Photoinduced short-range electron transfer in DNA with fluorescent DNA bases: lessons from ethidium and thiazole orange as charge donors†
Christa Prunkl, Sina Berndl, Claudia Wanninger-Weiß, Janez Barbaric and Hans-Achim Wagenknecht*
University of Regensburg, Institute for Organic Chemistry, Universitätsstr. 31, D-93053 Regensburg, Germany. E-mail: achim.wagenknecht@chemie.uni-regensburg.de; Fax: Int+49-941-943-4617; Tel: Int+49-941-943-4802
First published on 30th October 2009
Abstract
Charge transfer processes through the double helix of DNA cover a broad range of mechanistic models ranging from superexchange to hopping mechanisms. Over the last decade, these processes were studied by our group in a photoinduced fashion since (i) the starting time for the charge transfer is clearly defined by the absorption of the photon and (ii) photoexcitation delivers the necessary driving force to the DNA system. It is a prerequisite to modify oligonucleotides synthetically with suitable organic fluorophores that serve as photoinducable charge donors. In the first part of this perspective article we summarize our recent advances in the area of DNA-mediated reductive electron transfer processes over short ranges using synthetic DNA-donor–acceptor systems. The second part of this article focuses on ethidium as the photoinducable charge donor. Ethidium-modified DNA can be used to compare oxidative hole transfer with reductive electron transfer since the type of charge transfer can be controlled by choosing the right charge acceptor. Recent results showed that an efficient charge transfer through DNA using covalently bound ethidium is strongly influenced mainly by DNA dynamics but also by several other parameters that affect the electronic coupling between charge donor and acceptor.
 Christa Prunkl | Christa Prunkl was born in 1980 and studied chemistry at the University of Regensburg including a six-month stay at the University College Dublin, Ireland. In 2006 she finished her diploma thesis about the determination of ATP and Na+-K+-ATPase using luminescent lanthanide complexes under the supervision of Prof. Otto S. Wolfbeis. She is currently finishing her PhD thesis in the group of Prof. H.-A. Wagenknecht. Her work focuses on the synthesis of novel phenanthridinium-based fluorophores for DNA detection and on the investigation of photoinduced reductive electron transfer through DNA. |
 Sina Berndl | Sina Berndl was born in 1981 and studied chemistry with the main focus on organic chemistry and medicinal chemistry at the University of Regensburg. Her diploma work described the base mismatch detection by the DETEQ concept (Detection by Electron Transfer-controlled Emission Quenching) using thiazole orange-modified DNA. She received her diploma degree in 2006 and is now completing her PhD work under the supervision of Prof. H.-A. Wagenknecht. Her current work focuses on the study of thiazole orange as an artificial DNA/RNA base pair for molecular diagnostics and cell imaging. |
 Claudia Wanninger-Weiß | Dr Claudia Wanninger-Weiß was born in 1980. She studied chemistry at the Technical University of Munich and obtained her diploma in 2004. The diploma thesis about catalytic metathesis was supervised by Prof. Wolfgang A. Herrmann. In 2005 she started her PhD work at the University of Regensburg under the supervision of Prof. H.-A. Wagenknecht. For her work on the photoinduced charge transfer reactions in DNA she received her PhD in July 2008. She is currently working in the Faculty of Chemistry at the University of Regensburg. |
 Janez Barbaric | Janez Barbaric was born 1973 in Dornbirn (Austria). He studied chemistry at the Technical University Munich and received his diploma thesis about ethynylpyrene-modified nucleosides and DNA in 2005. Babaric obtained his PhD in 2009 from the University of Regensburg under the direction of Prof. H.-A. Wagenknecht. His thesis described DNA-based multichromophore systems and charge transfer reactions in DNA. Since 2009 he works as a freelancer in health care industry. |
 Hans-Achim Wagenknecht | Hans-Achim Wagenknecht, born in 1968, finished his chemistry studies at the University of Freiburg with a diploma thesis about glycosidase inhibitors (Prof. Jochen Lehmann). After completion of his PhD in the group of Prof. Wolf-D. Woggon (Basel) on synthetic porphyrin models of chloroperoxidase, he carried out postdoctoral research at the California Institute of Technology (Pasadena) in the group of Prof. Jacqueline K. Barton. In 2000, he started to set up an independent research group at the Technical University of Munich in the field of bioorganic and biophysical chemistry with DNA in order to obtain habilitation in 2003. In 2005, he moved as a professor of organic chemistry to the University of Regensburg. His current interest is developing functional DNA architectures for electron transfer, photocatalysis, molecular diagnostics and nanodevices. |
Introduction
Eley and Spivey proposed in the early 1960s that DNA is potentially suitable for charge migration.1 In the early 1990s Barton and co-workers started a highly controversial dispute about the question if DNA might serve as a molecular wire.2,3 Subsequently, they reported on a variety of donor–acceptor systems to study and describe this phenomenon.2–4 Later in the 1990s, the enormous interest in DNA-mediated charge transfer was motivated mainly by the mechanisms how oxidative DNA damages are formed, that may cause apoptosis, mutations, and cancer.5–9 The biological relevance of charge transfer as the precursor for DNA radicals on the road to DNA damages became obvious. Most recently, it has been proposed that charge transfer plays a role in sensing DNA damages to initiate repair.10 Over the last two decades the subject grew to an important research field including the vision of DNA as a charge-transporting biopolymer and material for potential applications in biotechnology and nanotechnology.11–14 Remarkably, DNA was considered to be a molecular wire,15 superconductor,16 conductor,17 semiconductor18 or insulator.19 This initial extreme controversy has been partially solved by the description of different charge transfer mechanisms and their experimental elucidation.20 It became clear that DNA-mediated charge transfer processes can occur on an ultrafast time scale and result in reactions over long distances. It is still a matter of debate, how far the charge can actually be transferred, especially through metallated DNA, and how DNA dynamics influence these processes.In various chemistry research groups, charge transfer in DNA was mainly studied in a photoinduced fashion:11–13 (i) The starting time for the electron transfer is clearly defined by the absorption of the photon. (ii) The photoexcitation pumps a significant amount of energy as a driving force into the system and shifts the endergonic process towards an exergonic situation. Accordingly, it is necessary to modify oligonucleotides with suitable organic fluorophores or inorganic luminophores that serve as photoinducable charge donors. We have focused our research on DNA-mediated reductive electron transfer processes over short range, and studied these processes using organic-synthetic DNA-donor–acceptor systems. This work will be summarized as a short review in the first part of this perspective article. In the second and main part, we focus on ethidium as photoinducable charge donor in DNA. In the corresponding DNA systems, the mode of charge transfer can be controlled by choosing the right charge acceptor. Hence, ethidium-modified DNA allows to compare oxidative hole transfer with reductive electron transfer in structurally very similar DNA constructs. In order to elucidate the important parameters that influence these processes we included some recent results from our laboratory into this part. These studies yielded important information about the chemistry and the dynamics of charge transfer through DNA.
