(CO2H)4–TriEG | 460 | 3.9 × 107 | 26 | 0.35 | 6.5 × 1010 |
| H2P(CO2H)4–TetraEG | 460 | 2.8 × 107 | 36 | 0.51 | 5.8 × 1010 |
| H2P(CO2H)4–HexaEG | 460 | 1.8 × 107 | 55 | 0.77 | 4.7 × 1010 |
| H2P(CO2H)4–HeptaEG | 440 | 1.5 × 107 | 67 | 0.83 | 3.9 × 1010 |
2.2 Particle size-dependent annihilation processes
In the excited state quenching process in these aggregated modes, the exciton–exciton annihilation process largely controls the actual lifetimes since exciton diffusion and exciton–exciton annihilation alternately occur within nanoparticles.72–75 Therefore, in order to observe the triplet–triplet (T–T) annihilation processes within nanoassemblies, further experiments are performed. When the laser power is increased at the same concentration of H2P(CO2H)4 nanoassemblies, the initial decay rate increased, with a change in decay kinetics from first order to a mixed order (first and second order) because of the triplet–triplet annihilation [eqn (1)].71,76| [3(H2P)*] + [3(H2P)*] → [1(H2P)*] + [H2P]. | (1) |
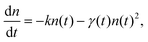 | (2) |
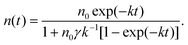 | (3) |
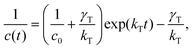 | (4) |
2.3 Photoelectrochemical properties of porphyrin nanoparticle-deposited electrodes
These self-assembled nanoparticles can be deposited as thin films on a nanostructured electrode using the electrophoretic technique for the fabrication of photoelectrochemical cells (Fig. 4). The fabrication method is as follows. A known amount (∼2 mL) of 5,15-bis(3,5-di-tert-butylphenyl)porphyrin (H2P) nanoparticle, which is clusterized in acetonitrile–toluene (9:1, v/v), is transferred to a cuvette in which an optically transparent electrode (OTE) and a TiO2-coated film electrode (OTE/TiO2) were kept at a distance of ∼6 mm using a Teflon® spacer. By applying an electric field between the two electrodes, the deposition of the film can be visibly seen as the solution becomes colorless with simultaneous browning of the OTE/TiO2 electrode [denoted as OTE/TiO2/(H2P)n].77 OTE/TiO2/(H2P)n is highly photoactive and capable of undergoing charge separation under visible light excitation. The photoelectrochemical performance of OTE/TiO2/(H2P)n was measured in a standard two-compartment electrolyte cell.77 Photoexcitation of the porphyrin film electrode assembly in a photoelectrochemical cell with visible light produces a relatively high photocurrent. A maximum photocurrent of 0.15 mA cm−2 and a photovoltage of 250 mV were attained using an I3−–I− redox couple under white light illumination. The incident photon-to-photocurrent generation efficiency (IPCE) was calculated by normalizing the photocurrent values for incident light energy and intensity using eqn (5).78| IPCE(%) = 100 × 1240 ×Isc/(Winλ), | (5) |
 | ||
| Fig. 4 (A) Illustrations of the preparation processes of porphyrin nanoparticles (left) and porphyrin–fullerene composite nanoparticles (right). (B) Comparison of the photocurrent response (IPCE values) of (a) OTE/SnO2/(H2P + C60)n, (b) OTE/SnO2/(C60)n, (c) OTE/SnO2/(H2P)n and (d) the sum of the IPCE response of OTE/SnO2/(C60)n (b) and OTE/SnO2/(H2P)n (c); an applied bias potential: 0.2 V vs. SCE, electrolyte: 0.5 M NaI and 0.01 M I2 in acetonitrile.79 | ||
Composite molecular nanoparticles of C60 (acceptor) and H2P (donor) prepared in a acetonitrile–toluene mixed solvent absorb over the entire spectrum of visible light. The highly colored composite nanoparticles can be assembled as a three dimensional array onto nanostructured SnO2 films using the same electrophoretic deposition approach [OTE/SnO2/(H2P + C60)n]. The OTE/SnO2/(H2P + C60)n film exhibits an IPCE as high as 17% at an applied potential of 0.2 V vs. SCE, which is significantly higher than the sum of the two respective IPCE values (porphyrin clusters: 1.6% and fullerene clusters: 5.0%) under the same photoelectrochemical conditions.79 The high IPCE observed with H2P and C60 composites demonstrates the synergy of these systems in yielding efficient photoinduced charge separation within these composite nanoassemblies. Thus, composite nanoparticles of porphyrins (donor) and fullerenes (acceptor) effectively work as photoactive materials in photovoltaic cells.
3. High-order supramolecular organization between porphyrins and fullerenes
3.1 Porphyrin alkanethiolate monolayer-protected gold nanoparticles
During the last decade, donor–acceptor combination systems, such as dyads, triads and tetrads, have been the popular choice for mimicking artificial photosynthesis, which demonstrates efficient photoinduced charge separation on a molecular scale.52,53,80,81 However, a very limited effort has been made to employ such systems for photovoltaic applications.82–86As discussed above, the molecular nanoassembly of porphyrins and fullerenes by simple blend is a useful way of constructing photovoltaic systems. However, in such a system, a serious problem is the limited IPCE value (∼17%). Organization of donor and acceptor moieties for efficient charge separation and carrier transport is required.
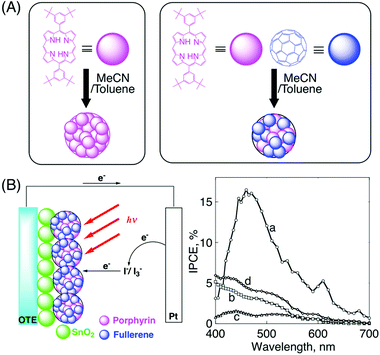
Therefore, a novel quaternary self-organization of porphyrin and fullerene moiety units by clusterization with gold nanoparticles is performed (Fig. 5). First, porphyrin alkanethiolate monolayer-protected gold nanoparticles (H2PCnMPC; where n is the number of methylene groups in the spacer) are prepared (secondary organization), starting from the primary component (porphyrin alkanethiol). These porphyrin-modified gold nanoparticles form complexes with fullerenes (tertiary organization) and they are further clusterized in an acetonitrile–toluene mixed solvent (quaternary organization).87 The composites are stable enough in solution for spectroscopic measurements (Fig. 6).
