DOI:
10.1039/B917189B
(Paper)
CrystEngComm, 2010,
12, 226-233
Crystallographic analysis of different water–halide cluster blends in cationic [(SNS)PdII] pincer complexes†
Received
19th August 2009
, Accepted 20th August 2009
First published on
4th September 2009
Abstract
X-Ray single-crystal crystallographic analysis of a series of Pd(II) pincer complexes with tridentate ligands comprising a secondary amine sandwiched by two thioether donors of general formula [PdX(RSC2H4N(H)C2H4SR)]X′·nH2O [X = X′ = Cl: R = Et, n = 1 (3); R = tBu, n = 1.5 (4), R = cychex, n = 2 (5); X = X′ = Br, R = iBu, n = 2 (6); X = Cl, X′ = NO3, R = iBu, n = 0 (7)] has been carried out at ambient or low temperature. All the cationic complexes with coordinated and uncoordinated halides (Cl or Br) invariably show hydrate aggregation and interaction with the halides to give water–halide cluster blends in the solid lattices. Extensive H-bonding is evident within these water-blends and between these clusters and the cations, giving different forms of hydrates that are dependent on the sulfur substituent and the halide. Complex 3 contains a water–chloride intercalating zigzag chain; 4 shows a wave-like water trimer–chloride; 5 (at 100 K) reveals a water–chloride tape-like structure formed from four disorder water molecules each with half occupancy; 6 (at 100 K) displays a step-like polymer blend formed by aggregation of water tetramers with orthogonal H-bonding interaction with bromides. No lattice hydration is evident when nitrate is the anion (viz. 7).
Introduction
The important chemical and biochemical role of hydrogen bonding in various life processes is well recognized.1 The ability of water molecules to aggregate into a directional network is one of the key phenomena in supramolecular recognition,1 such as those witnessed in bulk water, protein folding, and DNA pairing.2 It is also not uncommon to find water chains or aggregates in coordination frameworks.3 Many of these aggregates, or clusters, further interact with halides through H-bonding. Recent experimental and theoretical investigations also revealed the existence of H-bonded water molecules and hydronium ions trapped within the crystal structures.4 The immense significance of such secondary interactions is exemplified in anion receptors, ion transport in living systems,5 atmospheric chlorine dispersion,6etc. The relationship between water and the ion flow of halide is best exemplified in cell membranes. Structural analysis has linked the molecular basis of anion selectivity to the size of the chloride channel (∼3.0 Å),7a and revealed an intriguing environmentally-controlled switch between the cyclic hexamer and the helical conformations of a water–chloride cluster.7b This combinative aqua–aqua and aqua–halide H-bonding lead to new discoveries in water and halide channels in molecular lattices. For example, we have recently communicated the formation of chloride-stabilised tetrameric water clusters and their secondary interactions with skeletal amine protons (N–H) in the pincer complex [PdClL1]Cl·2H2O (L1 = bis(2-(ibutylthio)ethyl)amine).8a The use of pincers for water-based applications such as catalysis is another area of current interest.8 In this paper, we shall demonstrate the halide dependence of lattice water aggregation by providing crystallographic evidence of several new forms of water halide cluster blends, viz. water–chloride zigzag chain, water trimer–chloride chain and water tetramer–bromide hydrogen bond interactions.
Experimental
Materials and physical measurements
All solvents and chemicals were used as received without further purification. The synthesis of bis(2-(ibutylthio)ethyl)amine (L1), bis(2-(ethylthio)ethyl)amine (L3), bis(2-(tbutylthio)ethyl)amine (L4) and bis(2-(cyclohexylthio)ethyl)amine (L5) followed literature procedures.9 Elemental analyses (C, H, N) were performed on a Perkin–Elmer PE 2400 CHNS Elemental Analyzer. Infrared spectra were obtained on a Shimadzu IR–470 spectrometer from samples in KBr discs. The NMR spectra were measured at 25 °C using Bruker ACF 300 NMR and 500 NMR spectrometers. Electrospray ionisation mass spectrometric (ESI-MS) analysis was recorded in the positive ion mode using a thermo Finnigan LCQ spectrometer. Thermogravimetric analysis was carried out in a nitrogen stream using TA Instruments SDT 2960 Simultaneous DTA-TGA equipment with a heating rate of 10 °C min−1.