Photoinduced reductive electron transfer in DNA
Oxidative hole transfer vs. reductive electron transfer
Photochemically induced DNA-mediated charge transfer processes were categorized into oxidative-type processes called hole transfer, hole transport or hole migration and reductive types which were named excess electron transfer, electron transport or electron migration (Fig. 1).21,22 It is important to mention that both modes are in fact electron transfer reactions, but with different orbital control. During the oxidative hole transfer an electron is transferred from the DNA or the final acceptor to the photoexcited charge donor in a HOMO-controlled process. In case of the reductive electron transfer the photoexcited electron of the donor is injected into the DNA or transferred to the final electron acceptor in a LUMO-controlled fashion. This means that this chemical categorization is not just formalism about the different directions of the electron.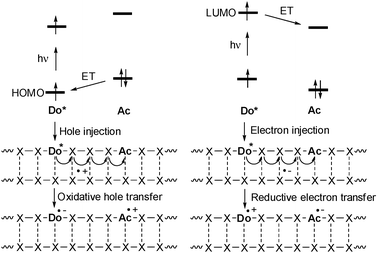 | ||
| Fig. 1 Comparison of photoinduced hole injection and oxidative hole transfer (HOMO-control) vs. electron injection and reductive electron transfer (LUMO-control) in DNA (Do = donor, Ac = acceptor, ET = electron transfer). | ||
Mechanisms of oxidative hole transfer processes and reductive electron transfer processes
With respect to the biological relevance of DNA damages, oxidative hole transfer processes in DNA were subject of intense studies especially in the 1990s.11–13 Excluding all mechanistic details that were elucidated by the different research groups, two major mechanisms were identified for describing DNA-mediated hole transfer reactions: (i) the superexchange mechanism (Fig. 2) and (ii) the hopping model (Fig. 3).20 A very simplified view on a given and static DNA-based donor–bridge–acceptor system shows that the energetic level of the DNA bridge medium relative to the levels of donor and acceptor (δEDo and δEAc) determines the hole transfer mechanism.20 | ||
| Fig. 2 Oxidative hole transfer (top) and reductive electron transfer (bottom) through a DNA bridge via the superexchange mechanism (Do = donor, Ac = acceptor, B = DNA base, HT = hole transfer, ET = electron transfer). | ||
 | ||
| Fig. 3 Photoinduced hole hopping (top) and electron hopping (bottom) in DNA (Do = donor, Ac = acceptor, HOP = hopping). | ||
In case of the superexchange mechanism (Fig. 2), the DNA bridge states lie above the level of the photoexcited donor. Consequently, the hole is transferred in one coherent jump to the acceptor and is never localized on the bridge. As a consequence, the rates are strongly distance-dependent. Since the results were often described according to the Marcus theory for nonadiabatic electron transfer, β was applied to characterize the exponential distance dependence of kHT:23

| kET = N−η |
In contrast to the broad knowledge about the oxidative hole transfer processes, the mechanistic details of reductive electron transfer remained unclear until the early 2000s. Due to the lacking experimental evidence the mechanisms of hole transfer were simply assigned to the problem of reductive electron transfer in DNA. Accordingly, a superexchange (Fig. 2) and a hopping mechanism (Fig. 3) were proposed.21 The pyrimidines cytosine C and T (or dU) are reduced more easily than the purines A and G.25,28 Hence, the pyrimidine radical anions C˙− and T˙− play the role of intermediate charge carriers during electron hopping. One of the major differences to hole transfer is that the electron transfer potentially involves all base pairs since pyrimidines are present in both types of base pairs (T–A and C–G).
Experimental elucidation of photoinduced electron hopping through DNA
Over the last few years we studied reductive electron transfer in DNA by photochemical and photophysical methods. Electron transfer does not occur without light. Hence, it was necessary to modify oligonucleotides with suitable organic chromophores. As mentioned in the previous section it was proposed that the level of the excited state potential of the electron donor primarily decides about the electron transfer mechanism (Fig. 4). Accordingly we used phenothiazine (Pz) and pyrene (Py) to photoreduce selectively the pyrimidine bases C and T in order to initiate an electron hopping process in the DNA. On the other hand, ethidium (Et) and thiazole orange (To) represent chromophores that are not able to reduce any of the DNA bases in order to initiate an electron hopping. Hence, a suitable acceptor has to be provided to study the superexchange mechanism.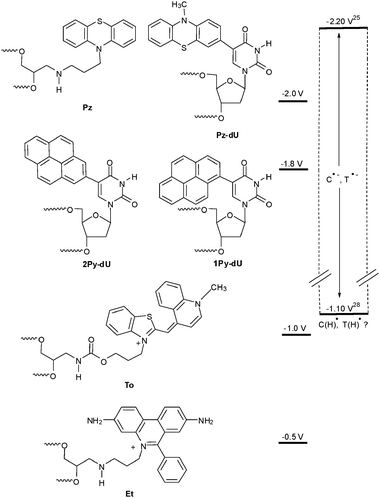 | ||
| Fig. 4 Estimated excited state reduction potentials of fluorophores that we applied for the investigation of reductive electron transfer, in comparison to the range of the reduction potentials of C and T25,28 (Et = ethidium, To = thiazole orange, Pz = phenothiazine, Py = pyrene). | ||
In order to apply Pz as a photoinducable electron donor for DNA we synthesized the Pz-modified 2′-deoxyuridine (Pz-dU) as a base modification29 as well as Pz as a base substitution30 and incorporated them into oligonucleotides. Especially Pz-dU was combined with brominated uridine (Br-dU) as the chemical electron trap yielding a complete DNA-donor–acceptor-system. After photoreduction, Br-dU spontaneously cleaves off bromide that yields a strand break of the corresponding oligonucleotide. Thus, it is possible to measure and compare electron transfer efficiences by chemical HPLC analysis. The electron acceptor was placed either two, three or four base pairs away from the Pz-dU group (Fig. 5). The intervening base pairs were either T–A or C–G to elucidate both the distance and sequence dependence of DNA-mediated electron transfer efficiency. Interestingly, the DNA duplexes with the T–A base pairs exhibit a significant higher cleavage efficiency compared to the DNA duplexes with the C-G base pairs indicating that T–A base pairs transfer electrons more efficiently than C–G base pairs. This implies the idea that the thymine radical anion T˙− represents the better electron carrier. Due to proton transfer processes that interfere with the electron transfer, as discussed below, the cytosine radical anion C˙− is not likely to play a major role as an electron transfer intermediate in a mixed DNA sequence.