 | ||
| Fig. 5 An illustration of the quaternary self-organization of porphyrin and C60 units with gold nanoparticles, and their TEM images of (A) H2PC11MPC, (B) (H2PC11MPC)m, and (C) (H2PC11MPC + C60)m.87,88 | ||
![(A) Absorption spectra of (a) (H2PC11MPC + C60)m in acetonitrile–toluene (3 : 1, v/v); [H2P] = 0.19 mM; [C60] = 0.31 mM, (b) H2PC11MPC in toluene, (c) C60 in toluene, and (d) (H2PC11MPC)m in acetonitrile–toluene (3 : 1, v/v). (B) Absorption spectra of (a) mixed H2PC15MPC (0.8 mM) and C60 (5.0 mM), (b) H2PC15MPC, and (c) C60 in o-dichlorobenzene. The charge-transfer (CT) absorption of H2PC15MPC (0.8 mM) and C60 (5.0 mM) (d) has been obtained by subtracting the absorption of (b) and (c) from (a).88](/image/article/2010/CP/b910564f/b910564f-f6.gif) | ||
| Fig. 6 (A) Absorption spectra of (a) (H2PC11MPC + C60)m in acetonitrile–toluene (3:1, v/v); [H2P] = 0.19 mM; [C60] = 0.31 mM, (b) H2PC11MPC in toluene, (c) C60 in toluene, and (d) (H2PC11MPC)m in acetonitrile–toluene (3:1, v/v). (B) Absorption spectra of (a) mixed H2PC15MPC (0.8 mM) and C60 (5.0 mM), (b) H2PC15MPC, and (c) C60 in o-dichlorobenzene. The charge-transfer (CT) absorption of H2PC15MPC (0.8 mM) and C60 (5.0 mM) (d) has been obtained by subtracting the absorption of (b) and (c) from (a).88 | ||
Since there are 57 porphyrin alkanethiolate chains on the gold surface of H2PC11MPC (vide supra),88 the C60 structure, which contains 60 carbons, with a diameter of 7.1 Å, can be used for the calculation of the structure of H2PC11MPC, with a 21 Å diameter, as the gold core.88 There are two type of C–C bonds in C60: 30 short ‘double bonds’(1.404 Å) and 60 long ‘single bonds’ (1.448 Å).88 The average distance between two gold atoms to which neighboring porphyrin alkanethiolates are attached is estimated as 4.24 Å by multiplying the average length of the C–C bonds (1.433 Å) by the ratio of the diameter of the gold core (21 Å) to the diameter of C60 (7.1 Å), as shown in Fig. 7. Using the schematic structure in Fig. 7A affords the center-to-center distances between two porphyrins in H2PC5MPC, H2PC11MPC and H2PC15MPC as 11.5 Å, 15.2 Å and 17.3 Å, respectively. On the other hand, the closest distance between a carbon of C60 and the center of a porphyrin ring has been reported as 2.856 Å by the X-ray crystal structure of the C60 complex with a jaw-like bis-porphyrin.89 Thus, the smallest center-to-center distance of two porphyrin rings which can accommodate a C60 between the rings is estimated as 12.8 Å by adding the diameter of C60 (7.1 Å) to twice the closest distance between a carbon of C60 and the center of a porphyrin ring (5.7 Å). The estimated distances between two porphyrins in H2PC11MPC (15.2 Å) and H2PC15MPC (17.3 Å) are long enough for the two porphyrins to accommodate a C60 between the rings, in contrast to H2PC5MPC (11.5 Å). The longer porphyrin–porphyrin distance in H2PC15MPC than in H2PC11MPC and H2PC5MPC may result in the accommodation of more C60 molecules between the porphyrin rings. The binding constants of H2PC15MPC, H2PC11MPC and H2PC5MPC are also calculated to be 64000, 47000, and 16000 M−1, respectively, by fluorescence quenching results. These trends are in good agreement with the IPCE results (vide infra).88
![(A) Illustration of a photoelectrochemical cell and the insertion of C60 between two porphyrin rings in H2PC15MPC. (B) OTE/SnO2/(H2PCnMPC + C60)m ([H2P] = 0.19 mM, [C60] = 0.31 mM); (a) n = 5, (b) n = 11, (c) n = 15, (d) [H2P] = 0.19 mM, [C60] = 0.38 mM, n = 15. Electrolyte: 0.5 M NaI and 0.01 M I2 in acetonitrile. (C) Current–voltage characteristics of (a) OTE/SnO2/(H2PC15MPC + C60)m and (b) OTE/SnO2/(H2P-ref + C60)m under visible light illumination (λ > 400 nm). Electrolyte: 0.5 M NaI and 0.01 M I2 in acetonitrile; input power: 11.2 mW cm−2. The chemical structure of H2P-ref is shown in Fig. 8.88](/image/article/2010/CP/b910564f/b910564f-f7.gif) | ||
| Fig. 7 (A) Illustration of a photoelectrochemical cell and the insertion of C60 between two porphyrin rings in H2PC15MPC. (B) OTE/SnO2/(H2PCnMPC + C60)m ([H2P] = 0.19 mM, [C60] = 0.31 mM); (a) n = 5, (b) n = 11, (c) n = 15, (d) [H2P] = 0.19 mM, [C60] = 0.38 mM, n = 15. Electrolyte: 0.5 M NaI and 0.01 M I2 in acetonitrile. (C) Current–voltage characteristics of (a) OTE/SnO2/(H2PC15MPC + C60)m and (b) OTE/SnO2/(H2P-ref + C60)m under visible light illumination (λ > 400 nm). Electrolyte: 0.5 M NaI and 0.01 M I2 in acetonitrile; input power: 11.2 mW cm−2. The chemical structure of H2P-ref is shown in Fig. 8.88 | ||
The absorption spectra of H2PC11MPC and C60 in neat toluene are compared to that of [(H2PC11MPC + C60)m] clusters in acetonitrile–toluene (3:1, v/v) in Fig. 6A. (H2PC11MPC + C60)m in the mixed solvent [spectrum (a)] exhibits a much broader and more intense absorption in the visible and near IR regions than those of the parent H2PC11MPC [spectrum (b)] and C60 [spectrum (c)], in toluene.88 This demonstrates that the composite clusters of H2PC11MPC and C60 are superior as light absorbers to the single component systems of H2PC11MPC or C60, since the composite assemblies absorb throughout the visible part of the solar spectrum.
Fig. 6B shows the charge-transfer (CT) absorption between H2PC15MPC and C60. The CT absorption is obtained by subtracting the spectrum of H2PC15MPC and C60 from that of composite H2PC15MPC and C60. The broad absorption in the visible and near IR regions is characteristic of the π-complex formed between porphyrins and fullerenes.90 A CT type interaction in the π-complex between porphyrin and fullerene molecules may be responsible for the long-wavelength absorption of the composite assemblies in Fig. 6B, where the spectrum of (H2PCnMPC + C60)m [spectrum (a) in Fig. 6A] is much broader than that of (H2PCnMPC)m [spectrum (d) in Fig. 6A]. Similar CT interactions leading to such an extended absorption have been observed for porphyrin–C60 dyads linked at close proximity.90,91 Thus, we can control the three-dimensional array of porphyrins and C60 molecules by using gold nanoparticles.
Additionally, the fluorescence lifetime, femtosecond transient absorption and electron spin resonance measurements also support the ultrafast photoinduced electron transfer between porphyrins and fullerenes.88 In particular, the growth of the absorption due to the porphyrin radical cation in (H2PC15MPC + C60)m is observed with a time constant of 375 fs by femtosecond transient absorption measurements.
Next, the highly colored composite assemblies can be assembled as three-dimensional arrays onto nanostructured SnO2 films (OTE/SnO2) to afford the OTE/SnO2/(H2PCnMPC + C60)m electrode using an electrophoretic deposition method. The evaluation of photovoltaic properties are performed using a standard two-compartment cell in the electrolyte (Fig. 7A). Fig. 7B shows the effect of the alkanethiolate chain lengths on the IPCE values. The action spectra indicate that a higher IPCE value and broader photoresponse are attained with a longer chain length of H2PCnMPC. In particular, OTE/SnO2/(H2PC15MPC + C60)m ([H2P] = 0.19 mM, [C60] = 0.38 mM) affords the highest IPCE value (54%) and a very broad photoresponse (up to ∼1000 nm) which extends to the near IR region.88 In OTE/SnO2/(H2PC15MPC + C60)m, a long methylene spacer of H2PC15MPC allows enough space for fullerene molecules to insert themselves between two neighboring porphyrin rings effectively, as compared to the assemblies with a shorter methylene spacer, leading to more efficient photocurrent generation (Fig. 7A). The power conversion efficiency (η) of the OTE/SnO2/(H2PC15MPC + C60)m electrode is calculated by eqn (6):92,93
| η = FFIscVoc/Win, | (6) |
3.2 Porphyrin dendrimers
