The influence of alkaline earth ions on the structural organization of acetone probed by the noncoincidence effect of the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O) band: experimental and quantum chemical results†
O) band: experimental and quantum chemical results†
Maria Grazia
Giorgini
*a,
Hajime
Torii
b and
Maurizio
Musso
c
aDipartimento di Chimica Fisica ed Inorganica, Università di Bologna, Viale del Risorgimento 4, I-40136 Bologna, Italy. E-mail: mariagrazia.giorgini@unibo.it; Fax: +39.051.2093690; Tel: +39.051.2093701
bDepartment of Chemistry, School of Education, Shizuoka University, 836 Ohya, Shizuoka 422-8529, Japan
cFachbereich Materialforschung und Physik, Abteilung Physik und Biophysik, Universität Salzburg, Hellbrunnerstraße 34, A-5020 Salzburg, Austria
First published on 7th November 2009
Abstract
We have investigated the Raman noncoincidence effect (NCE = ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) aniso − iso, where aniso and iso are the anisotropic and the isotropic Raman frequencies) of the ν(C
aniso − iso, where aniso and iso are the anisotropic and the isotropic Raman frequencies) of the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) band of acetone arising from the interactions of this solvent with the metal ions in acetone electrolytic solutions of alkaline earth metal (Mg, Ca, Sr, Ba) perchlorates. Assisted by the results of ab initio molecular orbital (MO) calculations carried out at the Hartree–Fock (HF) level with the 6-31+G(2df,p) and LanL2DZ basis sets, we have been able to attribute the anisotropic and isotropic components of this band to the formation of acetone–metal ion clusters, (acetone)nM2+, and to interpret its high and negative NCE, opposed to the positive NCE of the bulk liquid, as the consequence of the large separation between the higher frequency of the in-phase mode (active in the Raman isotropic spectrum) and the lower (average) frequency of the n − 1 out-of-phase modes (predominantly active in the Raman anisotropic spectrum). The negative sign of the NCE is compatible with the transition dipole coupling (TDC) mechanism. The comparison between the observed NCE for each electrolytic solution at the concentrations used in this study and those calculated for the different solvation numbers n of each (acetone)nM2+cluster gives a clear indication of the highest stability of the hexa-coordinated cluster for the Mg2+ ion, but leaving uncertain (n = 6 or 8) this conclusion for the acetone clusters of the remaining M2+ ions. We have interpreted the observed and calculated decrease of the magnitude of NCE with the ion size through the ion polarizing power in the light of the ion effective charge and its distance (M2+⋯OC) from the CO oscillators.
O) band of acetone arising from the interactions of this solvent with the metal ions in acetone electrolytic solutions of alkaline earth metal (Mg, Ca, Sr, Ba) perchlorates. Assisted by the results of ab initio molecular orbital (MO) calculations carried out at the Hartree–Fock (HF) level with the 6-31+G(2df,p) and LanL2DZ basis sets, we have been able to attribute the anisotropic and isotropic components of this band to the formation of acetone–metal ion clusters, (acetone)nM2+, and to interpret its high and negative NCE, opposed to the positive NCE of the bulk liquid, as the consequence of the large separation between the higher frequency of the in-phase mode (active in the Raman isotropic spectrum) and the lower (average) frequency of the n − 1 out-of-phase modes (predominantly active in the Raman anisotropic spectrum). The negative sign of the NCE is compatible with the transition dipole coupling (TDC) mechanism. The comparison between the observed NCE for each electrolytic solution at the concentrations used in this study and those calculated for the different solvation numbers n of each (acetone)nM2+cluster gives a clear indication of the highest stability of the hexa-coordinated cluster for the Mg2+ ion, but leaving uncertain (n = 6 or 8) this conclusion for the acetone clusters of the remaining M2+ ions. We have interpreted the observed and calculated decrease of the magnitude of NCE with the ion size through the ion polarizing power in the light of the ion effective charge and its distance (M2+⋯OC) from the CO oscillators.
1. Introduction
The performance of many industrial processes and technological devices and that of many living systems is based on the structure and dynamics of ion-clusterized molecules and biomolecules (so called ionophores). The transport of K+ ions across membranes, a process of vital importance for all organisms, is mediated by the formation of a complexon generated by the strong interactions of six CO bonds, present in the cavity of the large ring molecule of valinomycin, a carboxylic ionophore, pointing towards the central ion.1,2 The efficiency of rechargeable batteries relies upon the ionic conductivity of their electrolytic solutions which, in turn, depends on the structures of the solvated ionic species and their mobility. After the development of the rechargeable batteries based on the Li+ ion,3 more recently the efforts have been addressed to the research of high energy density secondary batteries based on Mg2+ technology.4
Computational5,6 and experimental7–15 approaches have been devised to address the issue regarding the interactions between alkali and alkaline earth metal ions and strong polar groups (like C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, CO, and SO) of organic solvents such as nitriles, ketones, amides, and sulfoxides, which, in the batteries technology, are the most suitable for their high permittivity and reasonably low viscosity. Among the experimental methodologies, vibrational spectroscopy has proved to offer an invaluable and versatile tool to shed light on the nature of these kinds of interactions.9–15 The spectral analysis of the ion field effects on the solvent is based on the appearance of new bands in the vibrational (IR and Raman) spectrum upon dissolution of the salt into the solvent. These new bands appear close in frequency to the most interaction-sensitive bands of the neat solvent, typically those referred to the CN, CO and SO stretching vibrations [denoted as ν(C≡N), ν(CO), and ν(SO) hereafter], and are caused by the modifications of these solvent normal modes by the ion field. Their frequency shifts, intensities, and concentration dependences help to assess the strength of the ion–solvent species (clusters) formed within the electrolytic solutions and to evaluate their solvation numbers, i.e., the number of solvent molecules clustering around each ion in the first solvation shell.12
N, CO, and SO) of organic solvents such as nitriles, ketones, amides, and sulfoxides, which, in the batteries technology, are the most suitable for their high permittivity and reasonably low viscosity. Among the experimental methodologies, vibrational spectroscopy has proved to offer an invaluable and versatile tool to shed light on the nature of these kinds of interactions.9–15 The spectral analysis of the ion field effects on the solvent is based on the appearance of new bands in the vibrational (IR and Raman) spectrum upon dissolution of the salt into the solvent. These new bands appear close in frequency to the most interaction-sensitive bands of the neat solvent, typically those referred to the CN, CO and SO stretching vibrations [denoted as ν(C≡N), ν(CO), and ν(SO) hereafter], and are caused by the modifications of these solvent normal modes by the ion field. Their frequency shifts, intensities, and concentration dependences help to assess the strength of the ion–solvent species (clusters) formed within the electrolytic solutions and to evaluate their solvation numbers, i.e., the number of solvent molecules clustering around each ion in the first solvation shell.12