 | ||
| Fig. 5 DNA-donor–acceptor system for the investigation of electron transfer in DNA based on Pz-dU as the electron donor and Br-dU as chemical electron trap (PCET: proton coupled electron transfer). | ||
Unfortunately, Pz-dU did not yield any interpretable transient absorption in time-resolved laser spectroscopy experiments. As an alternative we applied the locally excited state of pyrene (Py*) as a precursor state for an electron transfer to the DNA bases. We prepared pyrene-modified 2′-deoxyuridines (1Py-dU, 2Py-dU) and pyrene-modified cytidines (Py-dC) as nucleoside models for electron transfer especially with respect to the occurrence of proton-coupled electron transfer (Fig. 6).31–37 These studies revealed that the cytosine radical anion C˙− is protonated even in basic aqueous solution on a picosecond time scale (or faster). The time-resolved data at pH 5 show that the intramolecular electron transfer and protonation in 1Py-dU occurs within 4.7 ps, and in Py-dC within 40 ps. This combination of results suggests a proton-coupling of the electron transfer if the cytosine radical anion C˙− is involved as an intermediate in DNA, which was mentioned originally by Steenken.38,39
 | ||
| Fig. 6 Pyrene-modified nucleosides (dX = dU or dC; in 1Py-dU, 2Py-dU and Py-dC) as models for electron transfer in DNA (PCET = proton-coupled electron transfer). | ||
These nucleoside model studies provide strong evidence that protonation of the cytosine radical anion C˙− will occur rapidly and separates spin from charge. Such a proton transfer in DNA may limit electron transfer although it may be reversible on the ET time scale.
Subsequently, we used 1Py-dU as the photoinducable electron donor in DNA together with Br–dU to combine time-resolved spectroscospic studies with chemical probing using the radical clock (Fig. 7).40 The electron injection process in these functionalized duplexes occurs within 2 ps and shows only minor variations due to structural inhomogeneity in the covalently connected Py and dU moieties. However, the charge-separated state Py˙+–dU˙− which is formed after excitation exhibits a strong kinetic dispersion in its lifetimes. DNA is a flexible medium with a variety of conformational states exhibiting a wide range of dynamic flexibility. The electron shift from dU˙− to Br-dU through the base stack is much more sensitive to structural parameters and thereby characterized by a distribution of time constants around several hundred ps. Subsequent electron transfer steps will be faster since the Coulomb interaction between the excess electron and Py˙+ decreases drastically with distance. Hence, the measured rates provide a lower limit for the dynamics of reductive electron transfer in DNA.
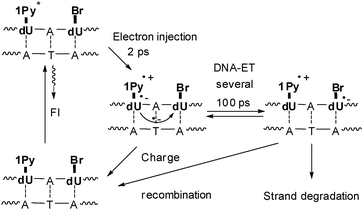 | ||
| Fig. 7 DNA-donor–acceptor system for the investigation of electron transfer in DNA based on 1Py-dU as the electron donor and Br-dU as the electron trap. | ||
Ethidium as a charge donor for comparison of hole and electron transfer in DNA
Ethidium as an artificial DNA base and charge donor
3,8-Diamino-5-ethyl-6-phenylphenanthridinium (“ethidium”, Et) represents a potent positively charged heterocyclic intercalator for DNA. Ethidium exhibits a strong fluorescence enhancement in presence of DNA.41 Beside the broad application as a staining agent for nucleic acids ethidium represents an interesting donor for charge transfer studies in DNA. Based on relative redox potentials, ethidium in the photoexcited state is not able to oxidize or reduce DNA in order to initiate hole or electron hopping, respectively: (i) The excited state oxidation potential of ethidium, E(Et*+/Et˙) ∼ 1.2 V,42 is lower than that of G, E(G˙+/G) ∼ 1.3 V.24 (ii) The excited state reduction potential, E(Et˙2+/Et*+) ∼−0.5 V,42 is less negative than that of T or C, E(T/T˙−) ∼ E(C/C˙−) ∼−1.8 V.28 Hence, 7-deazaguanine (Zg) that is known to quench the ethidium emission in DNA43 has been applied as hole acceptor in charge transfer studies,42,44–55 most extensively by the Barton group.42,52–55 For the latter investigations ethidium was covalently attached to the 5′-end via a linker. However, the site-specific intercalation of ethidium in DNA is crucial for a detailed study of the charge donor properties. Hence, we worked out synthetic protocols for the incorporation of the phenanthridinium heterocycle of ethidium as an artificial base into DNA (Fig. 8)56,57 and applied it for charge transfer studies.58,59 Due to the hydrolytic lability of the corresponding ethidium 2′-deoxyribofuranoside,60 the sugar moiety was replaced by an acyclic glycole linker between the phosphodiester bridges. | ||
| Fig. 8 Ethidium as a 5′-terminal modification, as a nucleoside, and as nucleoside analogs either with D-threolinol or (S)-aminopropanediol as the linker between the phosphodiester bridges. For the subsequently described charge transfer studies the latter DNA modification was used. | ||
Suitable charge acceptors for ethidium-modified DNA
As already mentioned above, suitable charge acceptors have to be provided in order to study charge transfer with ethidium-modified DNA (Fig. 9). For oxidative hole transfer reactions, 7-deazaguanine (Zg) exhibits a lower oxidation potential, E(Zg˙+/Zg) ∼ 1.0 V,42 compared to G24 and was used as a charge acceptor with ethidium in DNA.42,52–55,59,61 The redox potential of indole (In) is comparable, E(In˙+/In) ∼ 1.0 V, due to spontaneous deprotonation of the indole radical cation.62 Indole has been used as a hole trap not only in terms of a base surrogate63,64 but also as part of tryptophan in DNA-binding peptides65–68 or proteins.69 In contrast to 7-deazaguanine, indole as an artificial DNA base exhibits no preferential base pairing and behaves as a universal base analog.70,71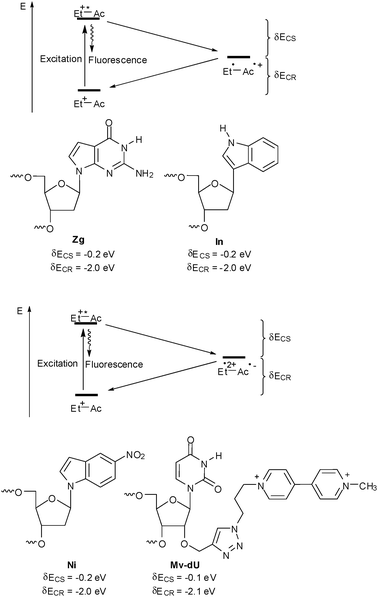 | ||
| Fig. 9 Charge acceptors for oxidative hole transfer studies (top) and reductive electron transfer studies (bottom) with ethidium-modified DNA (Do = donor: Zg = 7-deazaguanine, In = indole; Ac = acceptor: Ni = 5-nitroindole, Mv = methyl viologen, CS = charge separation, CR = charge recombination). | ||
For reductive ET, 5-nitroindole (Ni) exhibits a less negative reduction potential, E(Ni/Ni˙−) ∼−0.3 V,72 compared to C or T,28 and was applied as electron acceptor in DNA.61 Similarily to indole, 5-nitroindole as an artificial DNA base exhibits no preferential base pairing and behaves as a universal base analog.73 Methyl viologen (Mv) represents another interesting electron acceptor as it shows a reduction potential of E(Mv2+/Mv˙+) ∼−0.4 V that is quite similar to 5-nitroindole.74 Up to now it was used as a non-covalently bound electron acceptor.45–47,58
Titration experiments help to elucidate very quickly if a charge transfer between a donor and an acceptor takes place inside DNA. With respect to ethidium as the charge donor, these experiments can be performed in two different ways: Either (i) by titration of ethidium-modified DNA with potential charge acceptors, or (ii) by titration of DNA covalently modified by the charge acceptor with “free” ethidium. Some years ago we could show that the fluorescence of DNA bearing ethidium as an artificial DNA base was quenched significantly with an increasing amount of MV as a result of an electron transfer process which was predicted by the relative redox potentials (as described above).58 A similar experiment with indole failed due to the very weak binding of indole to ethidium-modified DNA.
