First-principles investigations of Ti-substituted hydroxyapatite electronic structure
Shuxia
Yin
ab and
Donald E.
Ellis
*abc
aInstitute for Catalysis in Energy Processes, Northwestern University, Evanston, IL 60208, USA. E-mail: don-ellis@northwestern.edu
bDept. of Physics and Astronomy, Northwestern University, Evanston, IL 60208, USA
cDept. of Chemistry, Northwestern University, Evanston, IL 60208, USA
First published on 10th November 2009
Abstract
The electronic structure of Ti-substituted hydroxyapatite is investigated using density functional theory within a periodic slab model. Two sorption mechanisms have been considered: i.e., Ti4+ and Ti(OH)22+ as the likely species to exchange with Ca2+. Ti4+ has a small ionic radius compared to Ca2+ and can dope into both distinct sites, showing no site preference; however, when two H were removed from the OH channel to obtain charge compensation, preferential site II substitution appears, accompanied with a large O shift forming a strong Ti–O bond. The species Ti(OH)22+ displays a strong site preference: substitution by Ti(OH)22+ on the hydroxyl channel (site II) is exothermic and favored strongly over the Ca column (site I). Ti(OH)22+ substitution for Ca2+ induces a large geometry relaxation and distortion, especially within the OH channel and Ca2+ column, with a considerable shift of Ti compared to the Ca sites in pure HA. These results are consistent with the experimental observation that material synthesis with high Ti doping (atomic ratio > 0.1) shows irregular particles formation with reduced crystallinity. The calculated cell shape and volume relaxations indicate that the volume and cell parameters both expand in all the substituted HA models. The site preference and volume expansion differences found are attributed to the metal ion shift caused in meeting the requirement of strong Ti–O coordination in site I and site II polyhedra.
1. Introduction
Hydroxyapatite (HA), with the chemical composition Ca10(PO4)6(OH)2, is the main mineral constituent of mammalian tooth enamel and bone and has become an important biomaterial with medical applications in the manufacture of artificial bone and implant composites. Its notably porous nature and highly active ion-exchange properties also play a multiple role in medical, environmental and catalytic applications. In recent years it has attracted increasing interest for use in environmental adsorbents and catalysts as well as biomaterials. HA is recognized as an uptaking trap for toxic heavy metals in waste and environmental contamination. Metal substitution on the Ca sites involves the biologically important iron, copper, and zinc as well as toxic Cd, Cr, Pb, etc. Modified apatites now are widely studied as a mineral support (adsorbent) for catalysts prepared by including different transition metal ions in the structure, for example, Ru–, Pd– and Ti–HA.1–4TiO2 has good catalytic activity for photo-oxidation of organic compounds and bioactive substances while its application has difficulties to overcome, such as the filtration of fine TiO2, fixation of catalyst particles and effective utilization of UV rays. Several attempts have been made to increase its photoefficiency by doping with other metals or adding a coadsorbent such as zeolite, activated carbon or clays. As HA shows an excellent affinity to biomaterials such as proteins, efforts have been devoted to develop a novel photocatalyst that contains a composite of photocatalytic materials and HA with a high affinity to biomaterials. In 2003 Wakamura et al.5 synthesized Ti-modified HA (called Ti–Hap by them) by coprecipitation and found it to possess a high affinity to organic molecules and bacteria as well as high photocatalytic activity for their oxidative decomposition. It was found that Ti-modified HA particles exhibit bactericidal functionality even in the dark and a Ti-doped HA adhesion filter demonstrates a high deodorization performance, which is exploited in air cleaning and bacteria removal. Experimentally, the concentration of Ti4+ is a key factor controlling Ti–Hap crystallinity and catalytic efficiency. Wakamura found that Ti–Hap with Ti/Ca atomic ratio of 0.1 exhibits the highest bactericidal effect and decomposition of acetaldehyde compared to TiO2, while with higher Ti concentration, the material shows poor crystallinity and poor photocatalytic efficiency. It was observed that increasing Ti concentration is accompanied by an equivalent decrease of Ca2+, from which it is inferred that Ti in crystalline Ti–Hap is present as a divalent ion, such as Ti(OH)22+ or Ti(HPO4)2+. Following Wakamura’s work, there have appeared several patents and only two other reports concerning Ti-modified HA. Hu and co-workers6 carried out similar experiments as Wakamura’s, obtaining similar results; while Sugiyama suggested that the Ti–Hap chemical compound was not obtained through the coprecipitation procedure.7a However, the Sugiyama group later claimed7b that Ti may be incorporated into HA by ion exchange and it is Ti(SO4)2 that was introduced, which converted to hydroxide under basic conditions. They suggested that TiO2 is the active species in the calcinated TiHA. Detailed compositions and crystal structures have not yet been reported for Ti-containing HA, so the Ti4+ sorption mechanism is still unclear.
