Enhancing the interactions between neutral molecular tweezers and anions†
Jose M.
Hermida-Ramón
,
Marcos
Mandado
,
Marta
Sánchez-Lozano
and
Carlos M.
Estévez
*
Departamento de Química Física, Facultade de Química, Universidade de Vigo 36310, Vigo, Spain. E-mail: cestevez@uvigo.es
First published on 9th November 2009
Abstract
Several structural modifications to the original molecular tweezers of the Klärner’s group were made with the aim of improving its binding capacity towards anions. The proposed modifications raise the molecular electrostatic potential inside the cavity and provide more conformational flexibility. The complexes of these new molecules with the halide anions Cl−, Br−, I− were optimized at the MPW1B95/6-31+G* level of theory. The molecular interactions were analyzed by single point density fitted local second-order Møller–Plesset perturbation theory (DF-LMP2) and DF-LMP2 spin-component-scaled MP2 (SCS-MP2), calculations were performed with the cc-pVTZ basis set. In view of the large magnitude of the interaction energies computed and the stability of the complexes in different solvents, this kind of molecule is a good candidate as molecular host for anion recognition.
1. Introduction
Molecular recognition of anions plays a significant role in biological and environmental processes such as the design of anion channels/transporters,1,2 membranes,3 treatment of pollutants,4etc. As a consequence, this area of supramolecular chemistry of anions is experiencing growing interest.5In recent years there have been a great number of studies of both theoretical and crystallographic determinations that describe non covalent interactions between the anion and the molecular guest.4,6–18 Among the different types, anion-π interactions are of particular interest since they present a series of advantages that improve their selectivity and directionality, namely the neutrality of the receptors, the variety in size and shape of the anions, the wide range of stability given by these interactions, etc.19 These characteristics turn anion-π interactions into a potential tailor-made solution for several problems involving anions. Most of the experimentally observed anion hosts that present anion-π interactions also showed other types of interactions such as hydrogen bonding, electrostatic interaction, coordination to a metal ion and ion pairing.12,20,21 Despite the number of solid state and theoretical studies of anion-π interactions, only a few studies have addressed this kind of interactions in solution.22,23
With the aim to contribute to the increasing knowledge about and to test the limits of the anion-π interactions, we have carried out several theoretical studies in gas phase and in solution on molecular systems that can be used as potential hosts for anion recognition. We have decided to study molecular tweezers similar to those synthesized by Klärner’s group24 since they are simple enough, from a computational point of view, and they present only interactions between the aromatic rings and the host, which provides a useful model to study anion-π interactions in depth. However, these molecular tweezers are reported to bind on electrodeficient aromatic and aliphatic substrates as well as organic cations. Complexes between these molecular tweezers and electron-rich aromatic, aliphatic or anionic substrates have not been observed. Recently, using theoretical models, we have shown the feasibility of this family of molecular tweezers to bind anions, provided the aromatic rings have enough fluorine substitution.25 A SAPT analysis of the interactions pointed out the relevance of the induction and dispersion terms. We were also able to quantify and characterize the individual anion-π interactions between halides and the fluorine substituted molecular tweezers, using density functional and MP2 models and the QTAIM theory.26 The interaction energies were close to those observed for molecular tweezer binding cations,27 even if the molecular electrostatic potentials inside the cavity were slightly negative. This fact points out the relevance of the exchange-repulsion, dispersion and inductive energies in the description of this type of binding.28
Here we present several structural modifications to the original molecular tweezer 1 (Scheme 1), which were made with the aim of improving its binding capacity towards anions. One of the limitations of molecular tweezer 1 is the fact that its molecular electrostatic potential inside the cavity is negative, despite the fluorine substitution and, due to the hydrogen atoms in the hinge, more positive outside than inside. We tried to change this by making slight changes to the structure. The main modification introduced to the structure was the substitution of the methylene bridged hinges by two dioxin rings in the pincers of the tweezer and changing the other two methylene bridges by two oxy bridges, yielding structure 2 (Scheme 1). The hydrogens of the aromatic rings were substituted by fluorine atoms, 2-F, or by cyanide groups, 2-CN. The proposed modifications raise the molecular electrostatic potential inside the cavity and provide more conformational flexibility. Using theoretical methods we have quantified the energetics involved in the process of anion recognition. We have studied the interactions of these proposed tweezers with several halide anions (Cl−, Br− and I−) since they are the most computationally manageable anions (we did not study the F− anion complexes due to their different interaction with the tweezers, as pointed out in our previous paper).26