On the other hand, titration experiments of ethidium were performed with DNA that has been modified with 7-deazaguanine and 7-deazaisoguanine,52 and with indole and methyl viologen by our group (Fig. 10). The latter results will be briefly presented here. For the synthetic incorporation of indole as an artificial DNA base into oligonucleotides,72 the indole moiety was attached via its C-3 position in order to maintain the possibility of a proton-transfer coupled hole trapping via the aromatic N–H group. This protocol72 was applied for the preparation of DNA1a. The titration of ethidium in buffer solution with DNA1a showed a small but detectable difference in comparison with the corresponding experiment using the unmodified DNA1b. It shows that the fluorescence of the intercalated ethidium molecules is partially quenched by the presence of the indole as an artificial base in DNA1a presumably by hole transfer.
 | ||
| Fig. 10 Titration curves for the spectrofluorimetric titrations (λexc = 510 nm) of ethidium (3 μM in 10 mM Na–Pi buffer, pH 7, 20 °C) with In-modified DNA1a (left) and Mv-modified DNA2a (right). The solutions used for the titrations contained 3 μM DNA (double stranded) and 3 μM ethidium in 10 mM Na–Pi buffer, pH 7, 20 °C. Titrations with unmodified duplexes DNA1b and DNA2b serve as references. | ||
For reductive electron transfer, methyl viologen represents a promising charge acceptor as the one electron-reduced radical shows a characteristic transient absorption at ∼600 nm.75 However, methyl viologen cannot be synthetically incorporated into oligonucleotides using the corresponding phosphoramidite as a DNA building block due to its instability during the strong basic conditions that are typically used for DNA workup.76 However, the “click” chemistry allows the postsynthetic modification of oligonucleotides that were presynthesized and modified by an alkyne group.77 Using the methyl viologen-substituted propyl azide,78 and the protocol for the Cu(I)-catalyzed “click” modification77 we were able to prepare the corresponding modified DNA2a (see ESI†). The titration of ethidium in aqueous buffer solution revealed a significant difference in the fluorescence enhancement in comparison with the corresponding experiments using the unmodified DNA2b. The twisted bipyridyl conformation of methyl viologen allows only binding into the minor groove but prevents intercalation into the base stack. A comparison of the melting temperatures between the indole-modified DNA1a and methyl viologen-modified DNA2a (see ESI†) revealed a higher duplex stability (ΔTm = +10 °C) of the latter one. Obviously, this structural difference of DNA2a compared to the indole-modified DNA1a allows a significantly higher part of the intercalated ethidium molecules to get involved in an electron transfer process resulting in a stronger fluorescence quenching.
Relevance of intercalation: comparison with thiazole orange
Thiazole orange (To) represents an interesting alternative to ethidium for charge transfer studies in DNA. For this purpose, it is crucial to covalently link thiazole orange to oligonucleotides. It was tethered to internucleotidic79 or terminal80,81 positions of oligonucleotides, to DNA-binding peptides82,83 and to PNA.84–86Similar to ethidium, the thiazole orange chromophore exhibits an oxidation potential of E(To+/To˙2+) = 1.4 V,87 that together with E00 = 2.4 V is not sufficient for the photoreduction of any of the DNA bases in order to initiate an electron transfer. Recently, we reported about the fluorescence of thiazole orange as a DNA base surrogate modulated by photoinduced short-range electron transfer (Fig. 11).88 Interestingly, an efficient reductive electron transfer from thiazole orange to 5-nitroindole as the acceptor was observed exclusively in the single strands. Obviously, the attachment of thiazole orange via its quinoline nitrogen prevents the intercalation of this dye and thus inhibits the electron transfer in the duplex completely. The absence of differences in the absorption spectra between the thiazole orange-modified single and double strands indicate the lacking intercalation.
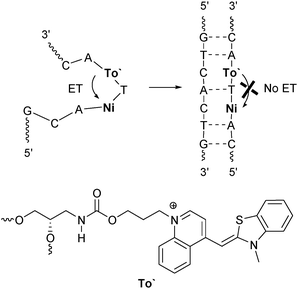 | ||
| Fig. 11 Electron transfer with To′ (incorporated via its quinoline nitrogen) as a photoinducable artificial DNA base and Ni as the acceptor occurs efficiently in the single strand and is inhibited in the duplex (ET = electron transfer). | ||
In order to improve the intercalation and charge transfer properties we introduced a structural and an electronic change. Thiazole orange was (i) attached via its benzothiazole nitrogen to (S)-1-aminopropane-2,3-diol as the linker between the phosphodiester bridges and (ii) combined with 7-deazaguanine (Zg) for hole transfer experiments (Fig. 12).
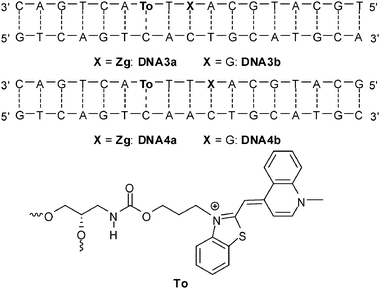 | ||
| Fig. 12 Sequences of DNA3a-DNA4b for hole transfer studies between thiazole orange and 7-deazaguanine (To = thiazole orange attached via its benzothiazole nitrogen, Zg = 7-deazaguanine). | ||
Thiazole orange in its photoexcited state (E(To+/To˙) = −1.0 V in combination with E00 = 2.4 V)87 reveals a borderline case concerning the oxidation of G, E(G˙+/G) ∼ 1.3 V.24 However, experiments with different neighbor bases clearly showed, that To is not able to oxidize DNA in order to initiate a CT process. Therefore, the acceptor 7-deazaguanine was provided in two different distances to the thiazole orange fluorophore (DNA3a and DN4a). The duplexes DNA3b and DNA4b served as references without charge transfer. In analogy to our experiments with ethidium (see next section), C was placed opposite to thiazole orange.54
As expected for the To chromophore, the fluorescence intensity increased upon duplex formation in all four cases (Fig. 13). The photoinduced hole transfer efficiency from thiazole orange to 7-deazaguanine was determined by the amount of fluorescence quenching (Fq) of DNA3a and DNA4a in comparison with the corresponding references DNA3b and DNA4b, respectively. Interestingly, hole transfer was observed both in single strands as well as in duplexes. Moreover, the hole transfer efficiency over the short distance in DNA3a could be improved by adding 250 mM NaCl (see Supporting Information).
