Carotenoids can act as antioxidants by oxidizing the superoxide radical anion†
Annia
Galano
a,
Rubicelia
Vargas‡
ab and
Ana
Martínez
*b
aDepartamento de Química, División de Ciencias Básicas e Ingeniería, Universidad Autónoma Metropolitana-Iztapalapa, San Rafael Atlixco 186, Col. Vicentina, Iztapalapa. AP POSTAL 55-534, México DF 09340, México
bInstituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior S. N., Ciudad Universitaria, CP 04510, México DF. E-mail: martina@iim.unam.mx
First published on 6th November 2009
Abstract
The electron transfer (ET) reaction between carotenoids and the superoxide radical anion is found to be not only a viable process but also a very unique one. The nature of the O2˙− inverts the direction of the transfer, with respect to ET involving other ROS: the O2˙− becomes the electron donor and carotenoids (CAR) the electron acceptor. Therefore the “antioxidant” activity of CAR when reacting with O2˙− lies in their capacity to prevent the formation of oxidant ROS. This peculiar charge transfer is energetically feasible in non-polar environments but not in polar media. In addition the relative reactivity of CAR towards O2˙− is drastically different from their reactivity to other ROS. Asthaxanthin (ASTA) is predicted to be a better O2˙− quencher than LYC and the other CAR. The CAR + O2˙− reactions were found to be diffusion controlled. The agreement with available experimental data supports the density functional theory results from the present work.
Introduction
The superoxide radical anion (O2˙−) is of great importance in biochemical processes.1 In addition to its potential role in the development of several disorders associated with oxidative stress, it is also a major source of other highly reactive oxygen species (ROS).2 Carotenoids (CAR), on the other hand, are naturally occurring organic compounds known for their antioxidant activity. Cardounel et al.3 demonstrated for the first time direct scavenging of superoxide anion by carotenoid derivatives. This pioneer work was followed by two other studies confirming the O2˙− scavenging activity of CAR.4,5There are three viable mechanisms generally accepted for the reactions of CAR with free radicals:6 electron transfer (ET), radical adduct formation, and hydrogen atom transfer. The ET mechanism is mostly thought of as an electron transfer from the CAR to the ROS, which is the case for most of the ROS involved in oxidative stress processes. Additionally the ET has been described to be favored by polar environments.7 However, O2˙− is a very peculiar free radical because in addition to its spin imbalance it is also a negatively charged species. Therefore its behavior when involved in charge transfer processes may significantly differ from those of other, non-charged, ROS. Actually it has been recently demonstrated that the nature of the reacting free radical plays an important role on the relative importance of the free radical scavenging mechanisms.8 As a result and due to the peculiar nature of O2˙−, the mechanism of CAR + O2˙− reactions is expected to be different from those of CAR with other ROS.
In this work the ET process between O2˙− and a large series of CAR has been studied by density functional theory (DFT). The two possible directions for the ET have been taken into account: the conventional one, from the CAR to the O2˙−:
| CAR + O2˙− → CAR˙+ + O22− path I |
| CAR + O2˙− → CAR˙− + O2 path II |
Computational details