It is worth to notice that, unlikely the ν(CN) mode which exhibits a blue shift in both the Raman and IR spectra upon cluster formation,9 the ν(CO) mode of carbonyl solvents such as acetone,12,13 γ-butyrolactone,14 and N,N-dimethylformamide15 appears blue shifted in the Raman and red shifted in the IR spectrum. This only apparent difference has found its justification in the light of the results reported in our previous paper16 by considering that isotropic and anisotropic profiles of these bands are affected by positive and negative frequency shifts, respectively, and that while a Raman spectrum is essentially composed of its isotropic component and then results blue shifted, its IR counterpart has a density of states which essentially reproduces that of the anisotropic Raman spectrum, thereby resulting red shifted due to the presence of the ion field. The physical origin of the noncoincidence of the IR and Raman profiles, and more specifically of the isotropic and anisotropic profiles of the ν(CO) Raman band of generic liquid carbonyl compounds is nowadays definitely established.17–20 This phenomenon is known as either the Raman noncoincidence effect, if expressed by the separation between the anisotropic and the isotropic first moments M (NCE = Maniso − Miso), or as the IR–Raman noncoincidence effect, given by the separation between the IR and the Raman first moments M (NCE = MIR − MR). It is brought about by the intermolecular coupling between identical oscillators of adjacent molecules in condensed phase systems, through their transition dipoles (called transition dipole coupling, TDC). This is the reason that makes NCE predominantly observable in strongly IR active bands such as the ν(CO) band of carbonyl compounds and the reason that makes it a sensitive probe of the condensed phase structure (molecular positions and orientations) of these molecular systems.21–24 In fact, as is well known from fundamental electrostatics, the value of a TDC constant (including the sign) is determined by the magnitudes and the relative orientations of the transition dipoles ![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) ki and kj (for the kth oscillator) of the interacting molecules (i,j), and is also proportional to the inverse cube of their separation rij. Along this line of reasoning, positive NCE values are expected,25 and indeed observed, in dipolar liquids predominantly structured by dipolar forces21–22 and negative NCE values in dipolar liquids predominantly structured by non-dipolar forces (typically, H-bonded systems).26,27
ki and kj (for the kth oscillator) of the interacting molecules (i,j), and is also proportional to the inverse cube of their separation rij. Along this line of reasoning, positive NCE values are expected,25 and indeed observed, in dipolar liquids predominantly structured by dipolar forces21–22 and negative NCE values in dipolar liquids predominantly structured by non-dipolar forces (typically, H-bonded systems).26,27
As first noticed by Kecki and Sokolowska,28 the ν(CO) band of acetone exhibits a spectacular NCE sign inversion from +5 cm−1 in the neat solvent to −16 cm−1 in acetone/MX electrolytic solutions. This phenomenon, carefully investigated in our previous paper with the joint use of quantum chemical calculations,16 has offered the interpretative key for its onset and for its most relevant peculiarities. Shortly, the large and negative values of the NCE observed in acetone-electrolytic solutions are essentially the consequence of the large splitting between the in-phase and the n − 1 out-of-phase ν(CO) vibrational modes generated by n CO groups pointing towards the central ion in the (acetone)nM+ cluster, with the in-phase mode (active in the isotropic Raman spectrum) much higher in frequency than the out-of-phase modes (active in the IR and in the Raman, predominantly anisotropic, spectrum). This result additionally explains the reason for the opposite shifts of the ν(CO) band observed in the Raman and IR spectra as reported in literature mentioned above,12–15 since the blue shift of the ν(CO) band observed in the Raman spectrum essentially mirrors the high-frequency shift of the isotropic Raman spectrum and the red shift of the IR spectrum is the consequence of the low-frequency shift of the out-of-phase modes.
The purpose of this paper is that of extending the investigations and methodologies reported in ref. 16 for alkali metal M+X− (M = Li, Na) acetone electrolytic solutions to those of alkaline earth metal (M = Mg, Ca, Sr, Ba) salts with the focus of finding out a correlation between the observed negative NCE of the ion-perturbed ν(CO) band and the increased ion-field strength of these ions. In analogy with the approach we followed in ref. 16, the interpretation of this spectroscopic observable will be based on the results of quantum chemical calculations of the vibrational frequencies for (acetone)nM2+ clusters with n = 4, 6, and 8.
2. Experimental
2.1 Sample preparation
The anhydrous salts, Mg(ClO4)2 and Ba(ClO4)2 (Sigma-Aldrich), conserved under dried atmosphere since their delivery, were used as received; Ca(ClO4)2 tetrahydrate (CHEMOS, GmbH) and Sr(ClO4)2 hexahydrate (Alfa Aesar, Johnson Matthey Co.) were slowly dehydrated in small quantities at 124 °C and maintained at this temperature for two days. Acetone was a Sigma-Aldrich solvent with purity better than 99.8% and water content less than 0.1%.All electrolytic solutions examined in this study were prepared in a dry box under inert (N2) atmosphere and here transferred into quartz stopcock cells appropriate for high vacuum operations, where they were held during all the experiments to warrant against the leakage of the inert atmosphere and prevent the access of ambient water. The solutions examined in this study were prepared with concentrations (mole fraction, xsalt) ranging from approximately 0.05 to 0.01. This choice was guided by the achievement of the best compromise between salt solubility in acetone, spectral intensity of the ν(CO) band in the cluster species, and an efficient inhibition of ion-pairs formation in the solutions. Since the solubility gets lower as the salt cation M2+ gets larger, the concentration of the electrolytic solutions of the salts with the smaller size cations, Mg2+ and Ca2+, could be as high as 0.05, whereas those with the larger cations, Sr2+ and Ba2+, could not be higher than 0.03 and 0.02, respectively. The presence of ion-pairs or solvent-shared ions was checked through the observation of the spectral modifications of the ClO4− symmetric stretching band at 932 cm−1. As will be mentioned in the Results and discussion section, the purpose of completely avoiding ion-pair formation could not be reached for all solutions.
2.2 Raman spectra
The Raman spectra were obtained by exciting the sample with the 514.53 nm line emitted from an Ar+ ion laser (Coherent Innova 300), by analyzing the scattered light in the 90° scattering geometry configuration by the use of a spectrometer (Spex 1404) and by detecting the dispersed scattered light by the use of a multichannel detector (LN2 CCD, 1024 × 256 pixels, Spex). To eliminate any parasitic horizontal component in the vertically (V) polarized exciting laser light, a Glan-Laser polarizer was placed in front of the sample. The selection of the vertically (V) and horizontally (H) polarized components of the Raman scattered light was done with the use of a polarization sheet placed along the optical axis of observation, alternately oriented in the two directions. To compensate for the polarization dependent reflectance of the gratings, the VV and VH polarized scattered light was depolarized by a polarization scrambler placed behind the polarization sheet, before being focused on the entrance slit (width 100 μm, height 10 mm) of the spectrometer. In the spectroscopic region of interest in this study (around 1700 cm−1, where the carbonyl band of acetone appears) the detector pixel separation amounts to 0.35 cm−1, the frequency coverage of the CCD active area to 220 cm−1, and the instrumental slit function (fwhm) to 1.4 cm−1. The effective polarization conditions were checked by measuring the Raman depolarization ratio of the three lowest CCl4 bands. In addition, for each solution we have corrected the intensity distribution reaching the CCD (instrument spectral response) using the fluorescence spectrum of a fluorescein solution as reference (relative intensity correction of the spectrometer response).For the frequency calibration of the spectra the collection of each VV and VH sample spectrum was followed by that of the corresponding spectrum of a Neon calibration lamp. The Ne atomic line at 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 678.28 cm−1 was adopted as reference for drift corrections in the ν(CO) region.
678.28 cm−1 was adopted as reference for drift corrections in the ν(CO) region.
The Raman measurements were performed at room temperature and atmospheric pressure. Integration times and accumulations were chosen to obtain high levels of signal/noise ratio in all experiments.