 | ||
| Fig. 13 Fluorescence spectra of DNA3a–DNA4b: 2.5 μM in 10 mM NaPi buffer, pH 7, 250 mM NaCl, λexc = 490 nm (ss = single stranded, ds = double stranded). | ||
In contrast to our previous experiments,88 both the absorption and charge transfer properties of thiazole orange attached via the benzothiazole nitrogen showed that intercalation of the dye can partially occur. This interpretation is further supported by the increased quantum yields (20–25%) (see ESI†). Assuming that To cannot photooxidize G, a comparison of the hole transfer of the thiazole orange/7-deazaguanine combination with the ethidium/7-deazaguanine pair is meaningful. The dynamics of the hole transfer in ethidium-modified DNA will be discussed more detailed in the next section. However what becomes obvious just by the steady-state fluorescence is the fact that with ethidium as the charge donor, the hole transfer efficiency is significantly higher (Fig. 14) due to the pronounced intercalation properties of this chromophore. In contrast, the hole transfer efficiencies with thiazole orange in DNA3a and DNA4a are significantly lower since this dye tends to bind in the minor groove. Thiazole orange can only be forced into intercalation by adding NaCl to the buffer resulting in a more effective hole transfer. These results show that intercalation of the redox active probe in DNA and the stacking interactions with the DNA bases represent the most critical issues for an efficient and fast charge transfer.89,90
 | ||
| Fig. 14 Fluorescence quenching of To-modified DNA3a-DNA4 and Et-modified DNA5a-DNA7b in comparison with results from the literature.62 | ||
Influence of DNA dynamics
Ethidium-modified DNA has been employed mainly for investigating oxidative hole transfer due to the favorable reduction potential of ethidium.42,53–55 In these studies, the intercalator ethidium was covalently bound to the 5′-end of the oligonucleotides (Fig. 8). Due to the flexibility of the alkyl chain as a linker, ethidium can intercalate into the base stack without significant restraints of its orientational motion. Using this “Caltech system”, the first time-resolved measurements with ethidium as a charge donor in DNA were published.54 In contrast, our ethidium-modified DNA duplexes contained the chromophor as a base surrogate in the base stack. Hence, it is considerably more rigid and the ethidium lacks the conformational freedom that is characteristic for the “Caltech system”. Using ethidium as a base substitution the chromophore can intercalate without any unwinding of the helix whereas the base stack has to be locally unwind to accommodate the intercalator in the “Caltech system”. 5-Nitroindole and 7-deazaguanine were applied as charge acceptors to directly compare reductive electron transfer and oxidative hole transfer in a structurally very similar DNA-based donor–acceptor system (Fig. 15).61 | ||
| Fig. 15 DNA-donor–acceptor system for the comparison of reductive electron transfer (ET) and oxidative hole transfer (HT) based on Et as the charge donor. | ||
We have combined femtosecond pump–probe and nanosecond fluorescence lifetime measurements to cover different time scales. In the DNA duplexes, where the charge acceptor is separated from ethidium by a single base pair, charge transfer takes place as indicated by a rapid transient decay component of 50 ps (22%) for reductive electron transfer, or 150 ps (27%) for oxidative hole transfer, respectively. For the DNA duplexes bearing the charge acceptor separated by more than one base pair, no ultrafast dynamics was observed. In those cases, fluorescence lifetime measurements confirmed that slower charge transfer (on the nanosecond to microsecond time scale) takes place. That means that the influence of distances larger than one base pair on the charge transfer rates is astoundingly dramatic (4–5 orders of magnitude). It became obvious that ethidium, when rigidly inserted as a base pair surrogate, does not facilitate long-range ultrafast charge transfer. Remarkably, this result is valid for both types, reductive electron transfer and oxidative hole transfer, and stands in contrast to the results with the “Caltech system” where a nearly distance independent and ultrafast hole transfer was observed over 2–4 intervening base pairs. This can be attributed to their inherently different dynamical properties. In the base surrogate system, nuclear motions are largely inhibited due to the short linker and the tight insertion mode. Thus “conformational sampling” of the accessible configurational space is disabled. In contrast, in the loosely tethered intercalator system nuclear motions and conformational sampling is favorable and warrants high charge transfer rates, even across a distance of several base pairs. These results underline the importance of conformational gating, for facilitating efficient charge transfer in DNA over long distances. The fact that both electron and hole transfer are characterized by similar rates and distance dependencies, suggests that conformational sampling may be a generic prerequisite for any electronic transfer process through π-stacked nucleic acids.
In the time-resolved studies that have been described in the previous paragraph, an abasic site analog was applied as a spacer in the counterstrand opposite to the ethidium base surrogat. The lacking counterbase should allow the ethidium to be inserted into the stack without structural distortions. However, for bioanalytical applications the ethidium-modified oligonucleotide strands are considered as probe strands that hybridize with the counterstrands as the analyte. In those counterstrands an abasic site is typically not present. Hence, we tested the electron transfer between ethidium and 5-nitroindole with the additional duplexes DNA5a-DNA7b in order to see how the electron transfer is effected by C as an exemplary “counterbase” to ethidium. The electron transfer was studied by the quenching of the steady-state fluorescence (Fig. 14) and by nanosecond fluorescence lifetime measurements (see Supporting Information). Compared to the previous results with the abasic site opposite to the ethidium chromophore no significant difference has been observed. Within the experimental error, the fluorescence quenching exhibits the same distance dependence.