There has been a considerable amount of work by several groups devoted to understand the physical and electronic properties of HA and substituted HA, involving divalent cations (Zn, Pb, Sr, …) and anionic (F−, Cl−, CO32−, VO43−, AsO43−) substitutions.8–19 These works have focused on the chemistry of metal ions, anions and the modified HA structure using structural characterization techniques and theoretical calculations by atomistic simulations and first-principles modeling. de Leeuw and co-workers have extensively studied the electronic structure of hydroxyapatite, fluorapatite and chlorapatite using density functional theory and atomistic potentials.8–12 They have established that ordered hydroxyl group positions in the crystal structure are energetically favored, with all hydroxyl groups lining up with O and H ions alternating in a column channel in the c-direction. In crystalline HA, Ca cations are identified as two distinct groups by Ca coordination around these sites. Ca(I) and phosphate O are coordinated in a metaprismatic structure with Ca(I) in the center, forming the Ca(I) column. Ca(II) ions are coordinated with hydroxyls and phosphate O, forming triangles with hydroxyl in the center and give rise to a hydroxyl channel with the unit cell repetition. Some arguments for the preferential site occupation have been proposed for divalent metal cations based on the ionic radius and electronegativity of cations as well as cation–cation distances (spatial effect). Occupancy at the hydroxyl channel (site II) is favored for most divalent cations: the large radius and greater electronegativity are said to account for site II preference for Pb; while the large change of polyhedron volume at the calcium column (site I) supposedly accounts for Zn substitution on site II.17 Cation–cation interaction was found to cause distortion on site II for Sr and contribute to its site I-preferential occupancy.18 In summary, there has not yet been established any universal argument that unambiguously predicts the energetic preference for ion substitution on Ca(I) versus Ca(II) sites.
In the present work, we carried out periodic density functional theory (DFT) calculations on crystalline Ti–HA to investigate the Ti-induced structural modification and site-preferential sorption mechanism of Ti4+. Ti4+ and Ti(OH)22+, as the most likely ion-exchange species with Ca2+, were first introduced to bulk HA through substitution at different sites. The energetic stability and microscopic crystal structure of Ti–Hap is clearly highly associated with the solid solution concentration and chemical environment of sites. Thus to fully resolve the adsorption mechanism, theoretical studies on a large range of Ti4+ concentration on different Ca2+ sites and their distribution at different atomic ratios are required, considering both bulk and surface models. In the present paper we focused on bulk models, mainly reporting results at 10 at% substitution concentration, at which TiHA is apparently well crystallized, and for which beneficial chemical activity is said to be maximized.
2. Computational details
2.1 Density functional theory
First-principles calculations were carried out for HA bulk and Ti–HA using periodic slab DFT in the pseudopotential framework with a plane-wave basis set implemented in the VASP program (Vienna ab initio simulation package).20–22 The projected augmented wave (PAW) basis set was employed and the generalized gradient approximation (GGA) within the PW91 scheme was chosen for the exchange and correlation potential. Automatically generated Monkhorst–Pack grids with a 2 × 2 × 2 k-points mesh were used to perform the Brillouin zone integrations for both the geometry and energy calculations. The unit cell parameters and crystal volume were first optimized. For HA we obtained the lattice constants a = b = 9.51 Å, c = 6.90 Å, which is in good agreement with the