2.3 Data treatment and fitting strategy
The experimental Raman spectra of the ν(CO) band in each acetone electrolytic solution were analyzed using the spectroscopy software GRAMS/386. After corrections of the raw spectra for frequency drifts and for the instrumental spectral response, the IVV and IVH spectra were combined to obtain the isotropic profiles (Iiso = IVV − 4/3IVH), the anisotropic ones being directly given by the IVH spectra (Iani = IVH).
To fit the experimental Raman spectra, we have used a definite number of mixed Gaussian/Lorentzian components for the isotropic and anisotropic profiles, following the criteria already adopted for the spectral analysis of the ν(CO) bands observed in the acetone solutions of alkali metal perchlorates.16 The band parameters entered in the fitting procedure were chosen to achieve the best compromise between the χ2 value of the fitting and the uncertainties of the optimized parameters.
The isotropic Raman spectra of acetone solutions (acetone/Mg(ClO4)2, acetone/Ca(ClO4)2, acetone/Sr(ClO4)2 and acetone/Ba(ClO4)2) were all analyzed by means of five components unless otherwise stated. To reproduce the main, blue-side asymmetric, ν(12CO) band (occurring around 1710 cm−1 in all solutions), two components with only slightly separated frequencies were necessary, the one at the higher frequency mimicking the band asymmetry. Of the remaining three components, one was required to model the shoulder (which is attributed to the carbonyl band of the solvent molecules clustering around the ion) present on the higher frequency side of the main ν(12CO) carbonyl band. Two additional components were included: one to reproduce the ν(13CO) band at 1676 cm−1, which is due to acetone-13CO present as natural impurity, and another at 1752 cm−1 to reproduce the combination band ν17 + ν19 intensified by a Fermi resonance. Additionally, limitedly to acetone/Mg(ClO4)2 solution it was necessary to include a sixth component in the fitting to account for the weak band at 1788 cm−1, clearly discernible only in the isotropic spectrum of this solution, which is due to the Mg2+ ion effect on the combination band ν17+ν19.
The main ν(12CO) band of the anisotropic Raman profile appears, in all these electrolytic solutions, to be structured by a lower frequency component, which appears remarkably separated only in the acetone/Mg(ClO4)2 solution. Then, the anisotropic Raman profiles were all analyzed by four components, one of them representing the symmetric main ν(12CO) band (corresponding to the anisotropic Raman band of neat acetone), and another modeling the lower frequency profile of the main ν(12CO) band (attributed to the carbonyl band of the solvent molecules clustering around the ion). Two others were included to reproduce the band present at 1752 cm−1 due to the combination band ν17 + ν19 and the ν(13CO) band at 1676 cm−1 due to acetone-13CO present as natural impurity.
The fitting procedure directly provides the central frequencies (Pi) of all these decomposed spectral components, their fit standard errors (σi) which we have taken as their uncertainties, and their integrated intensities (Ai). For the main ν(12CO) band in the isotropic Raman spectra of the neat solvent and solutions, we have evaluated the corresponding first moment (M) as their weighted averages (P1A1 + P2A2)/(A1 + A2) from the peak frequencies (P1, P2) and the integrated intensities (A1, A2) of the two component bands. The uncertainty of this first moment was analogously obtained as the weighted average of the standard errors of the two components. With regard to the bands consisting of a single spectral component (such as the bands of the solvent molecules clustering around the ion), the first moments are the same as the peak frequencies. The NCE values were calculated as the difference Maniso − Miso, with their uncertainties evaluated according to the error propagation law.
3. Computational procedure
Ab initio molecular orbital (MO) calculations were carried out for the following species: (1) (acetone)nMg2+ with n = 3, 4, and 6, (2) (acetone)nCa2+ with n = 6 and 8, (3) (acetone)nSr2+ with n = 6 and 8, and (4) (acetone)nBa2+ with n = 6 and 8. The calculations were performed at the Hartree–Fock (HF) level, with the 6-31+G(2df,p) basis set for the Mg2+ and Ca2+ clusters and with the Los Alamos effective core potential plus double-ζ (LanL2DZ) basis set29 for the Sr2+ and Ba2+ clusters. To check the consistency between the results obtained with these two basis sets, calculations were also carried out with the LanL2DZ basis set for (acetone)6Mg2+, (acetone)6Ca2+, and (acetone)8Ca2+.For each species, the geometry was optimized, and the vibrational frequencies, the IR and Raman intensities, and the depolarization ratios were calculated at the potential energy minimum. The locations of the carbonyl oxygen atoms in the optimized structures were found to have approximate triangular, tetrahedral, octahedral, and square antiprism symmetries for the clusters with 3, 4, 6, and 8 acetone molecules, respectively (Fig. 1a and 1b). In the square antiprism structure, each vertex has 4 approximate nearest neighbor vertices. We also tried calculations for (acetone)8Mg2+ (with the 6-31+G(2df,p) basis set), but an optimized structure with 8 CO⋯Mg2+ contacts could not be obtained.
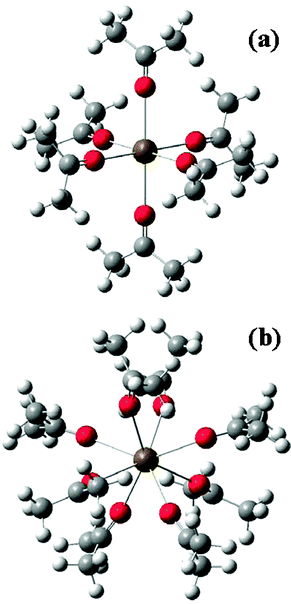 | ||
| Fig. 1 (a) Optimized structure of (acetone)6Mg2+ cluster (with no exact molecular symmetry). The six CO groups and the Mg2+ ion, however, fulfil a nearly octahedral structure (Oh molecular symmetry). (b) Optimized structure of (acetone)8Ba2+ (with no exact molecular symmetry). The oxygen atoms of the eight CO groups form a square antiprism in which each vertex has four nearest neighbor vertices. Note that the connections between Mg2+ and the CO groups are not covalent bonds but have ionic character. | ||
The number of the CO stretching modes is n for the clusters with n acetone molecules. The value of the NCE (NCE≡aniso–iso) was calculated from the intensity-weighted averages of frequencies iso = ∑nj = 1Iisojj/∑nj = 1Iisoj and aniso = ∑nj = 1Ianisojj/∑nj = 1Ianisoj,16,30 where j is the frequency of the jth CO stretching mode, and Iisoj and Ianisoj are its isotropic and anisotropic Raman intensities. All the calculated frequencies were first scaled by 0.9. Based on the comparison between the observed and calculated frequencies for the (acetone)4Li+ cluster [observed: −16.0 cm−1,16 calculated (scaled by 0.9): −19.6 cm−1 at HF/6-31+G(2df,p)16 and −17.1 cm−1 at HF/LanL2DZ], the frequencies calculated at HF/6-31+G(2df,p) were further multiplied by 0.816 (= 16.0/19.6), and those at HF/LanL2DZ by 0.936 (= 16.0/17.1).
These calculations were carried out by using the Gaussian 03 program31 on Dell PowerEdge 2950 and other servers in the laboratory of one of the authors (HT), and on Fujitsu PrimeQuest servers at the Research Center for Computational Science of the National Institutes of Natural Sciences at Okazaki.