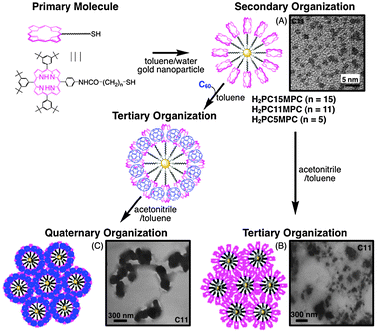
The above strategy can generally be applied to other systems, such as porphyrin dendrimers. Porphyrin dendrimers (DnPn) form supramolecular complexes with fullerenes in toluene and they are clusterized in an acetonitrile–toluene mixed solvent (Fig. 8). The organization of DnPn and C60 composite assemblies [denoted as (DnPn + C60)m (n = 4, 8, 16)] was performed by injecting a mixed toluene solution of DnPn and C60 into acetonitrile–toluene (3:1, v/v). The TEM image of (D8P8 + C60)m (Fig. 8) shows nanoassemblies of diameter ∼200 nm with a well-controlled size and shape. This is in sharp contrast to those of (H2P-ref + C60)m, which have irregular and smaller sizes. These results clearly show that the dendritic structure plays an important role in controlling the formation of molecular assemblies.94 The light energy conversion properties of OTE/SnO2/(DnPn + C60)m electrodes are evaluated in photoelectrochemical cells. The OTE/SnO2/(D4P4+C60)m system has a maximum IPCE value of 15%, as well as a broad photoresponse that extends well into the infrared region (up to 1000 nm).95 Such an effective photoenergy conversion is largely ascribed to the dendritic structure controlling the three-dimensional organization between porphyrin and C60. However, the IPCE value of OTE/SnO2/(D16P16 + C60)m is smaller than that of the reference system. This is likely caused by the steric effect of the porphyrin moieties in the higher generations of the dendrimers, which hamper the π–π interaction with C60.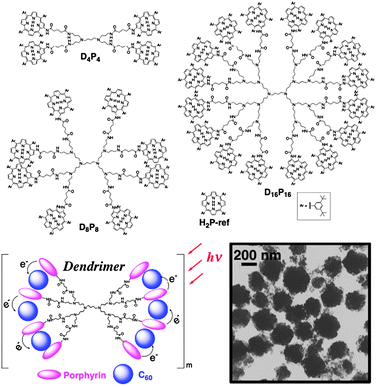 | ||
| Fig. 8 Porphyrin dendrimers and the reference compound employed in this study. The TEM image is of (D8P8 + C60)m.95,96 | ||
3.3 Porphyrin–peptide oligomers
As discussed above, in the case of composite nanoassemblies composed of porphyrin dendrimers and fullerenes, the light energy conversion property decreases with increasing dendritic generations because of the ‘rigid’ structure of the porphyrin dendrimers in the higher dendritic generations, which hampers effective interactions between the porphyrin units and the C60. Therefore, multi-porphyrin arrays with rather flexible structures, but without energy transfer quenching, are required to improve the energy conversion efficiency in the composite systems with C60. Fig. 9A shows the chemical structures of porphyrin–peptide oligomers with different numbers of porphyrins in the polypeptide unit (porphyrin functionalized α-polypeptides). Such porphyrin oligomers with polypeptidic backbones are flexible enough to accommodate C60 between the porphyrin units. Moreover, no undesirable and accelerated quenching of the excited state of the porphyrins is observed, which is in sharp contrast to the strong quenching of porphyrin units by gold nanoparticles in H2PCnMPC. Fig. 9A (right) also shows the preparation procedure and TEM image of (P(H2P)16 + C60)m. The sizes and shapes of (P(H2P)16 + C60)m are much more controlled as compared to those of (P(ZnP)1 + C60)m.97![(A) Chemical structures of porphyrin–peptide oligomers employed in this study (left). The right Figure shows an organization process of P(H2P)16 with C60 [denoted as (P(H2P)16 + C60)m]. TEM image is of (P(H2P)16 + C60)m. (B) The photocurrent action spectra (IPCE vs. wavelength) of (a) (P(H2P)16 + C60)m, (b) (P(H2P)8 + C60)m, (c) (P(H2P)4 + C60)m, (d) P(H2P)2 + C60)m and (e) (P(H2P)1 + C60)m on OTE/SnO2 electrodes. (C) The photocurrent action spectra of (a) (P(ZnP)16 + C60)m, (b) (P(ZnP)8 + C60)m, (c) (P(ZnP)4 + C60)m, (d) (P(ZnP)2 + C60) and (e) (P(ZnP)1 + C60)m on OTE/SnO2 electrodes.97](/image/article/2010/CP/b910564f/b910564f-f9.gif) | ||
| Fig. 9 (A) Chemical structures of porphyrin–peptide oligomers employed in this study (left). The right Figure shows an organization process of P(H2P)16 with C60 [denoted as (P(H2P)16 + C60)m]. TEM image is of (P(H2P)16 + C60)m. (B) The photocurrent action spectra (IPCE vs. wavelength) of (a) (P(H2P)16 + C60)m, (b) (P(H2P)8 + C60)m, (c) (P(H2P)4 + C60)m, (d) P(H2P)2 + C60)m and (e) (P(H2P)1 + C60)m on OTE/SnO2 electrodes. (C) The photocurrent action spectra of (a) (P(ZnP)16 + C60)m, (b) (P(ZnP)8 + C60)m, (c) (P(ZnP)4 + C60)m, (d) (P(ZnP)2 + C60) and (e) (P(ZnP)1 + C60)m on OTE/SnO2 electrodes.97 | ||
Fig. 9A shows the IPCE of the (P(H2P)n + C60)m (n = 1, 2, 4, 8, 16) modified electrode at a constant concentration ratio of porphyrin to C60 (vide supra). The IPCE value of the (P(H2P)n + C60)m system exhibits a remarkable increase with increasing the number of porphyrins in a polypeptide unit. In particular, the (P(H2P)16 + C60)m system has a maximum IPCE value of 48% at 600 nm, as well as a broad photoresponse, extending into the infrared region (up to 1000 nm).96,97 Such an effective light energy conversion is ascribed to the polypeptide structure controlling the three-dimensional organization between porphyrins and C60. The maximum IPCE of the (P(ZnP)n + C60)m system also exhibits a remarkable increase with an increasing number of porphyrins in a polypeptide unit. The maximum IPCE of (P(ZnP)16 + C60)m is determined to be 56% at 480 nm.97
The major reason for the increase of IPCE values with an increase of the number of porphyrins in a polypeptide unit is the charge-separation process between porphyrins and fullerenes. The UV–vis absorption and fluorescence spectral changes give us useful information on supramolecular formation between porphyrins and fullerenes. The binding constants between porphyrins and fullerenes are thereby evaluated quantitatively. The spectral changes indicate that C60 forms a 1:1 complex with the porphyrin moiety. Based on the reported equation,98 the apparent formation constant (K) between P(H2P)16 and C60 determined from the fluorescence quenching (8.2 × 104 M−1) is significantly larger than that determined from the UV–vis spectral change (1.4 × 104 M−1). This indicates that the excited energy migration between the porphyrin units occurs efficiently prior to the electron transfer. The K values of various types of porphyrin–peptide oligomers [P(H2P)n or P(ZnP)n] and C60 derivatives are listed in Table 2.97 The association constant between porphyrins and C60 increases with an increasing number of porphyrins in a polypeptide unit. This indicates that the interaction between porphyrin and C60 becomes stronger with an increasing number of porphyrins in the porphyrinic polypeptide. Such differences in the association constants are in good agreement with the IPCE results (Fig. 9B and C).97
97
The photodynamics of the composite molecular clusters was further measured by femtosecond time-resolved transient absorption spectra to examine the charge-separation process (Fig. 10). Upon a 387 nm laser pulse excitation, we observe a broad absorption in the 450–510 nm region as we populate the porphyrin singlet excited state (Fig. 10A). This transient quickly decays to form a triplet excited state. In addition, a strong transient bleaching (around 610 nm) arises from the fluorescence. In contrast to the (P(ZnP)1 + C60)m system, the strong absorption arising from singlet and triplet excited states of the porphyrins (450–510 nm) is missing in the case of (P(ZnP)8 + C60)m. Instead, a broad absorption at around 650 nm appears after the laser pulse excitation, as shown in Fig. 10B. Such broad absorption spectra are a clear indication of the formation of the porphyrin radical cation. It is also interesting to note that the bleaching originating from fluorescence (610 nm region) is absent in the transient spectra (Fig. 10B). Thus, different supramolecular formations between porphyrins and fullerenes have a great effect on the photoinduced electron transfer rate constants.