experimental values a = b = 9.43, c = 6.88,23,24 and with previous theoretical results of de Leeuw et al.8–10 using almost identical models and the ultrasoft ‘Vanderbilt’-like pseudopotential of a = b = 9.56 Å, c = 6.83 Å. The computed lattice parameters by different groups using DFT or HF methods and different basis sets as well as experimental values are presented in Table 1, showing that the DFT_GGA method has good accuracy in predicting lattice structure and thus the presently used PAW_PW91 method is applicable here. With cell dimensions initially fixed, all atomic positions were then allowed to relax in the Ti substitutions; later, the cell volume was also allowed to change. The self-consistent field was considered to be converged when forces on all atoms were smaller than 0.01 eV Å−1. Gaussian smearing of electronic levels was used for both ionic relaxation and density of states (DOS) calculations. Local DOS were obtained by projecting the plane-wave functions of the electronic eigenstates onto spherical harmonics within selected atomic spheres (RWS) of (0.9, 0.95, 0.75, 1.6, 0.5 Å) for (Ti, Ca, P, O, H), respectively.| a | b | c | α, β, γ | |
|---|---|---|---|---|
| This work | 9.51 | 9.51 | 6.90 | 90, 90, 120 |
| DFT-ultrasoft-GGA [ref. 10] | 9.56 | 9.56 | 6.83 | 90, 90, 120 |
| DFT-LDA [ref. 13] | 9.11 | 9.11 | 6.73 | 90, 90, 120 |
| DFT-ultrasoft-BFGS-GGA [ref. 14] | 9.418 | 9.418 | 6.875 | 90, 90, 120 |
| HF-LCCO [ref. 15] | 9.329 | 9.329 | 6.949 | 90, 90, 120 |
| Expt. [ref. 23 and 24] | 9.43 | 9.43 | 6.88 | 90, 90, 120 |
2.2 Crystal structure and unit cell
HA has hexagonal crystal structure with space group P63/m, and a unit cell of 44 atoms. The tetrahedral phosphate anion has strong covalent character and acts as a robust group in interaction with the surrounding environment. Ca cations occupy two distinct crystal sites, Ca(I) and Ca(II): thus the formula unit can be rewritten as Ca(I)4Ca(II)6(PO4)6(OH)2. Ca(I) is 9-fold coordinated to oxygen ions from six neighboring phosphate groups, while Ca(II) is 7-fold coordinated with oxygen ions, six from phosphate groups and one from an OH. Table 2 lists Ca–O bond lengths. Ca(I) lies along columns parallel to the crystal c-axis, two Ca(I) ions in each column. Ca(II) cations are distributed in two triangle bases, twisted 60° relative to each other with hydroxyl O in the center, forming a hexagonal channel with the OH array located in the center and parallel to the channel (also the lattice c-axis) (Fig. 1a and b).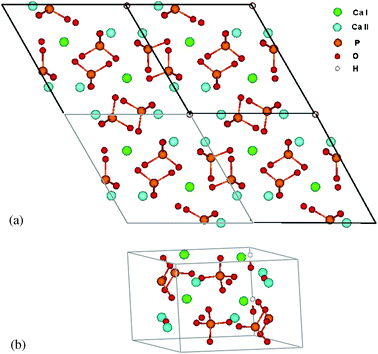 | ||
| Fig. 1 Structure of hydroxyapatite seen in top view along the c-axis. Unit cell is shown. (a) top view of 2 × 2 × 1 cell, centered on hexagonal c-axis channel; (b) unit cell looking along a and b-axis. | ||
| Structure | HA | Ti | Ti (H removal) | |||
|---|---|---|---|---|---|---|
| Site I | Site II | Site I | Site II | |||
| a ΔEsub = ETi–HA + ECa(OH)2 + 2EH2O − ETi(OH)4 − ECaHA. | ||||||
| E | −313.41 | −314.32 | −314.37 | −306.28 | −308.25 | |
| Ti(Ca)–O | Ca(I) | Ca(II) | 2.21 | 2.26 | 1.98 | 1.73 |
| 2.43 ×3 | 2.78, 2.51 | 2.28 | 2.25 | 2.01 | 2.04 | |
| 2.80 ×3 | 2.48, 2.36 | 2.31 | 2.34 | 2.03 | 2.04 | |
| 2.46 ×3 | 2.36, 2.39 | 2.32 | 2.20 | 2.06 | 2.14 | |
| — | 2.34 | 2.24 | 2.22 | 2.09 | 2.23 | |
| — | — | — | — | 2.17 | — | |
| CoordN | 9 | 7 | 5 | 5 | 6 | 5 |
| ΔEsuba | — | — | — | — | 2.70 | 0.73 |
DFT calculations were performed for Ti occupation on both Ca(I) and Ca(II) sites using a 1 × 1 × 1 unit cell to investigate distinct geometric modification. A single substitution thus represents a 10 at% solid solution of Ti. A major goal of this theoretical study is to determine the Ti substitution energetic preference for non-equivalent Ca(I) and Ca(II) cationic sites at some given concentration and the underlying mechanism for the preference. For lower substitution concentration, supercells are needed, which are briefly discussed below in the present report. The substitution behaviors of tetravalent Ti4+ ion and divalent Ti(OH)22+ were both explored. The substitution reactions can be described as, respectively,