4. Results and discussion
4.1 Observed spectra
In Fig. 2a we report the isotropic and anisotropic Raman spectra of the acetone/Mg(ClO4)2 solution in the ν(CO) mode region and, in Fig. 2b, for comparison purposes, the analogous spectra of neat liquid acetone. From a comparison of the two panels in Fig. 2, it is rather evident that, in the isotropic Raman profile of the electrolytic solution, a new spectral feature arises on the higher frequency side of the main ν(12CO) band (the B band, which is close in frequency to the analogous band in neat acetone reported in Fig. 2b). The former, which is separated from the latter by approximately 20 cm−1, is referred to as C band. In Fig. 2a the anisotropic profile appears as the overlap of two bands, one peaked at around 1713 cm−1, closely coincident in frequency with the analogous ν(12CO) profile in neat acetone and having approximately the same bandwidth (20 cm−1), and the other peaked at approximately 1695 cm−1. The six and four components, obtained by the numerical analysis of the measured isotropic and anisotropic Raman spectra of the ν(CO) mode region, respectively, of the acetone/Mg(ClO4)2 solution (Fig. 2a) are shown in Fig. 3a and 3b (see details in section 2.3 and in the legend of this figure).
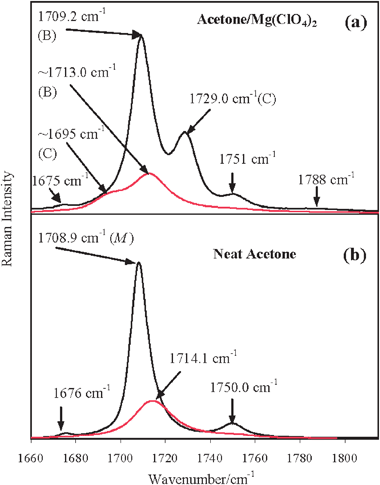 | ||
| Fig. 2 (a) Isotropic (black line) and anisotropic (red line) Raman spectra of acetone/Mg(ClO4)2 solution (xMg(ClO4)2 = 0.045) in the ν(CO) mode region and (b) of neat liquid acetone (for comparison purposes). In panel (a), in the isotropic Raman profile, the higher frequency component (denoted as C) is rather well separated from the main ν(CO) feature (denoted as B) peaked at approximately the same frequency as in neat acetone; in the anisotropic Raman profile the lower frequency component (denoted as C) results appreciably well separated from the main ν(CO) feature (denoted as B) peaked at approximately the same frequency as in neat acetone. In the isotropic Raman profile, additionally to these two features, two satellite bands at the higher (1751 cm−1) and lower (1675 cm−1) frequency sides are well evident, as in the case of neat acetone, and, only marginally, a band at 1788 cm−1. M indicates the spectral first moment of the corresponding band. | ||
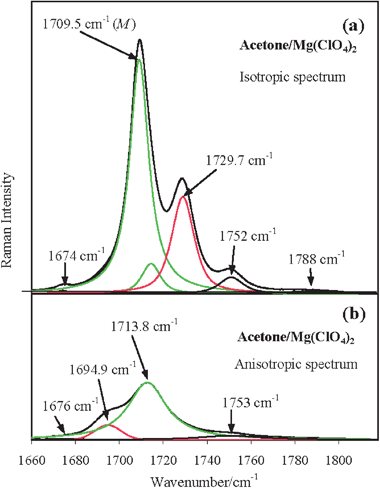 | ||
| Fig. 3 Spectral decomposition of the observed (a) isotropic and (b) anisotropic ν(CO) spectral Raman region of the acetone/Mg(ClO4)2 solution obtained by numerical fitting. The measured and fitted envelopes are indistinguishable since they are completely superimposed. In panel (a) the components at 1729.7 cm−1 (cluster – red line) and at 1709.5 cm−1 (bulk – superposition of the two green lines) refer to the ν(CO) band of acetone molecules involved and not involved, respectively, in the structuring effects of Mg2+. The asymmetric bulk profile peaked at 1709.5 cm−1 is compounded of a (symmetric) very strong and a (symmetric) very weak band entered in the fitting (see details in Section 2.3) to mimic its slight blue side asymmetry. The weak and the very weak components at 1750 cm−1 and at 1788 cm−1 (black lines) refer to the combination band ν17 + ν19 of acetone molecules involved and not involved, respectively, in the structuring effects of Mg2+. In panel (b) the components at 1694.9 cm−1 (cluster – red line) and 1713.8 cm−1 (bulk – green line) refer to the acetone molecules involved and not involved, respectively, in the structuring effects of Mg2+. M indicates the spectral first moment of the corresponding band. | ||
A qualitatively analogous situation holds for acetone/Ca(ClO4)2, acetone/Sr(ClO4)2 and acetone/Ba(ClO4)2 solutions, as displayed in Fig. 4a and 4b, in which collective views of the isotropic and anisotropic profiles, respectively, of the ν(CO) mode region of all the studied acetone electrolytic solutions are shown. For all solutions except acetone/Mg(ClO4)2 the anisotropic profiles are indicated as (B+C) because of the extended overlap of the two spectral components.
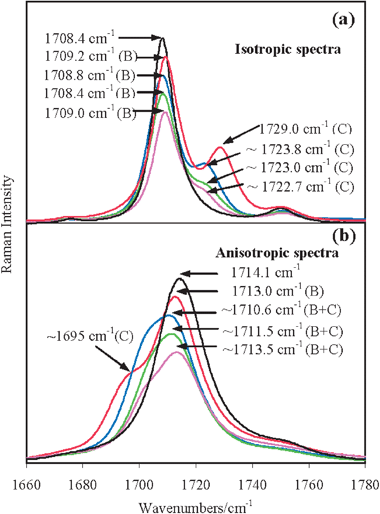 | ||
| Fig. 4 Collective views of the (a) isotropic and (b) anisotropic spectra of neat acetone (black line), acetone/Mg(ClO4)2 (red line, xsalt = 0.045), acetone/Ca(ClO4)2 (blue line, xsalt = 0.05), acetone/Sr(ClO4)2 (green line, xsalt = 0.02) and acetone/Ba(ClO4)2 (magenta line, xsalt = 0.01) solutions in the ν(CO) mode region. The heights of the isotropic (a) and anisotropic (b) spectra are normalized to unity and, for a better graphical clarity, scaled by 0.9 (red lines), 0.8 (blue lines), 0.7 (green lines) and 0.6 (magenta lines). The isotropic Raman spectra of all the solutions shown in panel (a) have the main ν(12CO) components (denoted as B) approximately at the same frequency as in neat acetone, while the shoulders (denoted as C) are located on the higher frequency side and are appreciably separated in frequency from the main ν(CO) features. In the anisotropic spectra shown in panel (b), each main component B and the corresponding lower frequency shoulder are significantly overlapped and thus collectively referred to as B+C. | ||
For the sake of completeness, we remark that the lower frequency wing of the isotropic Raman spectrum (and marginally also of the anisotropic Raman spectrum) of the ν(CO) band is featured in all acetone solutions, as in neat acetone, by the weak band around 1676 cm−1 due to the ν(13CO) mode of the acetone isotopic impurity present at natural abundance. On the other hand, the higher frequency wing of the isotropic Raman spectrum (and marginally also of the anisotropic Raman spectrum) is featured, in all acetone solutions (see Fig. 2a, 4a, and 4b), by the presence of the weak band around 1751 cm−1, very likely originating from the Fermi resonance combination ν17 + ν19 as observed in neat acetone at 1750 cm−1 (where ν17 is the antisymmetric C–C stretching at 1220 cm−1, and ν19 is the in-plane CO bending at 530 cm−1). Not surprisingly, even this band shows the effect of ion solvation and does so even more remarkably than the ν(CO) fundamental mode. The band appearing at 1788 cm−1, discernible only in the isotropic spectrum of the acetone/Mg(ClO4)2 solution (see Fig. 2a), results from the large perturbation produced by the Mg2+ cation on the ν17 + ν19 combination band (blue shift of 38 cm−1), probably because of the combined effects of Mg2+ ion on the two vibrations ν17 and ν19. Specifically, the addition of the Mg2+ ions to acetone is expected to have a remarkable effect on the in-plane CO bending, in analogy to that observed for the same mode in N,N-dimethylformamide (DMF)/Mg2+ solutions where it appears blue shifted by approximately 28 cm−1.32 Moreover, because the antisymmetric C–C stretching in acetone gets blue shifted by 8 cm−1 due to the interactions with Li+,12 its blue frequency shift due to the interactions with Mg2+ is expected to be larger. By using the intensity of this band it may be possible to obtain, in a future study, an estimate of the solvation numbers.33
Summarizing, in the studied acetone electrolytic solutions each isotropic and anisotropic Raman profile of the ν(12CO) band consists of a couple of bands. In the concentration range used in this investigation (xsalt < 0.05), the more intense of them is the one approximately coincident in frequency with the ν(12CO) band of neat acetone [1708.9 and 1714.1 cm−1 (first moments) in the isotropic and anisotropic Raman spectra, respectively], which is attributed to the solvent molecules left unaltered by the presence of the ions and hereafter called bulk component (B). The less intense band that occurs at a higher frequency in the isotropic Raman profiles (at 1729.7 cm−1 in acetone/Mg(ClO4)2 solution) and at a lower frequency in the anisotropic Raman profiles (at 1694.9 cm−1) is attributed to the ν(12CO) band of the solvent molecules affected by the presence of the ions and hereafter called cluster component (C).