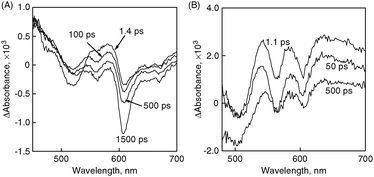 | ||
| Fig. 10 Time-resolved absorption spectra recorded following a 387 nm laser pulse excitation of (A) (P(ZnP)1 + C60)m and (B) (P(ZnP)8 + C60)m. All samples were prepared in argon-saturated acetonitrile–toluene (3:1, v/v) and measurements were made at 298 K.97 | ||
The fluorescence lifetime and electron spin resonance (ESR) results also support the results of the investigation of the photoinduced electron transfer process. The fluorescence decay curves are shown in Fig. 11A. The fluorescence decay of (P(H2P)16 + C60)m is strongly quenched in contrast to that of (P(H2P)16)m without C60. This indicates that efficient photoinduced electron transfer takes place from 1P(H2P)*16 to C60. The fluorescence lifetimes of the first major component [e.g., OTE/SnO2/(P(H2P)16 + C60)m: 70 ps and OTE/SnO2/(P(H2P)1 + C60)m: 135 ps] decrease with an increasing number of porphyrins in a polypeptide unit because of the acceleration of the forward electron transfer.97
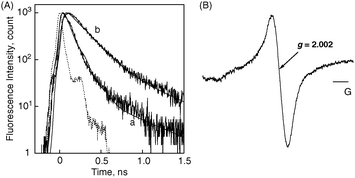 | ||
| Fig. 11 (A) Fluorescence decay curves of (a) (P(H2P)16 + C60)m and (b) (P(H2P)16)m modified electrodes. The fluorescence decays are observed at 650 nm by the time-correlated single-photon counting method. The excitation wavelength is 420 nm and IRF (instrument response function) curves are presented for each decay. (B) ESR spectrum of photoirradiated (P(ZnP)16 + C60)m in acetonitrile–toluene (3:1, v/v) under photoirradiation of a high-pressure mercury lamp, measured at 123 K.97 | ||
The formation of the radical cation of P(ZnP)16 and the radical anion of C60 in the (P(ZnP)16 + C60)m composite assemblies upon photoexcitation is confirmed by ESR measurements performed in frozen acetonitrile–toluene under photoirradiation. The resulting ESR spectrum of (P(ZnP)16 + C60)m upon photoirradiation in acetonitrile–toluene at 123 K is shown in Fig. 11B. The ESR signal (g = 2.002) is assigned to the overlap of two signals, due to the porphyrin radical cation and C60˙−.39 Thus, our supramolecular strategy using multi-porphyrin arrays and fullerenes for photovoltaics successfully contributes to the drastic improvement of light energy conversion properties.97 In addition to the above three systems, we have also observed similar drastic improvements in other systems, such as porphyrin-modified TiO2 nanoparticle/nanotubes and other dye molecules.78,92,93,99–105
3.4 Photocurrent generation mechanism in supramolecular composites between porphyrins and fullerenes
Photocurrent generation in the present systems (Scheme 1) is initiated by photoinduced charge separation from the singlet excited state of freebase porphyrin (1H2P*/H2P˙+ = −0.7 V vs. the normal hydrogen electrode, NHE)97 or zinc porphyrin (1ZnP*/ZnP˙+ = −1.0 V vs. NHE)97 to C60 (C60/C60˙− = −0.2 V vs. NHE)97 in the porphyrin–C60 complex, rather than direct electron injection to the conduction band of SnO2 (0 V vs. NHE) system.97 The reduced C60 injects electrons into the SnO2 nanocrystallites, whereas the oxidized porphyrin (H2P/H2P˙+ = 1.2 V or ZnP/ZnP˙+ = 1.0 V vs. NHE)97 undergoes electron-transfer reduction with the iodide (I3−/I− = 0.5 V vs. NHE)97 in the electrolyte system. In such a case, electron injection following charge separation occurs more effectively to a nanostructured SnO2 electrode.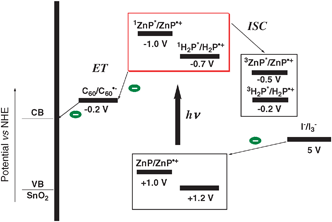 | ||
| Scheme 1 Schematic illustration of the photocurrent generation mechanism.97 | ||
4. Supramolecular assemblies of carbon nanotubes
4.1 SWCNT driven aggregation of protonated porphyrins
Single wall carbon nanotubes (SWCNTs) can provide an ideal network to promote charge transfer with dye molecules and to transport electrons to the collecting surface (e.g., photoelectrochemical cells) because of their π-conjugated cylinder structure. On the other hand, protonated porphyrins provide a convenient way to construct ordered molecular assemblies. Therefore, we have prepared a novel SWCNTs driven aggregation of protonated porphyrins to produce supramolecular assemblies in the form of macroscopic bundles (Fig. 12).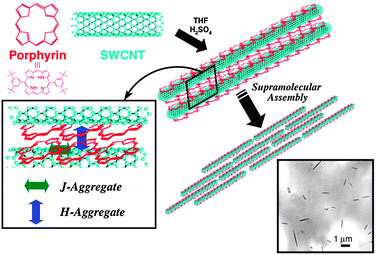 | ||
| Fig. 12 Illustration of a supramolecular assembly between protonated porphyrins and single wall carbon nanotubes (SWCNT) in this study.67 | ||
The general strategy and procedure of constructing a supramolecular assembly between 5,15-bis(3,5-di-tert-butylphenyl)porphyrin (H2P) and SWCNTs is shown in Fig. 12. 1 mg of purified SWCNTs was added to a 0.2 mM solution of porphyrin in THF containing 1.0% H2SO4 (v/v). The suspension was sonicated for 1 h at room temperature, followed by centrifugation of the suspension for 30 min at 10000 rpm to separate the SWCNTs–H4P2+ composite. The solid was resuspended in acidified THF with sonication (1 h) and centrifuged again to wash out any unbound porphyrin. The first step involves the binding of H2P on the surface of the SWCNTs. This is followed by Van der Waal’s interaction to form aligned macroscopic rods. The protonated form of H2P (H4P2+) and SWCNT composites form 0.5–3.0 μm sized rod-like structures.67
These rod-assemblies can be assembled onto nanostructured SnO2 films (OTE/SnO2) using a similar electrophoretic deposition method as in previous sections. These organized assemblies are photoactive and absorb strongly in the entire visible region. The IPCE of OTE/SnO2/SWCNTs–H4P2+ is ∼13% under an applied potential of 0.2 V vs. SCE.106 This value is much larger than those of the respective single component systems. Thus, the dual role of SWCNTs in promoting photoinduced charge separation and facilitating charge transport is presented.
4.2 Stacked-cup carbon nanotubes and others
Whereas conventional carbon nanotubes are made up of seamless cylinders of hexagonal carbon networks, the cup-stacked structure provides a hollow tubular morphology (Fig. 13).107,108 This stacked-cup morphology provides a large portion of exposed and reactive edges in the outer and inner surfaces of the hollow tubes. The availability of the inner and outer edges of these stacked-cups to chemical functionalization or surface modification opens up new avenues for their utilization in electronic and catalytic applications. Upon ball-milling, the longer stacked cups can be broken into smaller stacked cups and further increase the number of accessible active sites.109 These stacked-cup carbon nanotube (SCCNT) films on OTE/SnO2 undergo charge separation and deliver photocurrents under visible light irradiation. Its maximum IPCE of 17% at +0.2 V vs. SCE is much larger than those reported for pristine carbon nanostructures. For example, C60 films exhibit a maximum IPCE of ∼5% and SWCNT films exhibit a maximum IPCE of less than 0.2%.110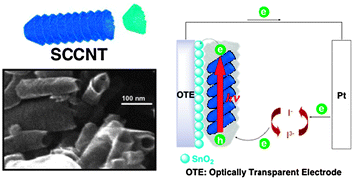 | ||
| Fig. 13 Illustration and SEM image of stacked-cup carbon nanotubes (SCCNT) and their application to the photoelectrochemical cell in this study.110 | ||
As stated above, the longer parent SCCNT can be broken into smaller ones upon ball-milling (vide supra). Three SCCNT samples with different tube-lengths (long type: L-SCCNT [>5 μm], medium type: M-SCCNT [1–5 μm], and short type: S-SCCNT [0.2–1.0 μm]) were electrophoretically deposited on OTE/SnO2 electrodes to probe the tube length dependence of photoelectrochemical behavior. The power conversion efficiencies (η) of SCCNT-modified electrodes increase with increasing tube-length. The maximum power conversion efficiency (η) of the OTE/SnO2/L-SCCNT electrode is determined to be 0.11%, which is about 6 times greater than that of the OTE/SnO2/S-SCCNT electrode (0.018%). As shown in a previous study,111 carbon nanotubes serve as conduits for transporting electrons efficiently. In this respect, the longer SCCNT tubes should transport the charge carriers over a longer distance without undergoing significant loss during the transit.
Moreover, molecular assemblies composed of S-SCCNT and 5,15-bis(3,5-di-tert-butylphenyl)porphyrin (H2P) prepared in acetonitrile–toluene (5:1, v/v) were deposited as three-dimensional arrays onto nanostructured SnO2 films to further improve their photoelectrochemical response in the visible region. The composite electrode of S-SCCNT–H2P (OTE/SnO2/S-SCCNT–H2P) exhibits an IPCE of 32% under an applied potential of 0.2 V vs. SCE.112 The observed increase in IPCE is greater than that of the additive effects observed from the single component systems (viz., OTE/SnO2/S-SCCNT or OTE/SnO2/H2P). In addition to SWCNT and SCCNT, we have also reported structural and photoelectrochemical properties of carbon nanohorn–porphyrin composites (IPCE: ∼6%).113 Thus, the organization of carbon nanotubes with dye molecules, such as porphyrins, demonstrates efficient light energy conversion properties.