| Ti(OH)4 + Ca10(PO4)6(OH)2 → TiCa9(PO4)6(O)2 + Ca(OH)2 + 2H2O | (1) |
| Ti(OH)4 + Ca10(PO4)6(OH)2 → Ti(OH)2Ca9(PO4)6(OH)2 + Ca(OH)2 | (2) |
3. Results and discussion
3.1 Substitution by Ti4+
We first considered Ti4+ substitution for Ca2+ on sites I or II; i.e., a bare Ti was placed on the metal site: the self-consistent electronic charge distribution of course differs from that of the nominal tetravalent d0s0 state due to covalency and charge transfer effects. When a Ca ion was directly replaced by Ti without any charge compensation, the relaxed structures for site I and II substitution have almost the same predicted total energy, with Ti showing only an in-plane shift of 0.25 Å relative to the bulk Ca. All the other atomic coordinates remain almost unchanged compared to HA. Both phosphate and OH groups that are bonded with Ti rotate very slightly.When Ca was replaced by Ti and two H were simultaneously removed from the hydroxyl channel to obtain charge compensation, the calculated results are very different: the relaxed structures of sites I and II becomes very different. The site II substituted structure exhibits an energy preference of 2.0 eV. The in-plane shift of Ti is 0.20 Å in site I and 0.40 Å for site II; the site I substituted structure shows a larger disorder due to the phosphate rotation. In both I and II substitution, there appears a large shift for O in the OH channel, compared to simple Ca replacement by Ti, in which OH remain in the channel as in the HA bulk. The site II preference may be ascribed to the O shift, resulting in strong Ti–O bonds around the hexagonal channel. Table 2 lists structural parameters, including the bond lengths and total energies. Following eqn (1), the substitution energy was calculated for the charge compensated substitution. Both sites display a positive substitution energy, indicating that this reaction is thermodynamically unfavorable.
In fact, for all these structures it was difficult to reach a fully self-consistent converged state, though the total energy and geometry become essentially unchanged after hundreds of optimization cycles. In experiments, the induced coupled plasma spectroscopy (ICP) assay has implied that Ti is present in crystalline HA as a divalent ion.5 Combining our calculation results and experimental suggestions, we hence extended our study to divalent substitutions.
3.2 Substitution by Ti(OH)22+
A broad conformation survey was made for Ti(OH)22+ substituting for Ca2+ on both sites I and II. In each conformation, the two OH groups may orient in different directions toward the OH channel and the Ca column. By relaxing the atomic positions from a variety of starting structures, some conformations could not be reached as a stable structure. In some cases phosphate became dissociated and HA dissociated into fragments. Finally two stable conformations were found at each site, with channel OH coordinated to Ti or phosphate O coordinated with Ti, as displayed in Fig. 2 and 3 and Table 3. Below we give a detailed description of the geometry and energetics for these stable conformations.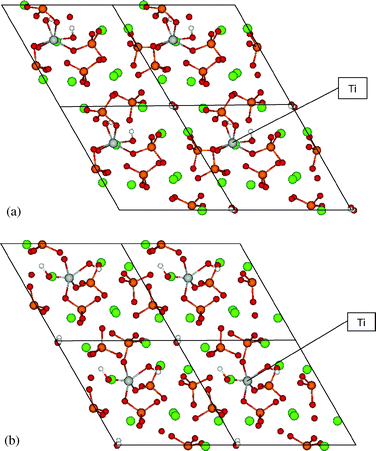 | ||
| Fig. 2 Calculated Ti(OH)2HA structures for Ti substituted at sites Ia and Ib, centered on hexagonal c-axis channel. | ||
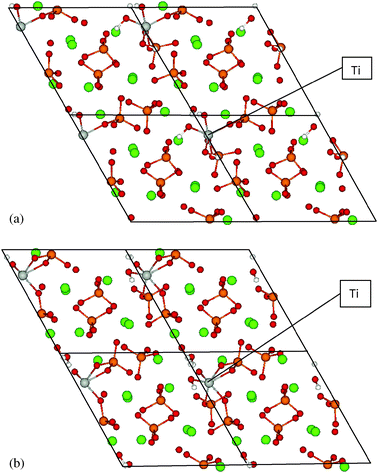 | ||
| Fig. 3 Calculated Ti(OH)2HA structures for Ti substituted at sites IIa and IIb, centered on hexagonal c-axis channel. | ||
| Structure | Site I | Site II | ||
|---|---|---|---|---|
| Ia | Ib | IIa | IIb | |
| a ΔEsub = ETi(OH)2HA + ECa(OH)2 − ETi(OH)4 − ECaHA. | ||||
| E | −335.54 | −335.83 | −337.59 | −337.89 |
| Ti–O | 2.05, 2.13 | 1.93, 2.01 | 1.98, 2.09 | 1.80, 1.99, 2.12, |
| 2.23, 2.30 | 1.95 | — | 2.24, 2.11 | |