4.2 Relation between spectral features and solvent configurations
The evaluated isotropic and anisotropic first moments and the NCE of the ν(CO) band of the bulk and cluster components for all electrolytic solutions and, for comparison purposes, those of the ν(CO) band in the neat solvent are reported in Table 1. We can see that in all the studied electrolytic solutions (a) the ν(CO) Raman band of the acetone molecules affected by the presence of cations (cluster components) features a large and negative NCE, as opposed to the unperturbed ν(CO) Raman band (bulk component) showing a smaller and positive NCE; (b) the negative NCE decreases with the increase of the cation size, being −34.8 cm−1 in acetone/Mg(ClO4)2 at x = 0.045, −22.8 (−23.0) cm−1 in acetone/Ca(ClO4)2 at x = 0.045 (0.02), −22.1 (−21.3) cm−1 in acetone/Sr(ClO4)2 at x = 0.02 (0.03), and −21.1 (−21.0) cm−1 in acetone/Ba(ClO4)2 at x = 0.02 (0.01), (c) the metal ion charge has a remarkable influence on the negative NCE, as revealed from a comparison of the values of NCE between the above four solutions and the acetone/LiClO4 and acetone/NaClO4 solutions [Table 1 in ref. 16], where the latter amount to −15.9 and −14.7 cm−1, respectively. As discussed in ref. 16, the negative sign of the NCE of the cluster component is compatible with the solvent structure formed around the cation, where the CO bonds of the solvent molecules are pointing toward the cation, and the TDC mechanism of vibrational coupling between the CO oscillators.
O) modes of the bulk and cluster components of acetone solvent in acetone electrolytic solutions and neat liquid acetone
| Mixture | (xsalt) | Bulk component | Cluster component | ||||
|---|---|---|---|---|---|---|---|
| M iso | M aniso | NCE , | M iso | M aniso | NCE , | ||
| a All the reported values are in cm−1 unit. b The numbers in parentheses are errors in the last digit. | |||||||
| Acetone | (0) | 1708.9 | 1714.1 | 5.2(1) | |||
| Acetone/Mg(ClO4)2 | (0.045) | 1709.5 | 1713.8 | 4.3(9) | 1729.7 | 1694.9 | −34.8(1) |
| Acetone/Ca(ClO4)2 | (0.02) | 1708.8 | 1713.1 | 4.4(5) | 1724.8 | 1701.8 | −23.0(4) |
| (0.045) | 1708.6 | 1712.6 | 4.0(9) | 1724.0 | 1701.2 | −22.8(3) | |
| Acetone/Sr(ClO4)2 | (0.02) | 1708.7 | 1713.1 | 4.4(2) | 1723.9 | 1701.8 | −22.1(3) |
| (0.03) | 1708.4 | 1713.5 | 5.1(1) | 1723.4 | 1702.1 | −21.3(1) | |
| Acetone/Ba(ClO4)2 | (0.01) | 1709.2 | 1714.0 | 4.8(2) | 1722.5 | 1701.3 | −21(1) |
| (0.02) | 1709.3 | 1713.7 | 4.4(2) | 1722.1 | 1701.5 | −21.1(9) | |
The comparison of the isotropic and anisotropic spectral profiles of the ν(CO) band for acetone/M(ClO4)2 (M = Ca, Sr, Ba) solutions at salt concentrations, xCa(ClO4)2 = 0.045 and 0.02, xSr(ClO4)2 = 0.02 and 0.03 and xBa(ClO4)2 = 0.01 and 0.02, indicate appreciable variations of the relative intensity of the two components, Iν(CO)cluster/Iν(CO)bulk, but a substantial invariance of the isotropic and anisotropic first moments and the NCE values as indicated in Table 1. This suggests that the structural organization of acetone molecules around each ion, i.e. the solvation number of the cluster, is left unaltered by dilution within this concentration range. Additionally, to ascertain the occurrence of contact ion pair formation in our solutions, we have used the ν1 band of the ClO4− anion at 933 cm−1. In Fig. 5 we report the VV Raman spectra of acetone/Mg(ClO4)2, acetone/Ca(ClO4)2, acetone/Sr(ClO4)2, and acetone/Ba(ClO4)2 solutions in the ν(ClO4−) totally symmetric stretching mode region. On the higher frequency side of the band originated by the ‘free’ anions peaked at approximately 933 cm−1, very weak satellites are only marginally evident in the low concentration regimes of these solutions. These bands are assigned to solvent-shared ion pairs and contact-ion pairs in ref. 34 and to a monodentate contact-ion pair in ref. 35. On the lower frequency side, a band separated from the free ClO4− band by approximately 6 cm−1 appears, very evident in the acetone/Sr(ClO4)2 and acetone/Ba(ClO4)2 solutions, quite weak in acetone/Ca(ClO4)2, and negligible or absent in acetone/Mg(ClO4)2 solutions. The intensity of this band relative to that of the free anion slightly increases with the salt concentration. This means that it must be very likely associated with some sort of ion-pairing. In the attempt of finding the origin of these low and high frequency satellites of the free-ClO4− anion, a supplementary quantum chemical calculation at the HF/3-21G* level has been carried out on possible contact ion pairs directly derived by substitution of one solvent molecule with one anion molecule in the (acetone)6 Sr2+ cluster species. [The 3-21G* rather than the LanL2DZ basis set is necessary to correctly represent the ClO4− anion.] It has been found that the ClO4− vibration of the bidentate mixed cluster species (acetone)5 (ClO4−) Sr2+ (see Fig. 6a) is located at ∼20 cm−1 lower than that of an isolated ion, reasonably accounting for the observed red shifted ClO4− vibration, whereas that of the monodentate form of the same cluster species (see Fig. 6b), which is obtained by constraining one of the Sr⋯O–Cl angles to 180° and is less stable, is upshifted by 15 cm−1 with respect to that of the free anion then accounting for the weak satellites observed in the higher frequency side of the main band of the free-ClO4−. Both species are categorized as contact-ion pairs according ref. 34. Then it is possible to interpret (at least qualitatively) the observed band profile in the 930 cm−1 spectral region by free ClO4− ions (dominant), bidentate contact ion pairs, and monodentate contact ion pairs (less stable).