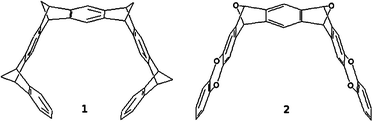 | ||
| Scheme 1 Original, 1, and modified, 2, molecular tweezer structures. | ||
2. Theoretical methods
Optimizations of the structures were performed with the Gaussian03 program,29 employing the density functional method with the MPW1B95 functional proposed by Thrular, which gives reasonable results on systems where dispersion energies are relevant.30 All atoms were described by the 6-31+G* basis set except the iodine atom represented by the basis set and effective core potential of LaJohn, Christiansen et al.31 a procedure that allow us to compare the result with those obtained in our previous studies.25,26 In all cases the anion is located in the center of the tweezer despite the fact that we have tried different initial positions only in the case of the Cl− complexes were we able to find other stationary points, but with energies slightly higher that the one reported. On the obtained optimized structures with minimum energy single point MP2 energy calculation were performed. To obtain the intermolecular interaction energy of the ion-tweezer complexes, a basis set superposition error (BSSE) correction for all the complexes was carried out using the counterpoise (CP) method of Boys and Bernardi.32 The BSSE corrected interaction energy for the complexation process is:| Eint = Ecomplex(AB) − |(EA(AB) + EB(AB))| | (1) |
| Edef = |EA(A) + EB(B) − (EAopt(A) + EBopt(B))| | (2) |
In order to improve the energetic description of the complexation procedure between the anion and the tweezer, single point calculations were performed at different levels: density fitted second-order Møller–Plesset perturbation theory (DF-MP2)33 and DF-MP2 spin-component-scaled MP2 (SCS-MP2)34 with the aug-cc-pVDZ basis set, and single point density fitted local second-order Møller–Plesset perturbation theory (DF-LMP2)35 and DF-LMP2 spin-component-scaled MP2 (SCS-LMP2) with the cc-pVTZ basis set. These methods proved to be accurate for the description of intermolecular interactions energies and essentially free of basis set superposition error.36 These calculations were done with the Molpro2006 program package.37 To estimate the effects of different solvents on the complexes, single point DFT calculations that include a polarizable continuum model PCM38 were also performed.
To shed more light on the nature of the binding energy between the anions and the tweezers, we used the symmetry-adapted perturbation theory combined with a density functional theory DFT-SAPT39 to obtain a physical interpretation of the interaction energy. In this method, the interaction energy is expressed as a sum of perturbative corrections in which each correction results from a different physical effect. The different intermolecular terms obtained from this method can be summarized in electrostatic, exchange-repulsion, induction, and dispersion contributions.28,40 The interaction energy, Eint, is given by eqn (3):
| Eint = Ees + Eexch + Eind + Edisp | (3) |
3. Results and discussion
The new proposed molecules have a structure that resembles that of molecular clips rather than molecular tweezers (see Scheme 1), with the two pincers almost linear (the dihedral angle C–O–O–C, with the carbons in trans, of the dioxin ring was in all cases near 180.0°), at least while not binding to a guest. Due to the small frequency of the butterfly vibrational mode of the dibenzo-p-dioxin fragment,41 the dioxin ring provides enough conformational flexibility so the pincers could slightly bend upon binding to the guest.Apart from the geometrical changes, the most significant effect caused in the tweezers by the introduced modifications was an increase of the molecular electrostatic potential inside the cavity (concave side), being noticeably positive in all cases and higher than the convex side. In Fig. 1 we show the molecular electrostatic potential of 2-F and 2-CN calculated at the MPW1B95/6-31+G* level on a solvent accessible surface (radius of solvent 1.5 Å). Since electrostatic interactions seem to play a significant role in the global interaction energy of anions with π systems, this distribution of the electrostatic potential should favor the inclusion of the anion host inside the cavity. This figure also shows larger positive values in the electrostatic potential corresponding to the 2-CN tweezer than those of the 2-F one. According to this, the larger interaction energies with anions will correspond to the former molecule.