We have elucidated the structural influence of the rigid insertion of ethidium as a base surrogate using the glycole linker prohibiting efficient charge transfer processes over more than two intervening base pairs. The question is what happens if the charge acceptor has been incorporated in a structurally similar way. It could facilitate the charge transfer due to increased conformational flexibility or block it due to the lost electron coupling. Accordingly, 7-deazaguanine was replaced by indole as nucleoside analog in the next set of duplexes. Following our published procedures63,70 we synthesized DNA8a-DNA10a (Fig. 16). In the duplexes DNA8a-DNA10a the distance between ethidium as the charge donor and indole as the charge acceptor has been varied from one intervening base pair to three base pairs. DNA5b-DNA7b serve again as reference duplexes lacking any charge acceptor. It is important to point out that we incorporated both chromophores, ethidium and indole, as artificial DNA base surrogates into the oligonucleotides using (S)-3-amino-1,2-propanediol as an acyclic linker that substitutes the 2′-deoxyribofuranoside between the phosphodiesters. As a first result, a significant duplex destabilization has been revealed in nearly all cases by measurement of the thermal DNA dehybridization. The melting temperatures (see ESI†) of the duplexes DNA8a-DNA10a carrying two non-nucleosidic modifications are significantly lower (ΔTm = 13–16 °C) compared to the duplexes DNA5b-DNA7b with only one modification. These Tm values form a contrast to our published ethidium/7-deazaguanin-modified duplexes that exhibit nearly the same melting temperatures as the corresponding ethidium-modified ones.62
 | ||
| Fig. 16 Sequences of DNA5a-DNA7b for electron transfer studies between Et and Ni, and of DNA8a-DNA10a for hole transfer experiments between Et and In′ (attached via an acyclic linker). | ||
The steady-state fluorescence spectra of DNA8a-DNA10a in comparison with the references DNA5b-DNA7b (Fig. 17) revealed only a very inefficient charge transfer in DNA8a over a distance of one intervening base pair. In the other two duplexes, DNA9a and DNA10a, the fluorescence intensity was significantly increased in comparison to the reference duplexes DNA6d and DNA7b, respectively. At the moment, we cannot explain this observation. But what becomes clear is that the additional structural perturbation of the DNA modified with ethidium and indole as two nucleoside analogs is so significant that the electronic coupling between ethidium and indole gets lost and is not sufficiently strong for a fast and efficient ET.
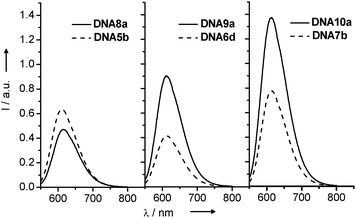 | ||
| Fig. 17 Fluorescence spectra of DNA8a-DNA10a and DNA5b-DNA7b (λexc = 530 nm, 5 μM in 10 mM Na–Pi buffer, 250 mM NaCl, pH 7, 20 °C). | ||
In conclusion, the results presented in this paragraph demonstrate clearly that charge transfer through the base stack of DNA cannot be reduced to a static picture of a “donor–bridge–acceptor” system with bridge = DNA. DNA must be understood as a dynamic medium with conformational motions and flexibility which occur on many time scales including those of electron transfer processes.
On the way to applications in bioanalytics
Based on the importance of the DNA dynamics in gating photoinduced charge transfer processes it can be assumed that charge transfer reacts very sensitive to structural alterations that are typically caused by single base mismatches. Hence, a base modification pair consisting of a fluorescent DNA base substitution and a charge acceptor as the fluorescence quencher 1–3 base pairs away should represent a promising approach for the fluorescent detection of genetic single base variations. In fact, such “detection by electron transfer-controlled emission quenching” (DETEQ) has been worked out by our group using ethidium or pyrene-modified guanine as DNA base substitutions and 7-deazaguanine and indole as charge acceptors (Fig. 18).59,63,91 From these experiments it became clear that combining the fluorescence properties of a suitable fluorophore as an artificial DNA base together with DNA-mediated charge transfer processes represents a new method for the homogeneous detection of DNA base mismatches and abasic sites. The advantage of this method is that changes of fluorescence are not limited to directly adjacent bases and therefore allow scanning a sequence of two or maybe three base pairs (representing a whole codon) in the future. Such an experimental setup could be used for the development of DNA assays for homogeneous detection or heterogenous detection of SNPs by single base mismatches. One could envision to immobilize probe strands consisting of fluorescence donor and quencher as two covalent labels around a questionable single codon on DNA microarrays and hybridize them with genetic material in order to read out the existence of SNPs by fluorescence. | ||
| Fig. 18 DNA donor–acceptor-systems for the analysis of single base mismatches using charge transfer according to the DETEQ concept. | ||
In addition to the DETEQ systems, we tested the applicability of charge transfer in a strand displacement experiment. We synthesized the duplexes DNA11a and DNA12a carrying the ethidium as the photoinducable donor and indole as the hole acceptor in two different arrangements, either diagonal or directly opposite to each in the two different strands (Fig. 19). The duplexes were only partially complementary. That means that duplexes are only formed in the neighborhood of the ethidium chromophore.
 | ||
| Fig. 19 Sequences DNA11a and DNA12a with ethidium (Et) and indole (In) for strand displacement experiments yielding DNA11b and DNA12b. | ||
The strand displacement by oligonucleotides that are fully complementary to the ethidium-modified strands of DNA11a or DNA12a, respectively, yields the full duplexes DNA11b and DNA12b and removes the indole nucleoside as the charge acceptor from the ethidium. As expected, a fluorescence increase is observed in the corresponding titration experiments (Fig. 20). Obviously, the diagonal arrangement of ethidium and indole as the donor–acceptor-couple in DNA11a is better for an efficient charge transfer and therefore the fluorescence enhancement due to the strand displacement is bigger. This system could be potentially applied in the bioanalytical detection of short oligonucleotide sequences within a genetic context.
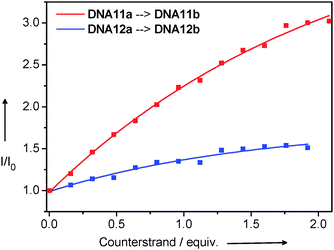 | ||
| Fig. 20 Strand displacement experiments followed by fluorescence spectroscopy (λexc = 530 nm). The semi-complementary DNA11a and DNA12a, respectively (2.5 μM dsDNA in 10 mM Na–Pi buffer, pH 7, 20 °C), are titrated with ssDNA (20 μM ssDNA and 2.5 μM dsDNA in 10 mM Na–Pi buffer, pH 7, 20 °C) that is perfectly complementary to the Et-modified strand in DNA11a or DNA12a. | ||
Conclusions
The central question for the consideration of DNA as a bioorganic and π-conjugated material is how well it transfers charges over nanometer distances. The planar heterocyclic aromatic DNA bases are π-stacked at a distance of 3.4 Å which initially brought up the idea that DNA could conduct electrons like a molecular wire. For the oxidative as well as the reductive mode of charge transfer in DNA it became clear that DNA does not behave like a molecular wire. However, DNA is able to transfer positive or negative charges along the helical axis. This transfer could occur via a superexchange mechanism over shorter distances or an incoherent charge hopping over longer distances (several base pairs). The main parameters that were described representatively for ethidium as a photoinducable charge donor are (i) the intercalation of both charge donor and acceptor and their stacking interactions with the DNA bases in order to couple them electronically, and (ii) the influence of the DNA dynamics up to conformational gating of the charge transfer. Despite this broad knowledge, the DNA research is still far away from a profound and clear understanding of the electronic properties and electronic interactions in DNA. However, charge transfer processes show high potential for applications in the development of new bioanalytical DNA assays and microarrays as well as DNA-inspired devices for nanotechnology.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (GK 640), the Fonds der Chemischen Industrie, and the University of Regensburg is gratefully acknowledged.References
- D. D. Eley and D. I. Spivey, Trans. Faraday Soc., 1962, 58, 411–415 RSC.