5. Supramolecular porphyrin nanorods
5.1 Sonication-assisted nanorods of porphyrins
Recently, bar-shaped assemblies of porphyrins have been widely reported. In particular, relatively strong interactions, such as ionic, coordination and hydrogen bonds, are popular ways for organization.29,43,114–116 On the other hand, ultrasonic waves are often utilized for the formation of crystal nuclei and growth in the preparation of molecular crystals but seldom favors the formation of an ordered assembly. We have constructed, in a sonicated solution medium, novel supramolecular nanorods and fibers controlled by intermolecular interactions such as the π–π stacking interaction of porphyrins, without other bonds/interactions or aiding surfactants.The porphyrin nanorod was prepared by the following procedure. First, a 3.5 mmol dm−3 5,15-bis(3,5-di-tert-butylphenyl)porphyrin (H2P) solution was prepared in toluene. Then, the toluene solution was simply mixed with 9 times its volume of acetonitrile (final concentration: 0.35 mmol dm−3 in a poor/good solvent = 9:1, v/v). The solution was sonicated (45 kHz) using our original set-up for 30 min at 15 °C to form macroscopic self-assembled porphyrins, such as rod-like structures.
Fig. 14A shows TEM images of H2P nanorods. The dimensions of H2P nanorods are 360 ± 130 nm in diameter and 5.02 ± 1.94 μm in length. To construct much longer fiber structures, the nanorod suspended solution is further allowed to stand for 6 days. Then, the crystallized porphyrins were stirred for 5 min at room temperature. The nanorod crystals finally grew to be longer fibers. In Fig. 14B, we can clearly see much longer fiber structures, measured as 890 ± 270 nm in diameter and 27.2 ± 6.9 μm in length. In the formation of the fiber structures, a large increase of length from 5.0 to 27.2 μm (∼6 times) relative to that of diameter (∼2 times) indicates that anisotropic crystal growth largely occurs in the length direction. Thus, we have successfully controlled the structures of porphyrin fibrous assemblies within a certain definite range. Additionally, the internal structures of the self-assembled porphyrins are investigated by XRD analysis. We can observe strong diffraction peaks of the a and c axes, such as (100) and (002), in the unit cell, whereas the diffraction intensity based on the b axis is very weak. Considering the unit cell structure (monoclinic structure: angles of 90° between axes a and b and b and c), the growth direction of the rod-crystals may be in the b axis direction.47
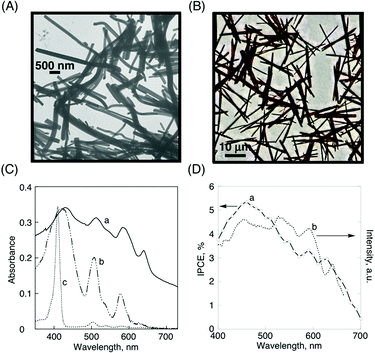 | ||
| Fig. 14 (A) TEM image of H2P nanorods, (B) optical microscope image of H2P fibers, (C) absorption spectra of (a) H2P nanorod film, (b) H2P drop-cast film on quartz plates, and (c) H2P (4 × 10−6 mol dm−3) in toluene, (D) (a) photocurrent (IPCE) action spectrum of H2P nanorods on an OTE/SnO2 electrode (electrolyte: 0.5 mol dm−3 LiI and 0.01 mol dm−3 I2 in acetonitrile), (b) excitation spectrum of H2P nanorod films (observed at 710 nm).47 | ||
In Fig. 14C, the absorption spectrum of H2P nanorods [spectrum (a)] exhibits a much broader and more intense absorption in the visible and near infrared regions than those of the corresponding simple drop-cast film prepared by H2P in toluene [spectrum (b)] and toluene solution [spectrum (c)]. This change of absorption property is likely because of the strong supramolecular π–π interactions between a porphyrin and its nearest neighbor in nanorod structures. The IPCE spectrum [spectrum (a) in Fig. 14D] also shows a broad photoresponse in the visible region (maximum IPCE: ∼5.5% at 460 nm),47 which parallels the corresponding absorption [spectrum (a) in Fig. 14C] and excitation spectra [spectrum (b) in Fig. 14D]. These experiments confirmed the role of H2P nanorods towards harvesting light energy and generating photocurrent during the operation of a photoelectrochemical cell.
5.2 Fullerene-encapsulated porphyrin hexagonal nanorods
To date, these bar-shaped porphyrin nanoassemblies are mainly composed of single molecular species.43,117–121 Considering the photovoltaic mechanism, a bar-shaped structure composed of two different molecules with separated inside and outside layers is a good candidate for efficient carrier generation (hole and electron). Therefore, we have succeeded in the construction of a new type of molecular composites: fullerene-encapsulated porphyrin hexagonal nanorods composed of zinc meso-tetra (4-pyridyl) porphyrin [ZnP(Py)4] and C60 [denoted as C60–ZnP(Py)4 nanorods], which are prepared by an aiding surfactant: cetyltrimethylammonium bromide (CTAB) in a DMF–acetonitrile mixed solvent (Fig. 15). The preparation method is as follows. A mixture of the appropriate ratio of ZnP(Py)4 and C60 in DMF solution (1. Basic monomers) was injected into 7.5 times its volume of a continuously stirred 0.20 mM CTAB acetonitrile solution at room temperature. The final concentrations of ZnP(Py)4 and C60 are 0.03 and 0.02 mM in DMF–acetonitrile (2:15, v/v), respectively. On injection, they largely form ZnP(Py)4 flake assemblies and C60-based nanoparticles (ca. 5–20 nm in diameter), separately (2. Building blocks and Fig. 15A). With the diffusion of DMF into acetonitrile, the Zn–N axial coordination of pyridyl N-atoms to the zinc atoms of ZnP(Py)4 promotes the growth of aggregates, which continue to grow into a flake structure. In this case, the organization process of the ZnP(Py)4 moieties is initially derived from coordination bonds, in contrast to the C60 assemblies, which are based on relatively weak π–π interactions. Therefore, once injected, two different types of assemblies are quickly observed. Fig. 15B further shows the build-up formations of ZnP(Py)4 and C60 after 3 min (3. Built-up process). Then, after several minutes, C60–ZnP(Py)4 nanorods are finally formed (Fig. 15C). The reference ZnP(Py)4 hexagonal nanotube without C60 was also prepared in the same manner for comparison [denoted as ZnP(Py)4 nanotubes]. It should be noted that the prepared samples were repeatedly centrifuged at 14000 rpm to remove CTAB from the DMF–acetonitrile solvent and filtrated to separate unbounded ZnP(Py)4 and C60.122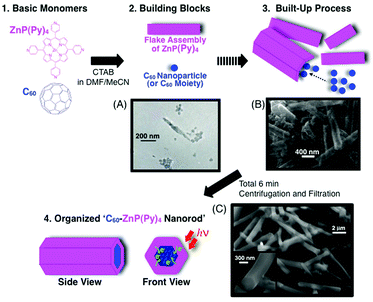 | ||
| Fig. 15 Schematic illustration of the organization process of C60 and ZnP(Py)4 with CTAB in this study. CTAB is omitted for clarify. The electron micrographs show the time-dependent formations: (A) 1 min, (B) 3 min and (C) 6 min after injection.122 | ||
ZnP(Py)4 nanotubes show a bar-like structure with a large hollow hole, whereas the hole is completely closed in C60–ZnP(Py)4 nanorods (Fig. 15C). C60–ZnP(Py)4 nanorods are measured to be 4.12 ± 0.94 μm in length and 490 ± 90 nm in outside diameter by SEM images. Compared to ZnP(Py)4 nanotubes, measured to be 2.13 ± 0.27 μm in length and 540 ± 30 nm in outside diameter, a large increase in the length direction from 2.13 to 4.12 μm, relative to unchanged diameters (∼500 nm), indicates that anisotropic crystal growth largely occurs in the length direction, due to π–π interactions of encapsulated C60 moieties within the ZnP(Py)4 assembly.122
The absorption spectrum of C60–ZnP(Py)4 nanorods exhibits much a broader and more intense absorption in the visible and near infrared regions than those of the corresponding monomers (ZnP(Py)4 or C60) in DMF because of aggregated interactions. Such a broad absorption property is useful for solar energy conversion. The picosecond fluorescence lifetime and nanosecond transient absorption measurements of C60–ZnP(Py)4 nanorods suggest efficient quenching of the singlet excited state by the C60 moiety because of photoinduced electron transfer (PET), which is in sharp contrast to the PET via3ZnP(Py)4* in the reference non-organized system prepared without CTAB. The photocurrent action spectrum of the OTE/C60–ZnP(Py)4 nanorods shows a broad photoresponse in the visible region, and the maximum IPCE (∼20%) is much larger than the sum of the two individual IPCE values (∼6.5%) of the OTE/ZnP(Py)4 nanotube and the OTE/C60 assembly under the same conditions.122 These results clearly indicate that an organized structure between C60 and ZnP(Py)4, as well as the excellent electron acceptor property of C60, play a role in the improvement of light energy conversion.