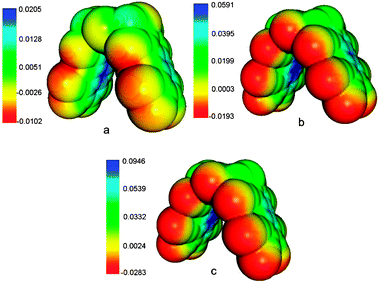 | ||
| Fig. 1 Electrostatic molecular potential (atomic units) of the tweezers: (a) tweezer 2-F, (b) tweezer 2-CN, (c) tweezer 2-CN in water. | ||
The interaction, binding and deformation energies of the complexation process in the gas phase are shown in Table 1. The interaction energies between our new 2-F compounds and the different halide anions are significantly higher than the parent molecular tweezers, 1.26 The interaction energies calculated at the DFT level are less negative than the MP2 and the SCS-MP2 energies with differences from 3 to 10% with respect to the SCS-MP2 values (Table 2).
In the process of binding, the 2-F molecule closes its pincers significantly to accommodate the anion, mainly by closing their hinges (see Fig. 2). There is also a grasping of the anion with the pincers which are no longer linear but slightly bent. The dihedral angle C–O–O–C of the dioxin ring is 166° for 2-F…Cl, 169° for 2-F…Br− and 166° for 2-F…I−. As a consequence of this change in geometry, the deformation energy of this process is slightly higher than in the original tweezers 1 (their values were less than 1 kcal mol−1),26 ranging from 2.79 kcal mol−1 in the Br− complex to 2.03 kcal mol−1 in the I− one. The anion is placed almost in the center of the cavity close to the two dioxin rings where, as can be seen in Fig. 1, the molecular electrostatic potential is more positive. We were able to locate some stationary points of the 2-F…Cl− complex with the anion located close to the terminal benzene rings of the pincers and close to the interior benzene rings but with higher energies than the one reported. We did not find these stationary points with the other anions probably because their size fits the tweezers’ cavity better.
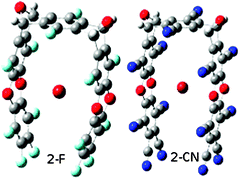 | ||
| Fig. 2 Optimized structures of the 2-F…Br− and 2-CN…Br− complexes. | ||
Substituted cyano benzenes and pyrazines are electron deficient arenes that have been described as potential anion binding sites with, in some cases, interaction energies higher than in the corresponding fluorinated arenes.11 In consequence, we have evaluated the interaction energies of the new molecular tweezers but with cyano groups instead of fluorine atoms (2-CN). The interaction energy (Table 1 and 2) of 2-CN with the anions is practically twice the value with 2-F. To our knowledge, these results are much larger than any value previously obtained with neutral arene receptors and halide anions,42 and also much larger than the interaction energies of cations with electron rich arenes.27
The closing of the 2-CN tweezers upon binding is more pronounced, but the bending of the dioxin ring is similar to if not less than that of 2-F. As a consequence of this bigger change in geometry the deformation energy is higher (6.9 kcal mol−1 for 2-CN…Cl, 7.2 kcal mol−1 for 2-CN…Br− and 5.6 kcal mol−1 for 2-CN…I−). Based on these results the chloride anion is the most stable inside the tweezer, both in the fluorine and the cyanide substituted tweezers.