All the modeled species have been fully optimized with the B3LYP hybrid HF-density functional10–12 and the 6-311G(d) basis set, frequency calculations were carried out at the same level of theory. Unrestricted calculations were used for open shell systems and local minima were identified by the number of imaginary frequencies (NIMAG = 0). All the electronic calculations were performed with Gaussian 0313 package of programs. The thermal corrections to Gibbs free energies of the B3LYP/6-311G(d) fully optimized stationary points plus the corresponding electronic energy, were used to obtain the adiabatic Gibbs free energy. The stationary points were first modeled in gas phase (vacuum), and solvent effects were included a posteriori by single point calculations using polarisable continuum model, specifically the integral-equation-formalism (IEF-PCM) at B3LYP/6-311+G(d) level of theory, with water and benzene as solvents for mimicking polar and non-polar environments, respectively. The absorption spectra of radical anion CAR have been computed with time dependent density functional theory (TDDFT) using the same level of theory on benzene solutions.The rate constants (k) were calculated using conventional transition state theory (TST)14–16 and 1M standard state as:
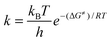 | (1) |
Since the studied reactions are electron transfers, the Marcus theory17 was used. It relies on the transition state formalism, defining the ET activation barrier (ΔG≠ET) in terms of two thermodynamic parameters, the free energy of reaction (ΔG0ET) and the nuclear reorganization energy (Λ):
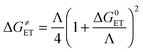 | (2) |
Some of the calculated rate constants (k) values are close to the diffusion-limit. Accordingly, the apparent rate constant (kapp) can not be directly obtained from TST calculations. In the present work we have used the Collins–Kimball theory for that purpose:18
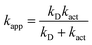 | (3) |
| kD = 4πRDNA | (4) |
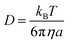 | (5) |
Results and discussion
The carotenoids studied in the present work are shown in Table 1. The Gibbs free energies of reaction (ΔG) has been computed for their reactions with superoxide radical anion through paths I and II, in polar and non-polar environments (Table 2). The first relevant finding that stands out is that path II is much more energetically favored than path I, regardless of the polarity of the environment. This means that while for most ROS the electron transfer takes place from the CAR to the ROS (path I),9,21–24 for the superoxide radical anion it occurs in the opposite direction, form O2˙− to the CAR (path II). In fact path I was found to be endergonic for all studied carotenoids, in both benzene and water solutions.| Code | Structure name |
|---|---|
| BC |
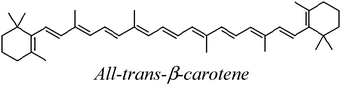
|
| BCRIP |

|
| ZEA |
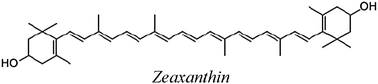
|
| LUT |

|
| 3dhLUT |
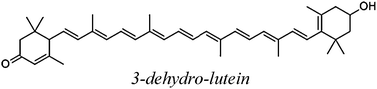
|
| 3hCAR |
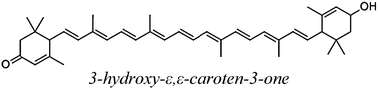
|
| 33CAR |
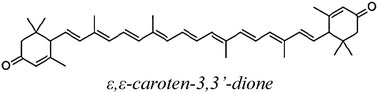
|
| OXO |
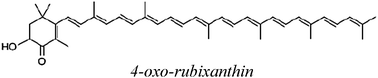
|
| ECH |
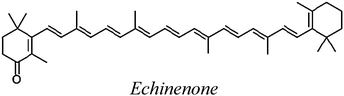
|
| 3hECH |
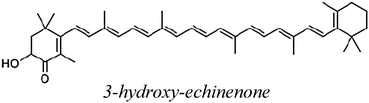
|
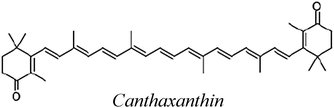
| CAN |
|
| ADO |

|
| ASTA |

|
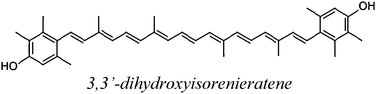
| DHIR |
|
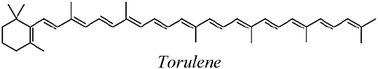
| TOR |
|
| LYC |
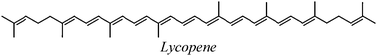
|
| BDOR |
|
| ahLUT |

|
| Benzene | Water | |||
|---|---|---|---|---|
| Path I | Path II | Path I | Path II | |
| BC | 153.7 | −0.2 | 48.0 | 20.3 |
| BCRYP | 154.8 | −1.1 | 48.8 | 19.8 |
| ZEA | 153.7 | −1.6 | 47.8 | 19.3 |
| LUT | 156.7 | −1.9 | 49.7 | 19.6 |
| 3dhLUT | 159.5 | −3.1 | 50.8 | 19.4 |
| 3hCAR | 159.4 | −1.1 | 51.5 | 20.6 |
| 33CAR | 160.5 | −4.3 | 50.9 | 19.0 |
| OXO | 158.0 | −10.2 | 50.4 | 12.5 |
| ECH | 157.7 | −7.9 | 50.8 | 13.5 |
| 3hECH | 157.2 | −8.9 | 49.8 | 13.8 |
| CAN | 161.1 | −11.4 | 53.1 | 11.8 |
| ADO | 161.5 | −13.4 | 52.7 | 10.7 |