| Ti–O(H) | 1.87, 1.80 | 1.73, 1.98 | 1.80, 1.90, 1.90 | 1.86 (H2O form) |
| Coord. no. | 6 | 5 | 5 | 6 |
| ΔEsuba | 1.98 | 1.69 | −0.07 | −0.37 |
| Ti shift | 0.28 | 1.11 | 0.57 | 0.80 |
Site I substitution. In the substitution structure Ia (Fig. 2 and Table 3), Ti was originally located on a Ca(I) position with OH groups extending up along the c-axis; after relaxation, Ti moved away from the Ca(I) column center with a shift of 0.28 Å compared to HA bulk. The Ca in another Ca(I) column and in the same layer with Ti also shows a lateral displacement of 0.32 Å, while the remaining two unsubstituted Ca(I) did not exhibit noticeable displacements. Meanwhile, six Ca(II) atoms around the OH channel maintained almost the same position as in HA. The channel OH groups became a bit tilted compared to the initial straight OH chain, forming a distorted H-bonding chain. Ti is here coordinated with six O atoms, two from OH groups (1.80, 1.87 Å), and four from phosphate groups (2.05, 2.13, 2.23, 2.30 Å).
Substituted structure Ib (Fig. 2 and Table 3) has a lower total energy than Ia by −0.3 eV. It displays a very distinct shift in position for Ti, Ca(I) and OH compared to structure Ia. Ti moved away from the Ca(I) column center toward phosphate groups by 1.11 Å as well as 0.51 Å for Ca(I) in its neighboring column at the same layer. All other Ca(I) have a similar small displacement (about 0.1 Å) as in structure Ia. The channel OH become almost perpendicular to the channel axis, a rotation of 90° compared to pure HA; the OH chain is thus totally disrupted. In this structure, Ti is 5-fold coordinated with oxygen, two from OH groups (1.73 Å, 1.98 Å), three from phosphate groups (1.93, 1.95, 2.01 Å). The Ti–O bond lengths in structure Ib are thus much shorter than in structure Ia, which results from the distinct shift of Ti toward phosphate groups. This also contributes to the energetically favored stability for structure Ib.
Site II substitution. For site II substitution, two stable Ti-modified HA configurations were also obtained (Fig. 3). In structure IIa, Ti substituted for one of six Ca2+ that form the hexagonal OH channel, initially located on the Ca position with one OH extending up and one extending down along the c-axis. Through a full relaxation, the optimized structure shows distinct changes compared to bulk HA. Ti displays a shift of 0.57 Å away from the pure HA Ca2+ triangle bases. All cations in sites I and II exhibit a displacement: (0.13, 0.30 Å) for Ca(I) and (0.14, 0.15, 0.21, 0.24, 0.27 Å) for Ca(II). One OH in the channel is coordinated to Ti, which is found in 5-fold coordination with two neighboring phosphate O (1.98, 2.09 Å) and three OH (1.80, 1.90, 190 Å). There are three Ti–OH bonds in structure IIa through bonding with channel OH and the original (OH)2 complexed to Ti. The OH group left in the channel is tilted with respect to the channel axis; its O atom shifts strongly toward Ti forming short Ti–O bonds.
Structure IIb has a lower total energy than structure IIa by −0.30 eV. Ti shows a larger shift of 0.85 Å from the bulk Ca(II) position, contracting toward the middle of the two triangle bases. Ca(I) atoms shift in a range of 0.14 and 0.29 Å. The shifts for Ca(II) atoms are 0.0, 0.23, 0.24, 0.32 and 0.48 Å. The predicted trend is thus that all atom shifts in structure IIb are larger than in IIa. Ti is here coordinated with six O in the form of Ti–O–PO3 (1.99, 2.11 Å) and (Ti–O)2–PO2 (2.12, 2.24 Å) through binding with phosphate, Ti–O–H (1.86 Å) and Ti–O (1.80 Å) from original Ti(OH)2 groups. The original Ti–O–H lost H, which is found bonded with OH in the channel to form a water molecule. Therefore there exists one OH and one H2O molecule in the hexagonal OH channel. The structural disorder in the OH channel along the c-axis is much more obvious in site II substitution than site I due to the strong bonding of Ti–OH.