The magnitude of the calculated interaction energies at the different levels of theory, with values up to 70 kcal mol−1, points out the collaborative effect of the modifications introduced and also sets this new class of tweezers as potential hosts for anion recognition.
To analyze the features of the intermolecular interactions for the complexation of the tweezers with anions, calculations using the DFT-SAPT method in the bromide–hexafluorobenzene, bromide-dioxane and bromide-tetrafluorodioxane systems were carried out. The results of these calculations are shown in Table 3. It can be seen that all energies increase drastically when the hydrogen atoms are replaced with fluorine atoms in the dioxane. This is due to the transfer of the charge density from the center of the ring to the fluorine atoms. Something analogous happens in the tweezers. Regarding the energies in C4O2F4 and hexafluorobenzene, the largest of the attractive contributions to the intermolecular energy is the induction term, about twice the electrostatic term. The dispersion term is not negligible and it is half of the electrostatic term. The exchange-repulsion energy is the largest. It is especially large in the C4O2F4 molecule, so the interaction energy is less attractive than for the hexaflorobenzene. However in the tweezer-anion complexes the anion is placed closer to the dioxin fragments than to the phenyl rings. This leads us to think that the charge density of the π system of the dioxin fragment in the tweezers is transferred to the neighboring rings (see Fig. 1) and because of that, the attractive interactions in the center of dioxin ring are favored, while the opposite effect occurs in the adjacent rings. This is indeed confirmed by the analysis of the dioxin deformation density caused by the substitution of the fluorine atoms in tetrafluorodioxin by fluorinated benzene rings to give the dibenzo-p-dioxin fragment. This deformation density is shown in Fig. 3. As one can see, there is a π-electron density depletion within the dioxin ring in the region closest to the phenyl rings. This π-electron density depletion is exclusively caused by a resonating effect with the phenyl ring, and is responsible for the enhancement of the interaction between the anion and the dioxin unit.
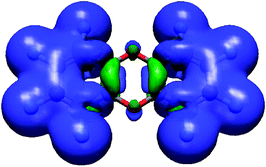 | ||
| Fig. 3 Isovalued (±0,005 au) surfaces of the dioxin deformation density caused by the substitution of the fluorine atoms in tetrafluorodioxin by fluorinated benzene rings to give the dibenzo-p-dioxin fragment. Blue: positive value, green: negative value. | ||
| Energy | C4O2H4…Br− | C4O2F4…Br− | C6F6…Br− |
|---|---|---|---|
| E int | −2.19 | −8.43 | −10.91 |
| E es | −0.45 | −14.88 | −12.46 |
| E exch | 7.94 | 43.35 | 28.15 |
| E ind | −6.89 | −29.39 | −20.14 |
| E disp | −2.79 | −7.50 | −6.45 |
To study the recognition process in solution we have performed single point DFT calculations that include a polarizable continuum model, PCM.38 The solvents chosen were: water, acetone, chloroform and DMSO. As can be seen in Fig. 1, the electrostatic potential inside the cavity is more positive in the water than in the gas phase. This effect will increase the ability of the tweezer to bind anions.25Table 4 gives the interaction energies with the halide anions. Since no significant variations are expected for the BSSE due to the solvent, these interaction energies were obtained using the BSSE in gas phase. The inclusion of the solvent model in the calculations modifies significantly the interaction energies, being positive in some cases. As expected, as the polarity of the solvent increases the interaction energy decreases. This is because in solvent the increase of the stability of the solvated anion gets larger than the increase of the complex stability when the polarity of the solvent rises. Although the fluorine substituted tweezers show less negative interaction energy than the cyanide tweezers in the absence of a solvent, this difference is strongly reduced with complexes solvated in solvents with a large dielectric constant.