- R. E. Holmlin, P. J. Dandliker and J. K. Barton, Angew. Chem., Int. Ed. Engl., 1997, 36, 2714–2730 CrossRef.
- E. M. Boon and J. K. Barton, Curr. Opin. Struct. Biol., 2002, 12, 320–329 CrossRef CAS.
- M. A. O’Neill and J. K. Barton, in Topics in Current Chemistry: Long Range Charge Transfer in DNA I, vol. ed. G. B. Schuster, Springer, Berlin/ Heidelberg, 2004, vol. 236, pp. 67–115 Search PubMed.
- P. O'Neill and E. M. Frieden, in Advances in Radiation Biology: DNA and Chromatin Damage Caused by Radiation, ed. J. T. Lett and W. K. Sinclair, Academic Press, New York, 1993, vol. 17, pp. 53–120 Search PubMed.
- B. Armitage, Chem. Rev., 1998, 98, 1171–1200 CrossRef CAS.
- C. J. Burrows and J. G. Muller, Chem. Rev., 1998, 98, 1109–1151 CrossRef CAS.
- D. Wang, D. A. Kreutzer and J. M. Essigmann, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1998, 400, 99–115 CrossRef CAS.
- S. Kawanashi, Y. Hiraku and S. Oikawa, Mutat. Res., Rev. Mutat. Res., 2001, 488, 65–76 CrossRef.
- E. J. Merino, A. K. Boal and J. K. Barton, Curr. Opin. Chem. Biol., 2008, 12, 229–237 CrossRef CAS.
- Longe-Range Charge Transfer in DNA I, ed. G. Schuster, Springer, Berlin, 2004 Search PubMed.
- Longe-Range Charge Transfer in DNA II, ed. G. Schuster, Springer, Berlin, 2004 Search PubMed.
- Charge Transfer in DNA—From Mechanism to Application, ed. H.-A. Wagenknecht, Wiley-VCH, Weinheim, 2004 Search PubMed.
- Charge Migration in DNA: Perspectives from Physics, ed. T. Chakraborty, Chemistry and Biology, Springer, Berlin, 2007 Search PubMed.
- Y. A. Berlin, A. L. Berlin and M. A. Ratner, Superlattices Microstruct., 2000, 28, 241–252 CrossRef CAS.
- A. Y. Kasumov, M. Kociak, S. Gueron, B. Reulet, V. T. Volkov, D. V. Klinov and H. Bouchiat, Science, 2001, 291, 280–282 CrossRef CAS.
- H.-W. Fink and C. Schönenberger, Nature, 1999, 398, 407–410 CrossRef CAS.
- D. Porath, A. Bezryadin, S. d. Vries and C. Dekker, Nature, 2000, 403, 635–638 CrossRef CAS.
- D. N. Beratan, S. Priyadarshy and S. M. Risser, Chem. Biol., 1997, 4, 3–8 CrossRef CAS.
- J. Jortner, M. Bixon, T. Langenbacher and M. Michel-Beyerle, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12759–12765 CrossRef CAS.
- B. Giese, Annu. Rev. Biochem., 2002, 71, 51–70 CrossRef CAS.
- H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2003, 42, 2454–2460 CrossRef.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–322 CAS.
- S. Steenken and S. V. Jovanovic, J. Am. Chem. Soc., 1997, 119, 617–618 CrossRef CAS.
- C. A. M. Seidel, A. Schulz and M. H. M. Sauer, J. Phys. Chem., 1996, 100, 5541–5553 CrossRef CAS.
- B. Giese, J. Amaudrut, A.-K. Köhler, M. Spormann and S. Wessely, Nature, 2001, 412, 318–320 CrossRef CAS.
- G. B. Schuster, Acc. Chem. Res., 2000, 33, 253–260 CrossRef CAS.
- S. Steenken, J. P. Telo, H. M. Novais and L. P. Candeias, J. Am. Chem. Soc., 1992, 114, 4701–4709 CrossRef CAS.
- C. Wagner and H.-A. Wagenknecht, Chem.–Eur. J., 2005, 11, 1871–1876 CrossRef CAS.
- C. Wagner and H.-A. Wagenknecht, Org. Biomol. Chem., 2008, 6, 48–50 RSC.
- N. Amann and H.-A. Wagenknecht, Synlett, 2002, 687–691 CrossRef CAS.
- E. Mayer, L. Valis, R. Huber, N. Amann and H.-A. Wagenknecht, Synthesis, 2003, 2335–2340 CAS.
- N. Amann, E. Pandurski, T. Fiebig and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2002, 41, 2978–2980 CrossRef CAS.
- R. Huber, T. Fiebig and H.-A. Wagenknecht, Chem. Commun., 2003, 1878–1879 RSC.
- M. Raytchev, E. Mayer, N. Amann, H.-A. Wagenknecht and T. Fiebig, ChemPhysChem, 2004, 5, 706–712 CrossRef CAS.
- A. Trifonov, I. Buchvarov, H.-A. Wagenknecht and T. Fiebig, Chem. Phys. Lett., 2005, 409, 277–280 CrossRef CAS.
- C. Wanninger-Weiß and H.-A. Wagenknecht, Eur. J. Org. Chem., 2008, 64–71 Search PubMed.
- S. Steenken, Free Radical Res. Commun., 1992, 16, 349–379 CrossRef CAS.
- S. Steenken, Biol. Chem., 1997, 378, 1293–12967 CAS.
- P. Kaden, E. Mayer-Enthart, A. Trifonov, T. Fiebig and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2005, 44, 1636–1639 CrossRef CAS.
- J. Olmsted, III and D. R. Kearns, Biochemistry, 1977, 16, 3647–3654 CrossRef.
- S. O. Kelley and J. K. Barton, Chem. Biol., 1998, 5, 413–425 CrossRef.
- L. J. P. Latimer and J. S. Lee, J. Biol. Chem., 1991, 266, 13849–13581 CAS.
- P. Fromherz, Chem. Phys. Lett., 1984, 109, 407–411 CrossRef CAS.
- P. Fromherz and B. Rieger, J. Am. Chem. Soc., 1986, 108, 5361–5362 CrossRef.
- D. A. Dunn, V. H. Lin and I. E. Kochevar, Biochemistry, 1992, 31, 11620–11625 CrossRef CAS.
- S. J. Atherton and P. C. Beaumont, J. Phys. Chem., 1987, 91, 3993–3997 CrossRef CAS.
- S. J. Atherton, J. Phys. Chem., 1995, 99, 12025–12029 CrossRef CAS.