| ASTA | 161.1 | −13.3 | 52.0 | 11.4 |
| DHIR | 156.5 | 2.3 | 51.9 | 19.8 |
| TOR | 147.8 | −0.9 | 46.5 | 17.2 |
| LYC | 155.5 | −2.0 | 50.0 | 18.9 |
| BDOR | 158.4 | −10.9 | 49.8 | 12.0 |
| ahLUT | 155.7 | −0.4 | 49.3 | 20.5 |
Path II, on the other hand, becomes exergonic when the ET takes place in benzene, with the exception of the DHIR + O2˙− reaction (Fig. 1, Table 2). This also means that the dependence of the ET feasibility with the polarity of the environment is also inverted with respect to other ROS. Since CAR are hydrophobic molecules, expected to be located mainly in the lipid phase of the membrane, its higher reactivity towards O2˙− in non-polar environments should play an important role in their ability to prevent lipid peroxidation.
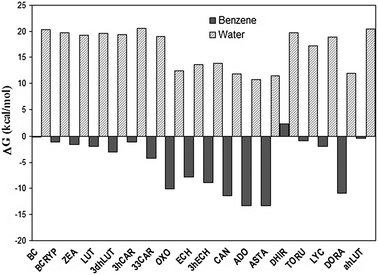 | ||
| Fig. 1 Gibbs free energies of ET reactions, through path II. | ||
All the above findings can be explained by the nature of the O2˙− species. Its negative charge enhances its electron donor capabilities, compared to those of non-charged ROS species, which favors path II over path I, i.e. O2˙− would be more likely to donate an electron than to accept one. When the ET reaction takes place through path I, O2˙− becomes a double-negative-charged species while it becomes a neutral species trough path II. CAR, on the other hand invariably evolves to a radical mono-charged ion. Therefore the stabilization of the CAR product by solvation is expected to be about the same magnitude, regardless of the path involved in ET (Table S1, ESI†). In addition the interaction with the solvent should be less important for CAR than for O2˙− since CAR are larger-sized species with a long conjugated chain that helps them cope with the charge imbalance. The presence of a polar environment, water in this case, increases the viability of path I, by stabilizing the O2˙−, with respect to non-polar environments. According to the data in Table 2 for path I, water lowers ΔG by about 100 kcal mol−1, compared to the equivalent values in benzene solution. This lowering, however, is not enough to overcome the reluctance of O2˙− to accept another electron, and path I remains endergonic for all the studied CAR. The donating character of O2˙− is so much higher than its accepting character that even though both studied paths are endergonic in water, the endergonicity of path II is lower than that of path I.
So far we have explained why the superoxide radical anion is more reactive through path II than it is through path I, and why path I is less endergonic in water solutions. However, the finding that path II is more feasible in benzene solutions than in water solutions, despite the fact that there are charged species involved, still needs to be explained. In order to answer this question we are going to focus on the nature of path II. In this case, the O2˙− donates an electron to CAR. Since O2˙− is the charge donator in all the studied reaction we first analyze its oxidation energy. It is significantly lower in benzene than in water: 2.4 vs. 3.9 eV, which means that O2˙− is a better electron donor in benzene than in water. It is explained by the fact that this mono-charged negative species becomes a neutral one through the electron donation process; therefore while the reactant is stabilized by strong interactions with a polar solvent, the product is not (Table S1†). The reduction energies of the CAR are also relevant to the studied process (path II). They are systematically lower when CAR are in benzene solution than when they are in water solution (Table 3) by about 0.5 eV. According to the above mentioned values the oxidation energy of O2˙− is 1.5 eV lower in benzene. Therefore the nature of O2˙− is the key factor in the higher feasibility of path II.