6. Concluding remarks
In this perspective, recent developments in supramolecular architectures for light energy conversion (i.e., organic photovoltaics), ranging from simple blend systems to highly organized assemblies using laborious synthetic chemistry, were described. The conventional photovoltaic cells composed of multilayer films are generally fabricated by top–down approaches, such as vacuum deposition and spin-coating techniques. Such methods are simply direct depositions of organic molecules as bulky assemblies onto surfaces, which hampers the ordered alignment of organic molecules at the molecular or nanometre level. On the other hand, in the cases of synthetic and supramolecular approaches, utilization of covalent and noncovalent bondings enables us to organize effectively. Therefore, considering photovoltaic processes, such as light-harvesting (and exciton diffusion), charge separation and carrier transport, we can systematically construct supramolecular photofunctional materials for light energy conversion. In the photoelectrochemical measurements, these highly organized molecular architecture-deposited electrodes quantitatively exhibit drastic enhancements of light energy conversion properties as compared to the reference non-organized system (maximum: ca. 50 times).As compared to the major conventional systems, such as dye-sensitized solar cells11 and solid-state photovoltaic cells,9 further improvements of the conversion efficiency are certainly required (maximum η: ∼2%). The current simple electrophoretic method is an easy method for molecular deposition. However, the relatively random orientation by electrophoresis has limitations for the more precise three-dimensional array of molecular assemblies, which hamper the efficient carrier transport and interfacial control between the organic layer and the electrode. Therefore, one of the new major strategies for the improvement of the light energy conversion property may be the precise alignment of these molecular assemblies onto the surface electrode using a patterning technique,120,123–128 which leads to the development of solid-state cells. Finally, by overcoming these problems, together with the development of new molecular architectures, such supramolecular strategies could pave the way for developing future photonics, electronics and light energy conversion systems.
Acknowledgements
The author thanks and expresses gratitude to his collaborators and coworkers, whose names appear in the references. This work was partially supported by Grant-in-Aids for Scientific Research (No. 18810014, 19710119 & 21710104) and special coordination funds for promoting science and technology from the Ministry of Education, Culture, Sports, Science and Technology, Japan. The author also thanks Dr Y. Araki (Tohoku Univ.) for useful discussion. All Figures, Schemes and Tables are reproduced from the related references with permission of the American Chemical Society, the Royal Society of Chemistry and Wiley-VCH.References
- M. I. Hoffert, K. Caldeira, G. Benford, D. R. Criswell, C. Green, H. Herzog, A. K. Jain, H. S. Kheshgi, K. S. Lackner, J. S. Lewis, H. D. Lightfoot, W. Manheimer, J. C. Mankins, M. E. Mauel, L. J. Perkins, M. E. Schlesinger, T. Volk and T. M. L. Wigley, Science, 2002, 298, 981–987 CrossRef CAS.
- M. I. Hoffert, K. Caldeira, A. K. Jain, E. F. Haites, L. D. D. Harvey, S. D. Potter, M. E. Schlesinger, S. H. Schneider, R. G. Watts, T. M. L. Wigley and D. J. Wuebbles, Nature, 1998, 395, 881–884 CrossRef CAS.
- M. E. Mann, R. S. Bradley and M. K. Hughes, Nature, 1998, 392, 779–787 CrossRef CAS.
- N. Myers and J. Kent, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4963–4968 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834–2860 CrossRef CAS.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef.
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737–18753 CAS.
- J. Y. Kim, K. Lee, N. E. Coates, D. Moses, T.-Q. Nguyen, M. Dante and A. J. Heeger, Science, 2007, 317, 222–225 CrossRef CAS.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef.
- M. Grätzel, Inorg. Chem., 2005, 44, 6841–6851 CrossRef.
- A. Hagfeldt and M. Grätzel, Chem. Rev., 1995, 95, 49–68 CrossRef CAS.
- H. Hoppe and N. S. Sariciftci, J. Mater. Res., 2004, 19, 1924–1945 CAS.
- P. Peumans, S. Uchida and S. R. Forrest, Nature, 2003, 425, 158–162 CrossRef CAS.
- V. Shrotriya, G. Li, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Adv. Funct. Mater., 2006, 16, 2016–2023 CrossRef CAS.
- X. Yang, J. Loos, S. C. Veenstra, W. J. H. Verhees, M. M. Wienk, J. M. Kroon, M. A. J. Michels and R. A. J. Janssen, Nano Lett., 2005, 5, 579–583 CrossRef CAS.
- R. J. Tseng, R. Chan, V. C. Tung and Y. Yang, Adv. Mater., 2008, 20, 435–438 CrossRef CAS.
- M. Hiramoto, T. Yamaga, M. Danno, K. Suemori, Y. Matsumura and M. Yokoyama, Appl. Phys. Lett., 2006, 88, 213105–213103 CrossRef.
- T. Hasobe, S. Fukuzumi and P. V. Kamat, Interface, 2006, 15, 47–51 Search PubMed.
- S.-S. Sun and N. S. Sariciftci, Organic Photovoltaics, Taylor & Francis, Boca Raton, 2005 Search PubMed.
- C. Brabec, V. Dyakonov, J. Parisi and N. S. Sariciftci, Organic Photovoltaics, Springer, Berlin, 2003 Search PubMed.
- G. Horowitz and M. E. Hajlaoui, Synth. Met., 2001, 122, 185–189 CrossRef CAS.
- G. Horowitz and M. E. Hajlaoui, Adv. Mater., 2000, 12, 1046–1050 CrossRef CAS.
- A. Di Carlo, F. Piacenza, A. Bolognesi, B. Stadlober and H. Maresch, Appl. Phys. Lett., 2005, 86, 263501–263503 CrossRef.
- V. Percec, M. Glodde, T. K. Bera, Y. Miura, I. Shiyanovskaya, K. D. Singer, V. S. K. Balagurusamy, P. A. Heiney, I. Schnell, A. Rapp, H. W. Spiess, S. D. Hudson and H. Duan, Nature, 2002, 417, 384–387 CrossRef.
- S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hägele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Angew. Chem., Int. Ed., 2007, 46, 4832–4887 CrossRef CAS.
- L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596–1608 CrossRef CAS.
- T. Seki, S. Yagai, T. Karatsu and A. Kitamura, J. Org. Chem., 2008, 73, 3328–3335 CrossRef CAS.
- F. J. M. Hoeben, M. Wolffs, J. Zhang, S. De Feyter, P. Leclere, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2007, 129, 9819–9828 CrossRef CAS.
- R. Madueno, M. T. Raisanen, C. Silien and M. Buck, Nature, 2008, 454, 618–621 CrossRef CAS.
- M. W. Hosseini, Acc. Chem. Res., 2005, 38, 313–323 CrossRef CAS.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS.
- Y. Gao, X. Zhang, C. Ma, X. Li and J. Jiang, J. Am. Chem. Soc., 2008, 130, 17044–17052 CrossRef CAS.
- R. F. Kelley, S. J. Lee, T. M. Wilson, Y. Nakamura, D. M. Tiede, A. Osuka, J. T. Hupp and M. R. Wasielewski, J. Am. Chem. Soc., 2008, 130, 4277–4284 CrossRef CAS.