From the geometric results discussed above, the bulk structure change is distinguished by an obvious OH chain deformation and a major metal ion shift compared to the original Ca. It is indicated that for Ti substitution on both I and II sites, the structures with a larger shift of Ti and O(H) position are energetically more stable, leading to a short Ti–O bond length (the Ti–O bond lengths in TiO2 bulk are 1.95 and 1.98 Å). To obtain good coordination with Ti, the corresponding phosphate groups rotate by a small angle and move a small distance compared to their original position. As a result, this also introduces an increase of disorder in the HA structure around phosphate sites as Ti is incorporated.
| ΔEsub = ETi(OH)2HA + ECa(OH)2 − ETi(OH)4 − EHA | (3) |
For the benchmark species Ca(OH)2 and Ti(OH)4, the calculations at the same level of theory and basis set as for HA show excellent agreement with experimental results and other theoretical calculations. For Ca(OH)2, the cell parameter is calculated to be a = b = 3.57 Å, and c = 4.90 Å, which compares favorably with the experimental values of a = b = 3.58, c = 4.87 Å.25 For Ti(OH)4, there are no experimental data available. Our calculated Ti(OH)4 complex gives the Ti–O distance of 1.88 Å and Ti–O–H angle of 119.0°, comparing well with hybrid functional B3LYP calculations.26
The substitution energies ΔEsub are listed in Table 3 for all Ti(OH)2HA configurations studied. ΔEsub is seen to be negative for all site II configurations and positive for site I substitutions; i.e., the substitution on site II is exothermic and on site I it is endothermic. The substitution energy difference between sites II and I is as great as 2.3 eV, indicating that the substitution of Ca2+ by Ti(OH)22+ on site II is highly preferred. This differential is strongly related to the structural difference between site I and site II. The Ca polyhedron on site I has a much bigger volume than the site II polyhedron. When Ca2+ is substituted by Ti(OH)22+ incorporation, Ti shows a large shift to form typical Ti–O coordination. Both sites show very different coordination environments. In the larger volume site I Ti polyhedron, Ti–O bond lengths are relatively much longer than on site II. While on site II, after a large Ti shift in position, a considerable number of Ti–O bonds become shorter (1.80–1.9 Å), similar to the Ti(OH)4 complex, showing the strong coordination bonding interaction.
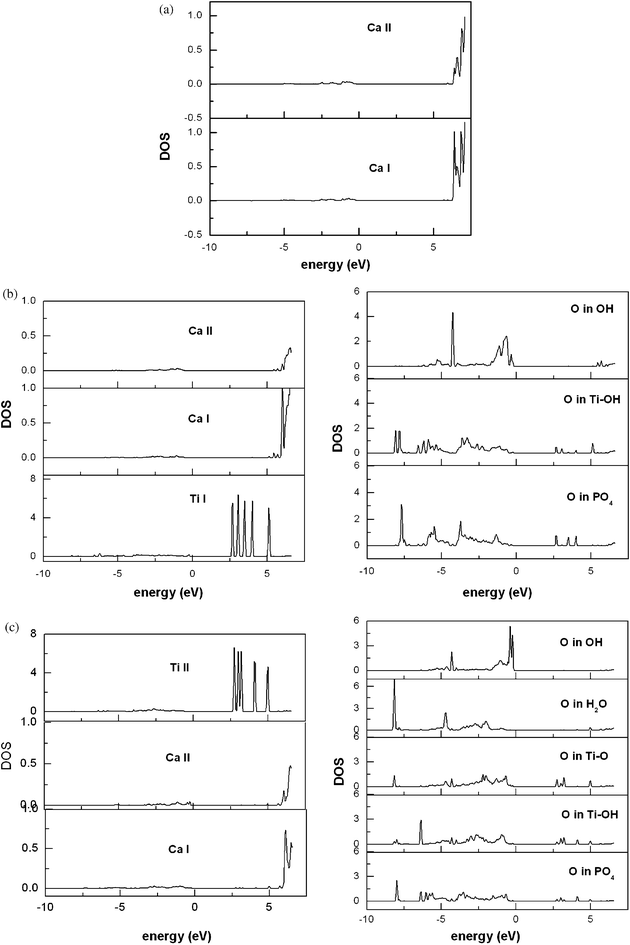 | ||
| Fig. 4 Projected local density of states (LDOS, arbitrary units): (a) bulk HA; (b) Ti(OH)2HA with substitution at site Ib; (c) Ti(OH)2HA with substitution at site IIb. | ||
3.3 Volume relaxation and lower substitution concentration
| a | b | c | α, β, γ | Volume | E 0 | E relaxed | |
|---|---|---|---|---|---|---|---|
| HA | 9.51 | 9.51 | 6.90 | 90.0, 90.0, 120.0 | 540.3 | −313.41 | — |
| Ti(OH)2HA-I | 9.59 | 9.76 | 7.05 | 85.6, 91.6, 119.8 | 570.2 | −335.83 | −336.95 |
| Ti(OH)2HA-II | 9.71 | 9.66 | 7.08 | 91.8, 87.7, 122.0 | 563.4 | −337.59 | −338.21 |
As described above, Ti(OH)22+ is incorporated in site I or II to form short Ti–O bonds. In the large polyhedron of site I, Ti and phosphate shift considerably from the original Ca2+ site to form higher Ti–O coordination. In site II, Ti(OH)22+ fits the smaller cation polyhedron well; thus Ti has a smaller shift (see Table 3) than in site I forming shorter Ti–O bonds and stronger complexation. This spatial difference and the consequent coordination are strongly associated with the site preference and volume change.