The interaction energies in solvent undergo a drastic reduction with respect to the gas phase energies. Almost all solvated complexes with the 2-F tweezer are unstable; however, and according to these results, most of the 2-CN halide complexes are stable in solution. This happens even in a solvent like water, with a large dielectric constant and where, to the best of our knowledge, stable complexes of an anion with a neutral receptor have not been found.42 The obtained interaction energies yield most of the complexes with iodine as unstable. Our calculations give a relative solvation energy of iodine in water regarding chloride and bromide of around −2 and 5 kcal mol−1, respectively, but the relative experimental solvation enthalpies are around 11 and 15 kcal mol−1.43 Even though a straightforward comparison between the solvation energy and enthalpy cannot be done, these results suggest an overestimation of the solvation energy of iodine by the PCM that will produce less stable interaction energies for the iodine complexes.
In order to give a qualitative picture of the path followed by the anion to get into the tweezers we have performed a numerical optimization procedure in solvent. For a fixed position of the anion, the two main dihedral angles (actually four angles but they are symmetric two by two) ruling the closing of the tweezers were used as the only variables to optimize in the fitting procedure of the complex energies to a quadratic equation. The anion was placed along two different lines: (i) the line corresponding to the C2 symmetry axis of the tweezer and (ii) the line that passes through the center of the tweezer cavity and is perpendicular to one of the symmetry molecular planes. Fig. 4 shows some of the profiles for the 2-CN tweezer when the bromide anion is moved along these lines in chloroform and in water. The plots show an asymptotic value of the energy at long distances. When the distance gets shorter, first the energy rises to a maximum and then it decreases to a minimum with the anion in the center of the tweezers. The energy of this minimum (at distance 0 Å) is the lowest energy for all complexes except for the 2-F…Br− complex in water. In most cases when the anion reaches the center of the tweezer it gets trapped in a depth energy well. Fig. 4a shows the 2-CN…bromide complex in chloroform increasing its energy when the anion is moved out from the tweezer, starting from the center of the cavity (taken as the energy reference) passing through a maximum and finally reaching an asymptotic value when both molecules are far apart (approx. 15 Å). The energy difference between isolated molecules (distance 15 Å, in Fig. 4a) and the complex (distance 0 Å) is around −22 kcal mol−1. This value agrees with that reported in Table 4 for this complex in chloroform, −21.61 kcal mol−1. The same comparison can be made for Fig. 4b–d. Thus, the energy differences obtained from Fig. 4b, c and d are around −5, −7 and 2.9 kcal mol−1, respectively; and the values reported in Table 4 for the same complexes are −1.81, −2.37 and 3.27 kcal mol−1. It should be pointed out that since most of the internal coordinates have been fixed to the complex geometry the energy differences between isolated molecules and the complex are underestimated. Taking this into account, we can see a qualitative agreement between the data in Table 4 and the energy differences given by profiles in Fig. 4.
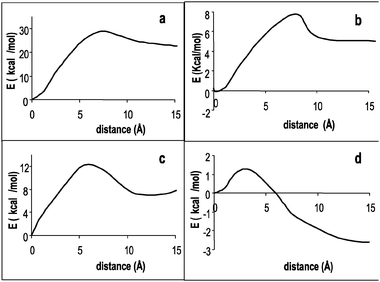 | ||
| Fig. 4 Energy profiles of the bromide complexes with: (a) 2-CN tweezer in chloroform, (b) 2-F tweezer in chloroform (c) 2-CN tweezer in water and (d) 2-F tweezer in water. The ion is moved out of the molecular tweezer along the axis perpendicular to one of the molecular symmetry planes (see text), except in b) where the ion is moved out of the molecular tweezer along the C2 symmetry axis. The origin of the profile corresponds to the anion placed at the center of the tweezer cavity. | ||
4. Conclusions
In summary, we present here neutral molecular tweezers built only with electrodeficient rings so only anion-π interactions are responsible for the binding of anions. The modifications introduced to the tweezers improved the intensity of their interactions with the different halide anions. In view of the large magnitude of the interaction energies computed and the stability of the complexes in different solvents (with values up to −3.84 kcal mol−1 in water and −21.61 kcal mol−1 in chloroform), and in most cases a deep energy well for the anion in the center of the cavity, this kind of molecule is a good candidate as a molecular host for anion recognition.Acknowledgements
The authors thank the Xunta de Galicia (PGIDIT06TMT31401PR) and the Ministerio de Ciencia e Innovación (CTQ2008-06767/BQU). The authors gratefully acknowledge the CESGA for computer time. M.M. is grateful to the Xunta de Galicia for financial support as a researcher in the Isidro Parga Pondal program.References
- V. Gorteau, G. Bollot, J. Mareda, A. Perez-Velasco and S. Matile, J. Am. Chem. Soc., 2006, 128, 14788–14789 CrossRef CAS.