| Benzene | Water | |||
|---|---|---|---|---|
| Oxidation energy | Reduction energy | Oxidation energy | Reduction energy | |
| BC | 4.9 | 2.3 | 4.5 | 2.9 |
| BCRYP | 4.9 | 2.3 | 4.5 | 2.9 |
| ZEA | 4.9 | 2.3 | 4.5 | 2.8 |
| LUT | 5.0 | 2.3 | 4.5 | 2.8 |
| 3dhLUT | 5.1 | 2.4 | 4.6 | 2.9 |
| 3hCAR | 5.1 | 2.3 | 4.6 | 2.8 |
| 33CAR | 5.2 | 2.5 | 4.6 | 2.9 |
| OXO | 5.1 | 2.8 | 4.6 | 3.2 |
| ECH | 5.1 | 2.6 | 4.6 | 3.1 |
| 3hECH | 5.1 | 2.7 | 4.6 | 3.2 |
| CAN | 5.2 | 2.8 | 4.7 | 3.3 |
| ADO | 5.3 | 2.9 | 4.7 | 3.3 |
| ASTA | 5.3 | 2.9 | 4.7 | 3.3 |
| DHIR | 4.9 | 2.2 | 4.5 | 2.9 |
| TOR | 4.8 | 2.5 | 4.4 | 3.0 |
| LYC | 5.0 | 2.4 | 4.6 | 2.9 |
| BDOR | 5.2 | 2.8 | 4.6 | 3.2 |
| ahLUT | 5.0 | 2.3 | 4.5 | 2.8 |
Logically the reactivity of CAR also changes when the ET reaction takes place through path II, compared to what has been previously reported for other ROS that react through path I. There are previous experimental reports on the order of reactivity of several CAR when they react through ET processes by donating one electron. Mortensen and Skibsted22 have studied the reactions of eight carotenoids with phenoxyl radicals. These authors reported the following order of reactivity: lycopene (LYC) > β-carotene (BC) > zeaxanthin (ZEA) > lutein (LUT) > echinenone (ECH). They also found that canthaxanthin (CAN) hardly reacts, while asthaxanthin (ASTA) does not react at all. Edge et al.23 have studied ET reactions between different pairs of carotenoids with similar outcomes. They28 found that LYC is the most easily oxidized followed by BC > ZEA > LUT > CAN > ASTA. A theoretical work from our group, performed at a similar level of theory than the one used in the present work, shows a perfect agreement with those experimental findings when non-charged free radicals are involved in the ET process.21,24 The reactivity order of CAR when the ET reaction involves O2˙− drastically changes. This particular species transform the reactivity order of the above mentioned CAR into: ASTA > CAN > ECH > LYC ≈ LUT > ZEA > BC. It is noticeable that ASTA and CAN, which are not very efficient as free radical scavengers, when they react with non-charged ROS though path I, become the most efficient O2˙− traps through path II, among this subset of CAR. This is an important finding, since path II is usually not considered when the antiradical capacity of CAR, or any other radical scavenger for that matter, is studied. Therefore their role as free radical scavengers might be underestimated by ignoring their ability to quench superoxide radical anions. The reactivity order found for the whole series of CAR studied in this work, when they react with O2˙− through path II is as follows: ADO ≈ ASTA > CAN > BDOR > OXO > 3hECH > ECH > 33CAR > 3dhLUT > LYC ≈ LUT > ZEA > BCRIP ≈ 3hCAR > TOR > anLUT ≈ BC > DHIR. As this series shows, concerning its reaction with O2˙−, lycopene is no longer the best free radical scavenger.
A direct relationship was found between the reduction energy of the studied carotenoids and their relative reactivity towards O2˙− through path II. It is has been plotted in Fig. 2 as the Gibbs free energy of reaction vs. the reduction energy of the studied CAR. The general, and logical, trend is that the higher the EA of the CAR, the more exergonic the ET (path II). The most reactive CAR were found to be ADO and ASTA, which are also those with the highest electron affinities. Therefore in addition to the general importance of the proposed electron transfer mechanism, our results also support the so far not-fully explained antiradical capacity of ASTA. For example its antioxidant activity has been reported to be 10 times stronger than that of ZEA, LUT, CAN and BC,25 while the conventional ET (path I) does not support that finding.23–25 In addition ASTA is one of the red pigments providing color to male birds, which females tend to prefer.26 It can be hypothesized that this natural selection is related to the health of the animal, i.e. redness is an honest signal of male quality27 that indicates the antioxidant status. Path II supports the antiradical activity of red CAR, particularly ASTA (Fig. 2).