- A. M. Brun and A. Harriman, J. Am. Chem. Soc., 1992, 114, 3656–3660 CrossRef CAS.
- A. I. Kononov, E. B. Moroshkina, N. V. Tkachenko and H. Lemmetyninen, J. Phys. Chem. B, 2001, 105, 535–541 CrossRef CAS.
- P. T. Henderson, E. Boone and G. B. Schuster, Helv. Chim. Acta, 2002, 85, 135–151 CrossRef CAS.
- H. Li, X. Peng and F. Seela, Bioorg. Med. Chem. Lett., 2004, 14, 6031–6034 CrossRef CAS.
- S. O. Kelley, R. E. Holmlin, E. D. A. Stemp and J. K. Barton, J. Am. Chem. Soc., 1997, 119, 9861–9870 CrossRef CAS.
- C. Wan, T. Fiebig, S. O. Kelley, C. Treadway, J. K. Barton and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6014–6019 CrossRef CAS.
- D. B. Hall, S. O. Kelley and J. K. Barton, Biochemistry, 1998, 37, 15933–15940 CrossRef CAS.
- R. Huber, N. Amann and H.-A. Wagenknecht, J. Org. Chem., 2004, 69, 744–751 CrossRef CAS.
- L. Valis and H.-A. Wagenknecht, Synlett, 2007, 2111–2115 CAS.
- N. Amann, R. Huber and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2004, 43, 1845–1847 CrossRef CAS.
- L. Valis, N. Amann and H.-A. Wagenknecht, Org. Biomol. Chem., 2005, 3, 36–38 RSC.
- N. Amann and H.-A. Wagenknecht, Tetrahedron Lett., 2003, 44, 1685–1690 CrossRef CAS.
- L. Valis, Q. Wang, M. Raytchev, I. Buchvarov, H.-A. Wagenknecht and T. Fiebig, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10192–10195 CrossRef CAS.
- W. A. Prütz and E. J. Land, Int. J. Radiat. Biol., 1979, 36, 513–520 CAS.
- C. Wanninger-Weiß, L. Valis and H.-A. Wagenknecht, Bioorg. Med. Chem., 2008, 16, 100–106 CrossRef.
- M. Pascaly, J. Yoo and J. K. Barton, J. Am. Chem. Soc., 2002, 124, 9083–9082 CrossRef CAS.
- H.-A. Wagenknecht, E. D. A. Stemp and J. K. Barton, J. Am. Chem. Soc., 2000, 122, 1–7 CrossRef CAS.
- H.-A. Wagenknecht, E. D. A. Stemp and J. K. Barton, Biochemistry, 2000, 39, 5483–5491 CrossRef CAS.
- E. Mayer-Enthart, P. Kaden and H.-A. Wagenknecht, Biochemistry, 2005, 44, 11749–11757 CrossRef CAS.
- L. Valis, E. Mayer-Enthart and H.-A. Wagenknecht, Bioorg. Med. Chem. Lett., 2006, 16, 3184–3187 CrossRef CAS.
- H.-A. Wagenknecht, S. R. Rajski, M. Pascaly, E. D. A. Stemp and J. K. Barton, J. Am. Chem. Soc., 2001, 123, 4400–4407 CrossRef CAS.
- C. Wanninger and H.-A. Wagenknecht, Synlett, 2006, 2051–1054 CAS.
- J. Barbaric, C. Wanninger-Weiss and H.-A. Wagenknecht, Eur. J. Org. Chem., 2009, 364–370 CrossRef CAS.
- G. Kokkinidis and A. Kelaidopoulou, J. Electroanal. Chem., 1996, 414, 197–208 CrossRef CAS.
- D. Loakes, F. Hill, D. M. Brown and S. A. Salisbury, J. Mol. Biol., 1997, 270, 426–435 CrossRef CAS.
- L. Michaelis and E. S. Hill, J. Am. Chem. Soc., 1933, 55, 1481–1494 CrossRef CAS.
- T. Watanabe and K. Honda, J. Phys. Chem., 1982, 86, 2617–2619 CrossRef CAS.
- S. T. Gaballah, C. E. Kerr, B. E. Eaton and T. L. Netzel, Nucleosides, Nucleotides Nucleic Acids, 2002, 21, 547–560 CrossRef CAS.
- S. Berndl, N. Herzig, P. Kele, D. Lachmann, X. Li, O. Wolfbeis and H.-A. Wagenknecht, Bioconjugate Chem., 2009, 20, 558–564 CrossRef CAS.
- S. Iida, N. Asakura, K. Tabata, I. Okura and T. Kamachi, ChemBioChem, 2006, 7, 1853–1855 CrossRef CAS.
- R. Lartia and U. Asseline, Chem.–Eur. J., 2006, 12, 2270–2281 CrossRef CAS.
- W. R. Algar, M. Massey and U. J. Krull, J. Fluoresc., 2006, 16, 555–567 CrossRef CAS.
- U. Asseline, M. Chassignol, Y. Aubert and V. Roig, Org. Biomol. Chem., 2006, 4, 1949–1957 RSC.
- C. Chen, B. Zhou and G. Xu, J. Photochem. Photobiol., A, 1995, 89, 25–29 CrossRef CAS.
- J. K. P. Mahon, M. D. Roy, J. R. Carreon, E. G. Prestwich, J. L. Rouge, S. Shin and S. O. Kelley, ChemBioChem, 2006, 7, 766–773 CrossRef.
- N. Svanvik, J. Nygren, G. Westman and M. Kubista, J. Am. Chem. Soc., 2001, 123, 803–809 CrossRef CAS.
- O. Köhler, D. V. Jarikote and O. Seitz, ChemBioChem, 2005, 6, 69–77 CrossRef.
- D. V. Jarikote, N. Krebs, S. Tannert, B. Röder and O. Seitz, Chem.–Eur. J., 2007, 13, 300–310 CrossRef.
- K. Hosoi, A. Hirano and T. Tani, J. Appl. Phys., 2001, 90, 6197–6204 CrossRef CAS.
- F. Menacher, M. Rubner, S. Berndl and H.-A. Wagenknecht, J. Org. Chem., 2008, 73, 4263–4266 CrossRef CAS.
- E. M. Boon, N. M. Jackson, M. D. Wightman, S. O. Kelley, M. G. Hill and J. K. Barton, J. Phys. Chem. B, 2003, 107, 11805–1181290 CrossRef CAS.
- T. Fiebig, C. Wan, S. O. Kelley, J. K. Barton and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1187–1192 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of methyl viologen-modified DNA and thiazole orange-modified DNA; experimental details about the spectroscopic measurements, the spectrophotometric/spectrofluorimetric titrations, and the strand displacement experiments, additional Fig. S1–S15 and additional Tables S1–S4. See DOI: 10.1039/b914487k |
| This journal is © the Owner Societies 2010 |