- S.-S. Li, B. H. Northrop, Q.-H. Yuan, L.-J. Wan and P. J. Stang, Acc. Chem. Res., 2009, 42, 249–259 CrossRef CAS.
- The Photosynthetic Reaction Center, ed. J. Deisenhofer and J. R. Norris, Academic Press, San Diego, 1993 Search PubMed.
- S. Fukuzumi, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 8, pp. 115–152 Search PubMed.
- S. Fukuzumi, Y. Endo and H. Imahori, J. Am. Chem. Soc., 2002, 124, 10974–10975 CrossRef CAS.
- D. Gust and T. A. Moore, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, CA, 2000, vol. 8, pp. 153–190 Search PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40–48 CrossRef CAS.
- C. M. Drain, G. Smeureanu, S. Patel, X. Gong, J. Garnod and J. Arijeloyea, New J. Chem., 2006, 30, 1834–1843 RSC.
- T. Kojima, R. Harada, T. Nakanishi, K. Kaneko and S. Fukuzumi, Chem. Mater., 2007, 19, 51–58 CrossRef CAS.
- Z. Wang, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2004, 126, 15954–15955 CrossRef CAS.
- A. D. Schwab, D. E. Smith, C. S. Rich, E. R. Young, W. F. Smith and J. C. dePaula, J. Phys. Chem. B, 2003, 107, 11339–11345 CrossRef CAS.
- P. A. J. de Witte, M. Castriciano, J. J. L. M. Cornelissen, L. M. Scolaro, R. J. M. Nolte and A. E. Rowan, Chem.–Eur. J., 2003, 9, 1775–1781 CrossRef CAS.
- S. J. Lee, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2008, 130, 9632–9633 CrossRef CAS.
- T. Hasobe, H. Oki, A. S. D. Sandanayaka and H. Murata, Chem. Commun., 2008, 724–726 RSC.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- V. D. Mihailetchi, J. K. J. van Duren, P. W. M. Blom, J. C. Hummelen, R. A. J. Janssen, J. M. Kroon, M. T. Rispens, W. J. H. Verhees and M. M. Wienk, Adv. Funct. Mater., 2003, 13, 43–46 CrossRef CAS.
- X. Ai, M. C. Beard, K. P. Knutsen, S. E. Shaheen, G. Rumbles and R. J. Ellingson, J. Phys. Chem. B, 2006, 110, 25462–25471 CrossRef CAS.
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Science, 1992, 258, 1474–1476 CrossRef CAS.
- H. Imahori, Y. Mori and Y. Matano, J. Photochem. Photobiol., C, 2003, 4, 51–83 CrossRef CAS.
- Y. Araki and O. Ito, J. Photochem. Photobiol., C, 2008, 9, 93–110 CrossRef CAS.
- X. Yang and J. Loos, Macromolecules, 2007, 40, 1353–1362 CrossRef CAS.
- Martijn M. Wienk, Jan M. Kroon, Wiljan J. H. Verhees, Joop Knol, Jan C. Hummelen, Paul A. van Hal and R. A. J. Janssen, Angew. Chem., Int. Ed., 2003, 42, 3371–3375 CrossRef CAS.
- H. Imahori, N. V. Tkachenko, V. Vehmanen, K. Tamaki, H. Lemmetyinen, Y. Sakata and S. Fukuzumi, J. Phys. Chem. A, 2001, 105, 1750–1756 CrossRef CAS.
- F. D’Souza, G. R. Deviprasad, M. E. El-Khouly, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 2001, 123, 5277–5284 CrossRef CAS.
- Y. B. Wang and Z. Lin, J. Am. Chem. Soc., 2003, 125, 6072–6073 CrossRef CAS.
- K. Tashiro and T. Aida, Chem. Soc. Rev., 2007, 36, 189–197 RSC.
- P. D. W. Boyd and C. A. Reed, Acc. Chem. Res., 2005, 38, 235–242 CrossRef CAS.
- Y. Zhang and S. Iijima, Phys. Rev. Lett., 1999, 82, 3472–3475 CrossRef CAS.
- H. Dai, Acc. Chem. Res., 2002, 35, 1035–1044 CrossRef CAS.
- D. M. Guldi, G. M. A. Rahman, F. Zerbetto and M. Prato, Acc. Chem. Res., 2005, 38, 871–878 CrossRef CAS.
- S. Barazzouk, S. Hotchandani, K. Vinodgopal and P. V. Kamat, J. Phys. Chem. B, 2004, 108, 17015–17018 CrossRef CAS.
- T. Ando, J. Phys. Soc. Jpn., 1997, 66, 1066–1073 CrossRef CAS.
- Y.-Z. Ma, J. Stenger, J. Zimmermann, S. M. Bachilo, R. E. Smalley, R. B. Weisman and G. R. Fleming, J. Chem. Phys., 2004, 120, 3368–3373 CrossRef CAS.
- T. Hasobe, S. Fukuzumi and P. V. Kamat, J. Am. Chem. Soc., 2005, 127, 11884–11885 CrossRef CAS.
- S. Jiang and M. Liu, J. Phys. Chem. B, 2004, 108, 2880–2884 CrossRef CAS.
- W. Xu, H. Guo and D. L. Akins, J. Phys. Chem. B, 2001, 105, 1543–1546 CrossRef CAS.
- X. Gong, T. Milic, C. Xu, J. D. Batteas and C. M. Drain, J. Am. Chem. Soc., 2002, 124, 14290–14291 CrossRef CAS.
- A. S. D. Sandanayaka, Y. Araki, T. Wada and T. Hasobe, J. Phys. Chem. C, 2008, 112, 19209–19216 CrossRef CAS.
- K. Yokoi and Y. Ohba, J. Chem. Phys., 1987, 86, 3318–3324 CrossRef CAS.
- T. Yago, Y. Tamaki, A. Furube and R. Katoh, Phys. Chem. Chem. Phys., 2008, 10, 4435–4441 RSC.
- A. Huijser, B. M. J. M. Suijkerbuijk, R. J. M. Klein Gebbink, T. J. Savenije and L. D. A. Siebbeles, J. Am. Chem. Soc., 2008, 130, 2485–2492 CrossRef CAS.
- A. Huijser, T. J. Savenije, A. Kotlewski, S. J. Picken and L. D. A. Siebbeles, Adv. Mater., 2006, 18, 2234–2239 CrossRef CAS.
- L. Valkunas, E. Åkesson, T. Pullerits and V. Sundström, Biophys. J., 1996, 70, 2373–2379 CrossRef CAS.
- T. Hasobe, H. Imahori, S. Fukuzumi and P. V. Kamat, J. Mater. Chem., 2003, 13, 2515–2520 RSC.
- T. Hasobe, H. Murata, S. Fukuzumi and P. V. Kamat, Mol. Cryst. Liq. Cryst., 2007, 471, 39–51 CrossRef CAS.
- T. Hasobe, H. Imahori, S. Fukuzumi and P. V. Kamat, J. Phys. Chem. B, 2003, 107, 12105–12112 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 1993, 26, 198–205 CrossRef CAS.
- S. Fukuzumi, Pure Appl. Chem., 2003, 75, 577–587 CrossRef CAS.
- H. Imahori, H. Norieda, H. Yamada, Y. Nishimura, I. Yamazaki, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 100–110 CrossRef CAS.
- H. Imahori, T. H., H. Yamada, Y. Nishimura, I. Yamazaki and S. Fukuzumi, Langmuir, 2001, 17, 4925–4931 CrossRef CAS.
- T. Hasobe, H. Imahori, H. Yamada, T. Sato, K. Ohkubo and S. Fukuzumi, Nano Lett., 2003, 3, 409–412 CrossRef CAS.
- H. Yamada, H. Imahori, Y. Nishimura, I. Yamazaki, T. K. Ahn, S. K. Kim, D. Kim and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 9129–9139 CrossRef CAS.
- H. Imahori, Y. Kashiwagi, T. Hasobe, M. Kimura, T. Hanada, Y. Nishimura, I. Yamazaki, Y. Araki, O. Ito and S. Fukuzumi, Thin Solid Films, 2004, 451–452, 580–588 CrossRef CAS.
- T. Hasobe, H. Imahori, S. Fukuzumi and P. V. Kamat, J. Am. Chem. Soc., 2003, 125, 14962–14963 CrossRef CAS.
- T. Hasobe, H. Imahori, P. V. Kamat, T. K. Ahn, S. K. Kim, D. Kim, A. Fujimoto, T. Hirakawa and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 1216–1228 CrossRef CAS.