- V. Gorteau, G. Bollot, J. Mareda and S. Matile, Org. Biomol. Chem., 2007, 5, 3000–3012 RSC.
- R. M. Fairchild and T. K. Holman, J. Am. Chem. Soc., 2005, 127, 16364–16365 CrossRef CAS.
- P. Gamez, T. J. Mooibroek, S. J. Teat and J. Reedijk, Acc. Chem. Res., 2007, 40, 435–444 CrossRef CAS.
- A. Bianchi, K. Bowman-James and E. García-España, Supramolecular Chemistry of Anions, Wiley & Sons Inc., 1997 Search PubMed.
- C. Estarellas, D. Quiñonero, A. Frontera, P. Ballester, J. Morey, A. Costa and P. M. Deya, J. Phys. Chem. A, 2008, 112, 1622–1626 CrossRef CAS.
- I. Alkorta, D. Quiñonero, C. Garau, A. Frontera, J. Elguero and P. M. Deya, J. Phys. Chem. A, 2007, 111, 3137–3142 CrossRef CAS.
- I. Alkorta, I. Rozas and J. Elguero, J. Am. Chem. Soc., 2002, 124, 8593–8598 CrossRef CAS.
- I. Alkorta, I. Rozas and J. Elguero, J. Fluorine Chem., 2000, 101, 233 CrossRef CAS.
- A. Frontera, D. Quiñonero, A. Costa, P. Ballester and P. M. Deya, New J. Chem., 2007, 31, 556–560 RSC.
- O. B. Berryman, V. S. Bryantsev, D. P. Stay, D. W. Johnson and B. P. Hay, J. Am. Chem. Soc., 2007, 129, 48–58 CrossRef CAS.
- H. Casellas, C. Massera, F. Buda, P. Gamez and J. Reedijk, New J. Chem., 2006, 30, 1561–1566 RSC.
- P. de Hoog, P. Gamez, H. Mutikainen, U. Turpeinen and J. Reedijk, Angew. Chem., Int. Ed., 2004, 43, 5815–5817 CrossRef CAS.
- C. Garau, D. Quiñonero, A. Frontera, D. Escudero, P. Ballester, A. Costa and P. M. Deya, Chem. Phys. Lett., 2007, 438, 104–108 CrossRef CAS.
- A. García-Raso, F. M. Alberti, J. J. Fiol, A. Tasada, M. Barcelo-Oliver, E. Molins, D. Escudero, A. Frontera, D. Quiñonero and P. M. Deya, Inorg. Chem., 2007, 46, 10724–10735 CrossRef CAS.
- D. Quiñonero, A. Frontera, D. Escudero, P. Ballester, A. Costa and P. M. Deya, ChemPhysChem, 2007, 8, 1182–1187 CrossRef CAS.
- D. Quiñonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Angew. Chem., Int. Ed., 2002, 41, 3389–3392 CrossRef CAS.
- D. Escudero, A. Frontera, D. Quiñonero and P. M. Deya, J. Comput. Chem., 2009, 30, 75–82 CrossRef CAS.