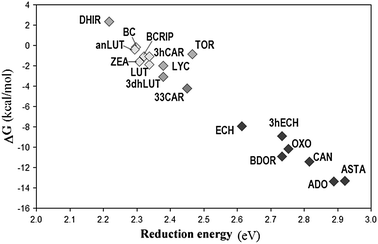 | ||
| Fig. 2 Reduction energy (in benzene solution) of CAR versus Gibbs free energies of ET reactions, through path II, in benzene solutions. | ||
Kinetic calculations have also been performed on the reactions proven as the most viable ones: those taking place through path II, in non polar environments (benzene solutions in this work). The calculated rate constants and the Gibbs free activation energies calculating according to eqn (2), at 298.15 K, are reported in Table 4. As the values in this Table show the lowest barriers among all the studied CAR + O2˙− reactions correspond to ASTA and ADO, in that order. However since the barriers are quite low for most of the studied reactions, they are predicted to be diffusion controlled. The only one of the studied carotenoids that reacts in a significantly slower way is DHIR. It also has the highest barrier, and its reaction with the superoxide radical anion is predicted to be endergonic. Therefore it is not expected to act as a good O2˙− scavenger, at least through electron transfer processes.
| ΔG≠ET | k 298 | |
|---|---|---|
| BC | 5.1 | 9.1 × 109 |
| BCRYP | 4.7 | 1.1 × 1010 |
| ZEA | 4.7 | 1.1 × 1010 |
| LUT | 4.5 | 1.1 × 1010 |
| 3dhLUT | 4.0 | 1.2 × 1010 |
| 3hCAR | 4.7 | 1.1 × 1010 |
| 33CAR | 3.5 | 1.3 × 1010 |
| OXO | 1.8 | 1.3 × 1010 |
| ECH | 2.4 | 1.3 × 1010 |
| 3hECH | 2.1 | 1.3 × 1010 |
| CAN | 1.3 | 1.3 × 1010 |
| ADO | 1.0 | 1.3 × 1010 |
| ASTA | 0.8 | 1.3 × 1010 |
| DHIR | 5.5 | 7.0 × 109 |
| TOR | 3.9 | 1.3 × 1010 |
| LYC | 4.2 | 1.2 × 1010 |
| BDOR | 1.8 | 1.3 × 1010 |
| ahLUT | 5.1 | 9.1 × 109 |
In addition, the absorption spectra of the studied CAR˙− have also been computed. The available experimental data is limited for these species; it has been reported for only 6 of the 18 modeled carotenoids. The details on the computed main vertical optical transitions are provided in Table 5. The calculated absorption maxima of CAR˙− are systematically blue shifted with respect to the experimental data by ∼50 nm, which represents a good agreement, taking into account that these species are anions, and that the size of the systems prevent calculations at higher levels of theory. In addition, and despite of any difference between calculated and experimental values for maxima of adsorption, the tendency is the same, with the order of wavelengths being: LUT < BC ≈ ZEA < LYC < CAN ≈ ASTA. Both agreements support the reliability of the level of theory used in the present work. The ratio λexpmax/λcalcmax is in all the cases around 1.07, which allows us to make an easy correction for the computed spectra of carotenoids, provided that they are calculated at B3LYP/6-31+G(d) level of theory. Since there are not previous reports on the UV-Vis absorption spectra of most of the studied CAR˙−, the corresponding correction has been applied to propose values (λcorrmax) that are expected to be almost identical to the experimental ones.