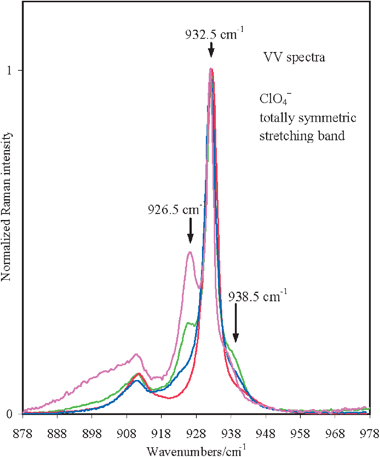 | ||
| Fig. 5 Normalized VV spectra around 930 cm−1 (ClO4− symmetric stretching) for the electrolytic solutions acetone/M(ClO4)2 (M = Mg (red), Ca (blue), Sr (green), and Ba (magenta)) at different concentrations allowed by salt solubilities. On the higher frequency side of the band peaked at 933 cm−1 typical of free ClO4− anion, there is a small band at approximately 940 cm−1. On the lower frequency side, a band at approximately 926 cm−1 becomes evident in acetone/Sr(ClO4)2, remarkable in acetone/Ba(ClO4)2, weak in acetone/Ca(ClO4)2, and negligible or absent in the acetone/Mg(ClO4)2 solution. | ||
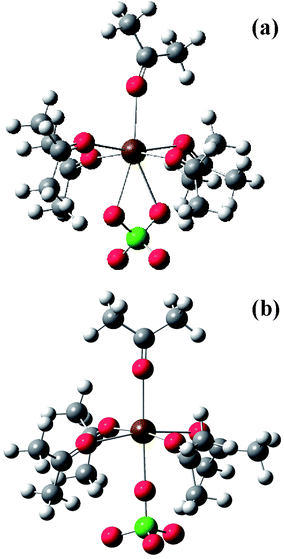 | ||
| Fig. 6 Optimized structures of the bidentate (a) and of the monodentate (b) structure of the (acetone)5 (ClO4−) Sr2+ cluster calculated at the HF/3-21G* level. | ||
The results of the quantum chemical calculations for the NCE of the ν(CO) band of different (acetone)nM2+ clusters, where n is the number of solvent molecules clustering around the metal cation M2+ (Mg2+, Ca2+, Sr2+, and Ba2+), are reported in Table 2. They indicate that (i) for all the studied cluster species, (acetone)nMg2+ with n = 3, 4, and 6, (acetone)nCa2+, (acetone)nSr2+ and (acetone)nBa2+ with n = 6 and 8, the average frequency of the ν(CO) anisotropic Raman profile (with significant contribution from the out-of-phase modes) is much lower than that of the ν(CO) isotropic one (generated by the in-phase mode), i.e. the ν(CO) Raman band has a large and negative NCE; (ii) for any given n, NCE gets less negative as the M2+ ionic radius (i.e. the M2+⋯OC distance) gets larger (see Table 3), being −34.53 cm−1 in (acetone)6Mg2+ with r(Mg2+⋯OC) = 2.64 Å, −28.20 cm−1 in (acetone)6Ca2+ with r(Ca2+⋯OC) = 3.01 Å, −24.64 cm−1 in (acetone)6Sr2+ with r(Sr2+⋯OC) = 3.17 Å, and −20.91 cm−1 in (acetone)6Ba2+ with r(Ba2+⋯OC) = 3.37 Å; (iii) as already observed in (acetone)nM+ clusters,16 for (acetone)nMg2+ the magnitude of the negative NCE increases with the number n of acetone molecules involved in the cluster, being −7.42, −29.17 and −35.08 cm−1 (at the HF/6-31+G(2df,p) level) for n = 3, 4, and 6; the same solvation number dependence of the negative NCE does not seem to be followed by the (acetone)nM2+ clusters with n = 6, 8 of the larger size cations.
| Cation (M2+) | n | NCE/cm−1 | |
|---|---|---|---|
| Calculated | Observed | ||
| a Calculated at the HF/6-31+G(2df,p) level, scaled by 0.7337 [=0.9 × (16.0/19.6)]. See text. b Calculated at the HF/LanL2DZ level, scaled by 0.8402 [=0.9 × (16.0/17.1)]. See text. c Evaluated for the lower concentration. d Evaluated for the higher concentration. | |||
| Mg2+ | 3 | −7.42a | |
| 4 | −29.17a | ||
| 6 | −35.08a/−34.53b | −34.8(1) | |
| Ca2+ | 6 | −29.93a/−28.20b | |
| 8 | −28.22a/−26.85b | −23.0(4)c/−22.8(3)d | |
| Sr2+ | 6 | −24.64b | |
| 8 | −22.64b | −22.1(3)c/−21.3(1)d | |
| Ba2+ | 6 | −20.91b | |
| 8 | −20.51b | −21(1)c/−21.1(9)d | |
| Clusters |
r(Mm+⋯OC)b/Å |
r(OC⋯OC)c/Å |
|---|---|---|
|
a Calculated at the HF/LanL2DZ level unless otherwise stated.
b Average separation between the central ion M+ or M2+ and the center of the OC bond.
c Average separation between the centers of the OC bonds.
d Taken from ref. 16. Calculated at the HF/6-31+G(2df,p) level.
|
||
| (acetone)4/6Mg2+ | 2.54/2.64 | 4.16/4.04 |
| (acetone)6/8Ca2+ | 3.01/3.12 | 4.61/4.63 |
| (acetone)6/8Sr2+ | 3.17/3.26 | 4.85/4.82 |
| (acetone)6/8Ba2+ | 3.37/3.45 | 5.16/5.11 |
| (acetone)4Li+ | 2.48d | 4.05d |
| (acetone)4/6Na+ | 2.87/2.95d | 4.69/4.52d |
The substantial agreement emerging between the quantum chemical and the experimental results of NCE would suggest the possibility to infer the cluster solvation number occurring in each solution. The best alignment of the observed NCE for acetone/Mg(ClO4)2 solution with the calculated NCE for the different clusters definitely indicates the highest stability of the n = 6 cluster in this solution. This conclusion merits a comment in the light of the results of an experimental study of the bond enthalpy and entropy differences in the gas-phase for (acetone)nMg2+ for n = 5, 6 and n = 6, 7.36 The increase of these thermodynamic quantities from n = 5 to n = 6 and their sharp drop observed from n = 6 to n = 7 suggest, in fact, a stronger stability of the n = 6 solvated species and the transition of the seventh acetone ligand to an outer solvation shell. The n = 6 seems to be the preferred solvation number for Mg2+; this solvation number, in fact, was deduced from the analysis of the CN stretching band intensity in the Raman spectrum of acetonitrile/Mg2+ solutions,37 from the analysis of the band intensity of the in-plane δ(O–C–N) bending vibration in N,N-dimethylformamide/Mg2+ and of the ν(N–CH3) stretching vibration in N,N-dimethylacetamide/Mg2+ solutions.32
The comparison between the experimental results of NCE for acetone/M(ClO4)2 solutions (M = Ca, Sr, Ba) and those calculated for (acetone)nM2+ clusters with n = 6, 8, does not allow to discriminate between hexa- and octa-coordinated ions. The best alignment of the experimental with the calculated NCE values (see Table 2) seems to be for the cluster with n = 8. In the case of M = Ca, however, the calculated NCE value is too high, but the precise reason for this result is not clear at present.