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2008, 37, 68–83 RSC.
- H. Schneider, K. M. Vogelhuber, F. Schinle and J. M. Weber, J. Am. Chem. Soc., 2007, 129, 13022–13026 CrossRef CAS.
- S. S. Zhu, H. Staats, K. Brandhorst, J. Grunenberg, F. Gruppi, E. Dalcanale, A. Lützen, K. Rissanen and C. A. Schalley, Angew. Chem., Int. Ed., 2008, 47, 788–792 CrossRef CAS.
- O. B. Berryman, F. Hof, M. J. Hynes and D. W. Johnson, Chem. Commun., 2006, 506–508 RSC.
- Y. S. Rosokha, S. V. Lindeman, S. V. Rosokha and J. K. Kochi, Angew. Chem., Int. Ed., 2004, 43, 4650–4652 CrossRef CAS.
- F.-G. Klärner, J. Benkhoff, R. Boese, U. Burkert, M. Kamieth and U. Naatz, Angew. Chem., Int. Ed. Engl., 1996, 35, 1130–1133 CrossRef.
- J. M. Hermida-Ramón and C. M. Estévez, Chem.–Eur. J., 2007, 13, 4743–4749 CrossRef CAS.
- M. Mandado, J. M. Hermida-Ramón and C. M. Estévez, THEOCHEM, 2008, 854, 1–9 CrossRef CAS.
- F. G. Klärner and B. Kahlert, Acc. Chem. Res., 2003, 36, 919–932 CrossRef.
- D. Kim, P. Tarakeshwar and K. S. Kim, J. Phys. Chem. A, 2004, 108, 1250–1258 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. V. Jr., K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2005, 109, 5656–5667 CrossRef CAS.
- L. A. LaJohn, P. A. Christiansen, R. B. Ross, T. Atashroo and W. C. Ermier, J. Chem. Phys., 1987, 87, 2812–2824 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553.
- M. Feyereisen, G. Fitzgerald and A. Komornicki, Chem. Phys. Lett., 1993, 208, 359–363 CrossRef CAS.
- S. Grimme, J. Chem. Phys., 2003, 118, 9095–9102 CrossRef CAS.
- H. J. Werner, F. R. Manby and P. J. Knowles, J. Chem. Phys., 2003, 118, 8149–8160 CrossRef CAS.
- J. G. Hill, J. A. Platts and H. J. Werner, Phys. Chem. Chem. Phys., 2006, 8, 4072–4078 RSC.
- H.-J. Werner, P. J. Knowles, R. Lindh, F. R. Manby, M. Schütz, P. Celani, T. Korona, G. Rauhut, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, C. Hampel, G. Hetzer, A. W. Lloyd, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklaß, P. Palmieri, R. Pitzer, U. Schumann, H. Stoll, A. J. Stone, R. Tarroni and T. Thorsteinsson, MOLPRO Search PubMed, version 2006.1, a package of ab initio programs, see http://www.molpro.net.
- E. Cancès, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef.
- G. Jansen and A. Hesselmann, J. Phys. Chem. A, 2001, 105, 11156–11157 CrossRef CAS.
- B. Jeziorski and K. Szalewicz, in Enciplopedia of Computational Chemistry, ed. P. v. Schleyer, N. L. Allinger, T. Clark, J. Gasteig, P. A. Kollman, H. F. I. Schaeferand and P. R. Screiner, Willey, Chichester, UK, 1998 Search PubMed.
- I. Ljubic and A. Sabljic, J. Phys. Chem. A, 2006, 110, 4524–4534 CrossRef CAS.
- P. Ballester, Struct. Bonding, 2008, 129, 127–174 CAS.
- I. Ripsand J. Jortner, J. Chem. Phys., 1992, 97, 536–546 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates and total energies of the compounds presented herein. See DOI: 10.1039/b915483c |
| This journal is © the Owner Societies 2010 |