| Exp.28 | Calculated | λ expmax/λcalcmax | λ corrmax/nm | ||
|---|---|---|---|---|---|
| λ max/nm | f | ||||
| a Oscillator strength. | |||||
| BC | 880 | 828 | 4.09 | 1.06 | 886 |
| BCRYP | 832 | 4.10 | 890 | ||
| ZEA | 880 | 831 | 4.11 | 1.06 | 889 |
| LUT | 870 | 807 | 4.02 | 1.08 | 863 |
| 3dhLUT | 768 | 3.84 | 822 | ||
| 3hCAR | 766 | 3.78 | 820 | ||
| 33CAR | 771 | 3.67 | 825 | ||
| OXO | 973 | 3.72 | 1041 | ||
| ECH | 943 | 3.79 | 1009 | ||
| 3hECH | 952 | 3.65 | 1019 | ||
| CAN | ≥1100 | 1050 | 4.44 | 1.05 | 1124 |
| ADO | 1060 | 4.41 | 1134 | ||
| ASTA | ≥1100 | 1081 | 4.52 | 1.02 | 1157 |
| DHIR | 856 | 4.34 | 916 | ||
| TOR | 862 | 3.51 | 922 | ||
| LYC | 950 | 885 | 4.59 | 1.07 | 947 |
| BDOR | 925 | 3.58 | 990 | ||
| ahLUT | 780 | 3.95 | 835 | ||
For the possible interactions between pairs of carotenoids, their relative order, in term of reduction potential, is shown in Fig. 3 based on the computed Gibbs free energies of ET reactions, through path II, in benzene solutions. This schematic representation, standing only for order in the ease of electron acceptance, is also in good agreement with the experimental results by Edge et al.28 The only difference between their results and ours is that they predicted BC and LUT to have about the same reduction potentials, while our results suggested that the reduction potential of BC is lower than that of LUT.
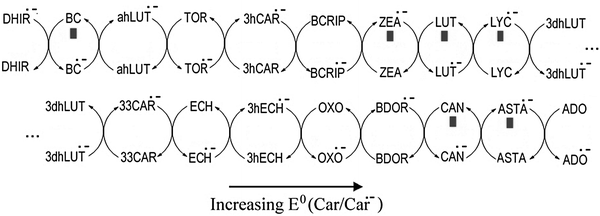 | ||
| Fig. 3 Relative order of carotenoids, in term of one-electron reduction potentials, in benzene solution. ■ ref. 28. | ||
According to the ordered one-electron reduction potentials shown in Fig. 3, ADO and ASTA are the most easily reduced carotenoids in non-polar environments. Therefore they are expected to repair other damaged carotenoids through redox reactions between pairs CAR2/CAR1˙−. Such repairing processes might be relevant to the combined antioxidant activity of carotenoids against the superoxide radical anion.
Conclusions
In summary, the electron transfer reaction between carotenoids and the superoxide radical anion in lipid media is not only a viable process but also a very unique one. The nature of the O2˙− inverts the direction of the transfer, with respect to ET involving other ROS: the O2˙− becomes the electron donor and the CAR the electron acceptor. Therefore the “antioxidant” activity of carotenoids when reacting with O2˙− lies in their capacity to prevent the formation of oxidant ROS. This peculiar charge transfer is energetically feasible in non-polar environments but not in polar media, which is very unique for ET reactions. The relative reactivity of CAR towards O2˙− is drastically different from their reactivity to other ROS, a significant example is that ASTA is better O2˙− quencher than LYC. These results support the so far not-fully explained antiradical capacity of ASTA. Kinetic calculations have been performed, and it was found that the CAR + O2˙− reactions are diffusion controlled. UV-Vis spectra have also been computed and show a good agreement with the available experimental data reported for the CAR˙−. The wavelength for the absorption maxima is reported for the first time for several of the studied CAR. The agreement between computed and experimental data supports the reliability of the results from the present work.Acknowledgements
This study was made possible due to funding from the Consejo Nacional de Ciencia y Tecnología (CONACyT), as well as resources provided by the Instituto de Investigaciones en Materiales IIM, UNAM. The work was carried out, using the KanBalam supercomputer, provided by DGSCA, UNAM and the facilities at Laboratorio de Supercómputo y Visualización en Paralelo of UAM Iztapalapa. The authors would like to acknowledge both Oralia L. Jiménez A and María Teresa Vázquez for their technical support. A. M. is grateful for financial support from DGAPA-UNAM-México, and R. V. for the financial support from CONACYT, through project 49057-F.References
- D. Salvemini, Z. Wang and J. L. Zweier, Science, 1999, 286, 304 CrossRef CAS.