We have considered the possibility that NCE in these clusters could be primarily determined by the ion polarizing power defined as Zeff/r, as previously suggested.11 Here r is the ion radius and Zeff is the effective ion charge, i.e. the nuclear charge shielded by the electron shell, calculated as Z/S, where the shielding factor S is evaluated by an empirical relation S = 5Z1.27/(r1/2I) according to ref. 38 with the ion charge Z and the ionization potential I. In Fig. 7 are reported the experimental NCE values and those for the tetra-solvated (acetone)4M+ (M = Li, Na) and the hexa-solvated (acetone)6M+ (M = Na) clusters calculated at the HF/6-31+G(2df,p) level (see Table 2 in ref. 16), and for the hexa-solvated (acetone)6M2+ (M = Mg, Ca, Sr, Ba) and for the octa-solvated (acetone)8M2+ (M = Ca, Sr, Ba) (see Table 2) calculated at the HF/LanL2DZ level. In the evaluation of the polarizing power, the ion radius has been taken as the Mm+⋯OC (m = 1,2) distance calculated at each theoretical level (see Table 4 in ref. 16 and Table 3 of this study), and the ionization potential has been extracted from ref. 39. A linear relation seems to emerge between the NCE values for all these clusters and the polarizing power of the central cations, a conclusion which extends the evidence reached in ref. 11 for Mg2+ to the remaining cations. This result is a further confirmation that the NCE observed in these acetone/Mm+ (m = 1,2) solutions can be considered a phenomenological manifestation of the splitting (degeneracy removal) of the vibrational energy of the n CO oscillators, which arises from the clustering of the oscillators around the central ion because of the ion field.
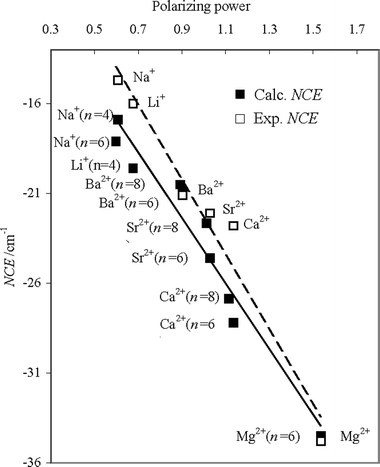 | ||
| Fig. 7 Dependence of NCE (experimental, open squares; calculated, closed squares) on the polarizing power. Calculated NCE values are obtained at the HF/6-31+G(2df,p) level for the (acetone)4M+ with M = Li, Na and the (acetone)6M+ cluster with M = Na, and at the HF/LanL2DZ level for the (acetone)6M2+ clusters with M = Mg, Ca, Sr, and Ba and for the (acetone)8M2+ clusters with M = Ca, Sr, and Ba. The polarizing power is defined as Zeff/r, where r is the ion radius and Zeff is the effective ion charge.11 The latter quantity is expressed as Z/S, where the shielding factor S is evaluated by an empirical relation S = 5Z1.27/(r1/2I) with the ion charge Z and the ionization potential I.39 The ion radius r was evaluated as the M2+⋯OC distance calculated at each theoretical level. | ||
4.3 Symmetry considerations
The arrangement of the six carbonyl groups in the optimized geometry obtained by quantum chemical calculation of the (acetone)6M2+ clusters (Fig. 1a) is very close to octahedral. This finding is confirmed by the analysis of the spectroscopic results (vibrational frequencies, Raman and IR intensities, and Raman depolarization ratios) obtained from the quantum chemical calculations for these clusters. For an octahedral arrangement of the six CO groups (Oh molecular point group), we expect, on the basis of symmetry considerations, that one of the six ν(CO) stretching modes belongs to the totally symmetric A1g species (in-phase mode, only active in the Raman spectrum and with Raman depolarization ratio ρ ∼ 0), and the remaining five stretching modes are distributed between the doubly degenerate non-totally symmetric Eg species (out-of-phase modes, only active in the Raman spectrum and with Raman depolarization ratio ρ = 0.75) and the triply degenerate non-totally symmetric T1u species (only active in the IR spectrum).
The analysis of the results of the quantum chemical calculations (at the HF/LanL2DZ level) for (acetone)6Mg2+ indicates that three of the six ν(CO) stretching normal modes are Raman active: that at 1680.6 cm−1 has the highest Raman intensity and a very low Raman depolarization ratio ρ = 0.025, thus contributing to the isotropic Raman component, and a couple of strictly coincident frequencies at 1639.8 cm−1, well separated from the first, with depolarization ratio ρ = 0.75, thus contributing to the anisotropic Raman component. In between there is a group of three stretching normal modes with closely coincident vibrational frequencies with separations less than 0.5 cm−1, at 1649.5, 1649.7 and 1649.7 cm−1, with zero Raman intensity (but strongly IR active). These findings confirm that the six carbonyl groups closely fulfil the octahedral coordination around the Mg2+ ion in (acetone)6Mg2+, the two lowest frequencies being correlated with the Eg, the highest with the A1g, and the three in between with the T1u symmetry species of the Oh molecular symmetry point group.
The vibrational analysis of the results of the quantum chemical calculations for the remaining clusters, (acetone)6M2+ (M = Ca, Sr, and Ba), replicates this overall symmetry picture, the only notable difference being the frequency separation between the three different vibrational species that gets progressively smaller with the increase of the ion radius. It is mainly due to the relevant systematic frequency decrease (up to 15 cm−1) of the totally-symmetric A1g vibrational mode and to the more modest decrease of the T1u vibrational modes (up to 4 cm−1), while the frequency of the lowest Eg vibrational modes remaining approximately unaltered. Considering that a vibrational frequency splitting must always be traced back to the degeneracy removal induced by a vibrational coupling, we can argue that the calculated progressive reduction of the frequency separation between the different CO oscillators is strongly suggestive of a vibrational coupling that progressively decreases with the ion radius.