It is also noticed that in sites I and II, the in-plane expansion of a and b is anisotropic. We found this to be related to the position of the metal ion that was replaced. The site I cation is located nearly along the b-axis (see Fig. 1), thus the b vector is largely affected by the substitution and shows a distinctly larger expansion than the a vector. The site II cation, on the other hand, is located near the origin of the hexagonal cell and in the [110] direction, thus a and b vectors show similar variation under the substitution induced volume relaxation.
Experimentally, XRD patterns have shown that the crystallinity became poor with increasing Ti/Ca ratio. Wakamura found XRD patterns characteristic of HA at Ti atomic ratio up to 50 at%, with increasing signs of disorder above 10 at%. No comment was made about possible lattice parameter changes with composition. Analyzing the figure given in that work, we noticed that the large-intensity XRD peaks showed a small shift toward increasing θ angle with increasing Ti/Ca ratio. The quality of the data are probably not sufficiently good to permit a quantitative fit. In the work of Sugiyama, it was stated that no systematic change of lattice constants and no evident shift of the XRD peaks with maximum intensity were observed. They thus concluded that Ti4+ was not incorporated into HA through substitution with Ca2+. Obviously, the structure of Ti-modified HA is not yet very clear experimentally; this may be due in part to variables of synthesis procedures. Further accurate measurements of the Ti–HA crystal structure are desirable to further characterize the metal sites and related structural changes that are predicted in the present theoretical work.
4. Conclusions
The present work provides an atomic-level analysis of Ti-induced structural modification of hydroxyapatite using periodic slab density functional theory. At 10 at% Ti-concentration, a strong preference for Ti(OH)22+ occupancy on site II is found. The divalent hydroxy complex on site II, with strong and short Ti–O coordinate bonds, is energetically favored by 2.0 eV over substitutions on site I. As to Ti4+occupancy, whose ionic radius is relatively much smaller than Ca2+, no noticeable preference is found on both sites I and II: both are endothermic. However, when H+ was removed from the OH channel to maintain the formal charge, the site II substitution appears preferential due to the large O shift forming a strong and short Ti–O bond. The Ti4+ substitution and occupation is not thermodynamically favored on either site; while the divalent Ti(OH)2 complex substitution on site II has a strong thermodynamic favorability. These results are consistent with the experimental ICP analysis suggesting divalent ion substitution in Ti-modified HA. Thus according to the thermodynamics of the substitution reaction, it is probable that crystalline Ti-modified HA can be synthesized under some moderate conditions. This prediction of Ti-incorporated HA crystallinity at low concentration agrees well with Wakamura’s experimental results.DFT calculations show that the stable substitution of Ti(OH)22+ for Ca2+ is distinguished by a large shift of Ti from undoped cation sites, which is highly related to the site preference difference as well as the volume expansion difference between sites I and II. The largest shift of Ti is found on site I, ascribed to the tendency to form short Ti–O coordination bonds in the larger polyhedron of that site. Still, this requires more energy for compensation and results in unfavorable energetics at site I. The full lattice relaxation of the 10 at% Ti composition shows noticeable cell parameter expansions and ∼5% volume change induced by Ti incorporation. There is a larger volume expansion in site I substituted HA, correlated with the largest shift of Ti in that site. The calculated cell parameter expansion is anisotropic along a and b lattice vectors, directly related to the position of the replaced cation in the crystal cell. Site I substitution leads to a larger cell parameter expansion along b and c, while a and b show nearly uniform expansion for the site II substitution. Experimental reports about crystal structure of Ti-modified HA by XRD are contradictory, or at least their interpretation is uncertain. The present prediction of Ti-induced cell and volume change may inspire some further experiments to verify the relationship of the site preference for Ti substitution and the corresponding cell parameter expansions. This will be important to an eventual understanding of the synergistic adsorbance and catalytic response of Ti–HA and its optimization.