- See for example: (a) M. T. Lin and M. F. Beal, Nature, 2006, 443, 787 CrossRef CAS; (b) S. Cuzzocrea, D. P. Riley, A. P. Caputti and D. Salvemini, Pharmacol. Rev., 2001, 53, 135 CAS.
- A. J. Cardounel, C. Dumitrescu, J. L. Zweier and S. F. Lockwood, Biochem. Biophys. Res. Commun., 2003, 307, 704 CrossRef CAS.
- B. J. Foss, H.-R. Sliwka, V. Partali, A. J. Cardounel, J. L. Zweierb and S. F. Lockwood, Bioorg. Med. Chem. Lett., 2004, 14, 2807 CrossRef CAS.
- H. L. Jackson, A. J. Cardounel, J. L. Zweierb and S. F. Lockwood, Bioorg. Med. Chem. Lett., 2004, 14, 3985 CrossRef CAS.
- G. W. Burton and K. U. Ingold, Science, 1984, 224, 569 CrossRef CAS.
- A. El-Agamey and D. J. McGarvey, J. Am. Chem. Soc., 2003, 125, 3330 CrossRef CAS.
- A. Galano, R. Alvarez-Diduk, M. T. Ramirez-Silva, G. Alarcon-Angeles and A. Rojas-Hernández, Chem. Phys., 2009, 363, 13 CrossRef CAS.
- A. Martinez, M. A. Rodriguez-Girones, A. Barbosa and M. Costas, J. Phys. Chem. A, 2008, 112, 9037 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision E.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- H. Eyring, J. Chem. Phys., 1935, 3, 107 CrossRef CAS.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1935, 31, 875 RSC.
- D. G. Truhlar, W. L. Hase and J. T. Hynes, J. Phys. Chem., 1983, 87, 2664 CrossRef CAS.
- (a) R. A. Marcus, Annu. Rev. Phys. Chem., 1965, 16, 155; (b) R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599 CrossRef CAS; (c) R. A. Marcus, Pure Appl. Chem., 1997, 69, 13 CrossRef CAS.
- F. C. Collins and G. E. Kimball, J. Colloid Sci., 1949, 4, 425 CAS.
- M. Smoluchowski, Z. Phys. Chem., 1917, 92, 129 Search PubMed.
- (a) A. Einstein, Ann. Phys., 1905, 322, 549 CrossRef; (b) G. G. Stokes, Mathematical and Physical Papers, Cambridge University Press, Cambridge, 1903, vol. 3, esp. sect. IV, p. 55 Search PubMed.
- A. Martinez, R. Vargas and A. Galano, J. Phys. Chem. B, 2009, 113, 12113 CrossRef CAS.
- A. Mortensen and L. H. Skibsted, J. Agric. Food Chem., 1997, 45, 2970 CrossRef CAS.
- R. Edge, E. J. Land, D. McGarvey, L. Mulroy and T. G. Truscott, J. Am. Chem. Soc., 1998, 120, 4087 CrossRef CAS.
- A. Galano, J. Phys. Chem. B, 2007, 111, 12898 CrossRef CAS.
- W. Miki, Pure Appl. Chem., 1991, 63, 141 CAS.
- G. E. Hill, Nature, 1991, 350, 337 CrossRef.
- G. E. Hill, in A Red Bird in a Brown Bag, Oxford University Press, New York, 2002 Search PubMed.
- R. Edge, A. El-Agamey, E. J. Land, S. Navaratnam and T. G. Truscott, Arch. Biochem. Biophys., 2007, 458, 104 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Free energies of solvation; optimized structures. See DOI: 10.1039/b917636e |
| ‡ On sabbatical leave at Instituto de Investigaciones en Materiales, UNAM |
| This journal is © the Owner Societies 2010 |