5. Concluding remarks
The ν(CO) Raman band of acetone in electrolytic solutions of alkaline earth salts, acetone/M(ClO4)2 with M = Mg, Ca, Sr, and Ba, is featured by a large and negative value of the Raman noncoincidence effect, NCE, in contrast to the small and positive NCE in neat acetone. Guided by the idea of solvent cluster formation around each cation, we have interpreted this observation on the basis of the frequencies and intensities of the ν(CO) vibrational modes of the (acetone)nM2+ clusters formed within the solutions obtained from ab initio quantum chemical calculations. With this approach we have been able to explain the most relevant aspects of this phenomenon. The large and negative values of the NCE are essentially the consequence of the large frequency splitting between the in-phase and the n − 1 out-of-phase ν(CO) stretching vibrational modes generated by n CO groups in the (acetone)nM2+ cluster, with the in-phase mode (active only in the isotropic Raman spectrum) being much higher in frequency than the out-of-phase modes (active predominantly in the anisotropic Raman spectrum). This is compatible with the TDC mechanism of vibrational coupling between the CO oscillators.
The effect of ion charge on the negative NCE has been analyzed in terms of ion polarizing power: the increase of the calculated (and observed) negative NCE from the (acetone)4Na+ cluster to the (acetone)6Mg2+ cluster (from the acetone/Na+ to the acetone/Mg2+ electrolytic solution) through the remaining acetone/Mm+ electrolytic solutions (M = Li, with m = 1, M = Ba, Sr, Ca, with m = 2) is paralleled by an analogous increase of the ion polarizing power.
Acknowledgements
M.G.G. heartily thanks Prof. L. Tassi of the Department of Chemistry of the University of Modena and Reggio Emilia (Italy) for his valuable assistance in the preparation of anhydrous Ca(ClO4)2 and Sr(ClO4)2. M.G.G. gratefully appreciates the financial support to this study given by the Italian Ministry of Education and Research (MIUR) within its activity PRIN 2007 for the project 2007NZLYE5, Transfer of energy, charge and molecules in complex systems.References
- B. C. Pressman, Annu. Rev. Biochem., 1976, 45, 501 CrossRef CAS.
- E. Gouaux and R. MacKinnon, Science, 2005, 310, 1461 CrossRef CAS.
- J. McBreen, H. S. Lee, X. Q. Yang and X. Sun, J. Power Sources, 2000, 89, 163 CrossRef CAS; K. Xu, Chem. Rev., 2004, 104, 4303 CrossRef CAS.
- D. Aurbach, Y. Gofer, Z. Lu, A. Schechter, O. Chusid, H. Gizbar, Y. Cohen, V. Ashkenazi, M. Moshkovich, R. Tungerman and E. Levi, J. Power Sources, 2001, 97–98, 28 CrossRef CAS.
- D. Spångberg and K. Hermasson, Chem. Phys., 2004, 300, 165 CrossRef CAS.
- R. Fischer, J. Richardi, P. H. Fries and H. Krienke, J. Chem. Phys., 2002, 117, 8467 CrossRef CAS.
- W. Kunz, P. Calmettes, M.-C. Bellissent-Funell, G. Jannink, T. Cartailler and P. Turq, in Lectures Notes in Physics, Springer-Verlach Publisher, 1993, vol. 415, p. 371 Search PubMed.
- V. I. Chizhik, I. S. Podkoritov, A. P. Kaikkonen and V. I. Mikhailov, J. Magn. Reson., Ser. A, 1996, 123, 1 CrossRef CAS.
- A. K. Mollner, P. A. Brooksby, J. S. Loring, I. Bako, G. Palinkas and R. W. Fawcett, J. Phys. Chem. A, 2004, 108, 3344 CrossRef CAS.
- S. A. Hyodo and K. Okabayashi, Electrochim. Acta, 1989, 34, 1557 CrossRef CAS.
- J. Bukowska, J. Mol. Struct., 1983, 98, 1 CrossRef CAS.
- Z. Deng and D. E. Irish, J. Chem. Soc., Faraday Trans., 1992, 88, 2891 RSC.
- M. K. Wong, W. J. McKinney and A. I. Popov, J. Phys. Chem., 1971, 75, 56 CrossRef.
- J. Wang, X. Xuan, J. Lu, N. Pei and Y. Mo, Z. Phys. Chem., 2001, 215, 437 CrossRef CAS.
- D. W. James and R. E. Mayes, J. Phys. Chem., 1984, 88, 637 CrossRef CAS.
- M. G. Giorgini, H. Torii, M. Musso and G. P. Venditti, J. Phys. Chem. B, 2008, 112, 7506 CrossRef CAS.
- G. Fini and P. Mirone, J. Chem. Soc., Faraday Trans. 2, 1973, 69, 1243 RSC.
- P. Mirone and G. Fini, J. Chem. Phys., 1979, 71, 2241 CrossRef CAS.
- C. H. Wang and J. L. McHale, J. Chem. Phys., 1980, 72, 4039 CrossRef CAS.
- D. E. Logan, Chem. Phys., 1986, 103, 215 CrossRef CAS.
- C. Czeslik and J. Jonas, J. Phys. Chem. A, 1999, 103, 3222 CrossRef CAS.
- H. Torii, M. Musso, M. G. Giorgini and G. Döge, Mol. Phys., 1998, 94, 821 CrossRef CAS.
- M. G. Giorgini, Pure Appl. Chem., 2004, 76, 157 CrossRef CAS.
- H. Torii, Computational methods for analyzing the intermolecular resonant vibrational interactions in liquids and the noncoincidence effect of vibrational spectra”, Novel Approaches to the Structure and Dynamics of Liquids: Experiments Theories and Simulations, ed. J. Samios and V. A. Durov, Kluwer, 2004, pp. 343–360 Search PubMed.
- H. Torii and M. Tasumi, J. Chem. Phys., 1993, 99, 8459 CrossRef CAS.
- T. W. Zerda, H. D. Thomas, M. Bradley and J. Jonas, J. Chem. Phys., 1987, 86, 3219 CrossRef CAS.
- M. Musso, H. Torii, P. Ottavini, A. Asenbaum and M. G. Giorgini, J. Phys. Chem. A, 2002, 106, 10152 CrossRef CAS.
- Z. Kecki and A. Sokolowska, J. Raman Spectrosc., 1994, 25, 723 CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- H. Torii, J. Phys. Chem. A, 1999, 103, 2843 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision D.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- M. Asada, T. Fujimori, K. Fujii, R. Kanzaki, Y. Umebayashi and S.-i. Ishiguro, J. Raman Spectrosc., 2007, 38, 417 CrossRef CAS.
- Y. Umebayashi, Y. Mune, T. Tsukamoto, Y. Zhang and S. Ishiguro, J. Mol. Liq., 2005, 118, 45 CrossRef CAS.
- D. W. James and P. Cutler, Aust. J. Chem., 1986, 39, 149 CAS; D. W. James and R. E. Mayes, Aust. J. Chem., 1982, 35, 1775 CAS.
- M. Chabanel and K. Touaj, J. Chem. Soc., Faraday Trans., 1996, 92, 4207 RSC.
- M. Peschke, A. T. Blades and P. Kebarle, J. Am. Chem. Soc., 2000, 122, 10440 CrossRef CAS.
- J.-N. Cha, B.-S. Cheong and H.-G. Cho, J. Phys. Chem. A, 2001, 105, 1789 CrossRef CAS.
- L. H. Ahrens, Nature, 1954, 174, 644 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC, Boca Raton, FL, 72nd edn, 1991–92 Search PubMed.
Footnote |
| † This article was submitted as part of a web theme issue highlighting papers from the Italian national meeting on Raman spectroscopy and non linear effects. |
| This journal is © the Owner Societies 2010 |