- D. Sun, F. S. Tham, C. A. Reed, L. Chaker, M. Burgess and P. D. W. Boyd, J. Am. Chem. Soc., 2000, 122, 10704–10705 CrossRef CAS.
- N. V. Tkachenko, C. Guenther, H. Imahori, K. Tamaki, Y. Sakata, S. Fukuzumi and H. Lemmetyinen, Chem. Phys. Lett., 2000, 326, 344–350 CrossRef CAS.
- F. D’Souza, S. Gadde, M. E. Zandler, K. Arkady, M. E. El-Khouly, M. Fujitsuka and O. Ito, J. Phys. Chem. A, 2002, 106, 12393–12404 CrossRef CAS.
- T. Hasobe, S. Hattori, P. V. Kamat and S. Fukuzumi, Tetrahedron, 2006, 62, 1937–1946 CrossRef CAS.
- T. Hasobe, S. Hattori, P. V. Kamat, Y. Urano, N. Umezawa, T. Nagano and S. Fukuzumi, Chem. Phys., 2005, 319, 243–252 CrossRef CAS.
- T. Hasobe, P. V. Kamat, M. A. Absalom, Y. Kashiwagi, J. Sly, M. J. Crossley, K. Hosomizu, H. Imahori and S. Fukuzumi, J. Phys. Chem. B, 2004, 108, 12865–12872 CrossRef CAS.
- T. Hasobe, Y. Kashiwagi, M. A. Absalom, K. Hosomizu, M. J. Crossley, H. Imahori, P. V. Kamat and S. Fukuzumi, Adv. Mater., 2004, 16, 975–979 CrossRef CAS.
- T. Hasobe, P. V. Kamat, V. Troiani, N. Solladie, T. K. Ahn, S. K. Kim, D. Kim, A. Kongkanand, S. Kuwabata and S. Fukuzumi, J. Phys. Chem. B, 2005, 109, 19–23 CrossRef CAS.
- T. Hasobe, K. Saito, P. V. Kamat, V. Troiani, H. Qiu, N. Solladié, K. S. Kim, J. K. Park, D. Kim, F. D'Souza and S. Fukuzumi, J. Mater. Chem., 2007, 17, 4160–4170 RSC.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- T. Nakanishi, T. Kojima, K. Ohkubo, T. Hasobe, K. Nakayama and S. Fukuzumi, Chem. Mater., 2008, 20, 7492–7500 CrossRef CAS.
- T. Hasobe, H. Murata, P. V. Kamat and S. Fukuzumi, Jpn. J. Appl. Phys., 2008, 47, 1223–1229 CrossRef CAS.
- K. Okamoto, T. Hasobe, N. V. Tkachenko, H. Lemmetyinen, P. V. Kamat and S. Fukuzumi, J. Phys. Chem. A, 2005, 109, 4662–4670 CrossRef CAS.
- T. Hasobe, S. Hattori, P. V. Kamat, Y. Wada and S. Fukuzumi, J. Mater. Chem., 2005, 15, 372–380 RSC.
- T. Hasobe, S. Hattori, H. Kotani, K. Ohkubo, K. Hosomizu, H. Imahori, P. V. Kamat and S. Fukuzumi, Org. Lett., 2004, 6, 3103–3106 CrossRef CAS.
- S. Fukuzumi, T. Hasobe, K. Ohkubo, M. J. Crossley, P. V. Kamat and H. Imahori, J. Porphyrins Phthalocyanines, 2004, 8, 191–200 CrossRef CAS.
- H. Imahori, T. Hasobe, H. Yamada, P. V. Kamat, S. Barazzouk, M. Fujitsuka, O. Ito and S. Fukuzumi, Chem. Lett., 2001, 30, 784–785 CrossRef.
- T. Hasobe, S. Fukuzumi and P. V. Kamat, J. Phys. Chem. B, 2006, 110, 25477–25484 CrossRef CAS.
- M. Endo, Y. A. Kim, T. Hayashi, T. Yanagisawa, H. Muramatsu, M. Ezaka, H. Terrones, M. Terrones and M. S. Dresselhaus, Carbon, 2003, 41, 1941–1947 CrossRef CAS.
- M. Endo, Y. A. Kim, T. Hayashi, Y. Fukai, K. Oshida, M. Terrones, T. Yanagisawa, S. Higaki and M. S. Dresselhaus, Appl. Phys. Lett., 2002, 80, 1267–1269 CrossRef CAS.
- Y. A. Kim, T. Hayashi, Y. Fukai, M. Endo, T. Yanagisawa and M. S. Dresselhaus, Chem. Phys. Lett., 2002, 355, 279–284 CrossRef CAS.
- T. Hasobe, S. Fukuzumi and P. V. Kamat, Angew. Chem., Int. Ed., 2006, 45, 755–759 CrossRef CAS.
- A. Kongkanand, R. MartinezDominguez and P. V. Kamat, Nano Lett., 2007, 7, 676–680 CrossRef CAS.
- T. Hasobe, H. Murata and P. V. Kamat, J. Phys. Chem. C, 2007, 111, 16626–16634 CrossRef CAS.
- G. Pagona, A. S. D. Sandanayaka, T. Hasobe, G. Charalambidis, A. G. Coutsolelos, M. Yudasaka, S. Iijima and N. Tagmatarchis, J. Phys. Chem. C, 2008, 112, 15735–15741 CrossRef CAS.
- H. Nobukuni, Y. Shimazaki, F. Tani and Y. Naruta, Angew. Chem., Int. Ed., 2007, 46, 8975–8978 CrossRef CAS.
- H. Matsui and R. MacCuspie, Nano Lett., 2001, 1, 671–675 CrossRef CAS.
- J. S. Hu, Y. G. Guo, H. P. Liang, L. J. Wan and L. Jiang, J. Am. Chem. Soc., 2005, 127, 17090–17095 CrossRef CAS.
- R. Harada, Y. Matsuda, H. Okawa and T. Kojima, Angew. Chem., Int. Ed., 2004, 43, 1825–1828 CrossRef CAS.
- Z. Wang, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2004, 126, 16720–16721 CrossRef CAS.
- Y. Kim, M. F. Mayer and S. C. Zimmerman, Angew. Chem., Int. Ed., 2003, 42, 1121–1126 CrossRef CAS.
- R. van Hameren, A. M. van Buul, M. A. Castriciano, V. Villari, N. Micali, P. Schon, S. Speller, L. Monsu Scolaro, A. E. Rowan, J. A. A. W. Elemans and R. J. M. Nolte, Nano Lett., 2008, 8, 253–259 CrossRef CAS.
- M. C. Lensen, J. A. A. W. Elemans, S. J. T. v. Dingenen, J. W. Gerritsen, S. Speller, A. E. Rowan and R. J. M. Nolte, Chem.–Eur. J., 2007, 13, 7948–7956 CrossRef CAS.
- T. Hasobe, A. S. D. Sandanayaka, T. Wada and Y. Araki, Chem. Commun., 2008, 3372–3374 RSC.
- I. I. Smalyukh, O. V. Zribi, J. C. Butler, O. D. Lavrentovich and G. C. L. Wong, Phys. Rev. Lett., 2006, 96, 177801–177804 CrossRef.
- S. Vyawahare, K. M. Craig and A. Scherer, Nano Lett., 2006, 6, 271–276 CrossRef CAS.
- J. V. Barth, J. Weckesser, C. Cai, P. Günter, L. Bürgi, O. Jeandupeux and K. Kern, Angew. Chem., Int. Ed., 2000, 39, 1230–1234 CrossRef CAS.
- M. A. Ray, H. Kim and L. Jia, Langmuir, 2005, 21, 4786–4789 CrossRef CAS.
- H. O. Jacobs, A. R. Tao, A. Schwartz, D. H. Gracias and G. M. Whitesides, Science, 2002, 296, 323–325 CrossRef CAS.
- R. van Hameren, P. Schon, A. M. van Buul, J. Hoogboom, S. V. Lazarenko, J. W. Gerritsen, H. Engelkamp, P. C. M. Christianen, H. A. Heus, J. C. Maan, T. Rasing, S. Speller, A. E. Rowan, J. A. A. W. Elemans and R. J. M. Nolte, Science, 2006, 314, 1433–1436 CrossRef CAS.
| This journal is © the Owner Societies 2010 |