Acknowledgements
This work was supported by the US Department of Energy through the Institute for Catalysis in Energy Processes at Northwestern University, Grant no. DE-FG-02-03ER15457. We thank Alexandre Rossi and Joice Terra for helpful discussions.References
- Z. Opre, J. D. Grunwaldt, M. Maciejewslki, D. Ferri, T. Mallat and A. Baiker, J. Catal., 2005, 230, 406 CrossRef CAS.
- S. Wuyts, D. E. De. Vos, F. Verpoort, D. Depla, R. De. Gryse and P. A. Jacobs, J. Catal., 2003, 219, 417 CrossRef CAS.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657 CrossRef CAS.
- A. Crosman, G. Gelbard, G. Poncelet and V. I. Parvulescu, Appl. Catal., A, 2004, 264, 23 CrossRef CAS.
- M. Wakamura, K. Hashimoto and T. Watanabe, Langmuir, 2003, 19, 3428 CrossRef CAS.
- A. M. Hu, M. Li, C. K. Chang and D. L. Mao, J. Mol. Catal. A: Chem., 2007, 267, 79 CrossRef CAS.
- (a) S. Sugiyama, S. Tanimoto, K. Fukuda, K. Kawashiro, T. Tomida and H. Hayashi, Colloids Surf., A, 2005, 252, 187 CrossRef CAS; (b) S. Sugiyama, S. Tanimoto, K. Fukuta and K. Sotowa, Phosphorous Res. Bull., 2006, 20, 141 Search PubMed.
- N. H. de Leeuw, Chem. Commun., 2001, 1646 RSC.
- N. H. de Leeuw, Chem. Mater., 2002, 14, 435 CrossRef CAS.
- N. H. de Leeuw, Phys. Chem. Chem. Phys., 2002, 4, 3865 RSC.
- D. Mkhonto and N. H. de Leeuw, J. Mater. Chem., 2002, 12, 2633 RSC.
- S. Peroos, Z. M. Du and N. H. de Leeuw, Biomaterials, 2006, 27, 2150 CrossRef CAS.
- L. Calderin, M. J. Stott and A. Rubio, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 134106 CrossRef.
- D. Haverty, S. A. M. Tofail, K. T. Stanton and J. B. McMonagle, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 094103 CrossRef.
- M. Corno, C. Busco, B. Civalleri and P. Ugliengo, Phys. Chem. Chem. Phys., 2006, 8, 2464–2472 RSC.
- K. J. Zhu, K. Yanagisawa, R. Shimanouchi, A. Onda and K. Kajiyoshi, J. Eur. Ceram. Soc., 2006, 26, 509 CrossRef CAS.
- X. Y. Ma and D. E. Ellis, Biomaterials, 2008, 29, 257 CrossRef CAS.
- J. Terra, E. R. Dourado, J. G. Eon, D. E. Ellis, G. Gonzalez and A. M. Rossi, Phys. Chem. Chem. Phys., 2009, 11, 568 RSC.
- D. E. Ellis, J. Terra, O. Warschkow, M. Jiang, G. Gonzalez, J. S. Okasinski, M. J. Bedzyk, A. M. Rossi and J. G. Eon, Phys. Chem. Chem. Phys., 2006, 8, 967 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1993, 47, 558 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- K. Sudarsanan and R. A. Young, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 1534 CrossRef CAS.
- R. M. Wilson, J. C. Elliott and S. E. P. Dowker, Am. Mineral., 1999, 84, 1406 CAS.
- D. M. Henderson and H. S. Gutowsky, Am. Mineral., 1962, 47, 1231 CAS.
- I. S. Ignatyev, M. Montejo and J. J. L. Gonzalez, J. Phys. Chem. A, 2007, 111, 7973 CrossRef CAS.
| This journal is © the Owner Societies 2010 |