Synthesis of [PdClL3]Cl·H2O (3), [PdClL4]Cl·1.5H2O (4) and [PdClL5]Cl·2H2O (5)
Complexes 3, 4 and 5 were prepared by a common procedure as follows: an aqueous solution of K2[PdCl4] (326 mg, 1 mmol) was mixed with an EtOH solution of L3 (192 mg, 1 mmol), L4 (248 mg, 1 mmol) or L5 (302 mg, 1 mmol). Upon standing for ∼3 weeks, yellow prismatic crystals suitable for X-ray diffraction experiments were collected. Complex 3 could also be obtained from the reaction of PdCl2(MeCN)2 with L3 in MeCN. For 3: Yield: 214 mg (55%). 1H-NMR (500 MHz, CDCl3, 25 °C): δ = 2.81 − 3.54 (m, 12H, CH2), 1.60 (s, 6H, CH3). 13C-NMR (125.77 MHz, CDCl3, 25 °C): δ = 13.9 (s, CH3), 34.5 (s, SCH2CH3), 37.3 (s, CH2CH2S), 54.5 (s, NHCH2CH2). Elemental analysis calcd C8H21Cl2NPdOS2 (388.68): C, 24.72; H, 5.45; N, 3.60%. Found: C, 24.67; H, 5.28; N, 3.45%. ESI-MS (m/z, %): [3 – H2O − Cl−]+ (336, 100). IR (KBr)/cm−1νN − H: 3472. For 4, Yield: 363 mg (80%). 1H-NMR (300 MHz, CDCl3, 25°C): δ = 2.66 − 3.56 (m, 8H, NHCH2CH2S), 1.75 (s, 18H, CH3). 13C-NMR (75.48 MHz, CDCl3, 25 °C): δ = 30.4 (s, CH3), 34.5 (s, C(CH3)3), 53.8, 56.0 (s, NHCH2CH2S). Elemental analysis calcd C24H60Cl4N2Pd2O3S4 (907.58): C, 31.76; H, 6.66; N, 3.09%. Found: C, 31.53; H, 6.57; N, 3.20%. ESI-MS (m/z, %): [4 − 1.5(H2O)−Cl−]+ (392, 100). IR (KBr)/cm−1νN − H: 3438. For 5: Yield: 438 mg (85%). 1H-NMR (300 MHz, CDCl3, 25 °C): δ = 3.49 − 3.57 (b, 2H, CH2CH2SCH), 3.26 − 3.37 (m, 4H) and 2.72 − 2.91 (m, 4H) for NHCH2CH2S, 2.40 (b, 4H), 1.78 − 1.97 (m, 10H), and 1.23 − 1.49 (m, 6H) for SCHC5H10. 13C-NMR (75.48 MHz, CDCl3, 25 °C): δ = 24.8 (s), 26.4 and 26.5 (d), 32.4 and 32.6 (d), 34.2 (s) for SC6H11, 52.5 (s), 54.1 (s) for NHCH2CH2S. Elemental analysis calcd C16H35Cl2NPdOS2 (514.87): C, 37.32; H, 6.85; N, 2.72%. Found: C, 37.35; H, 6.57; N, 2.65%. ESI-MS (m/z, %): [5 − 2(H2O)−Cl−]+ (444, 100). IR (KBr)/cm−1νN − H: 3439.
Synthesis of [PdBrL1]Br·2H2O (6)
An MeCN solution (20 mL) of PdBr2(MeCN)2 (348 mg, 1 mmol) was added to a MeCN solution (5 mL) of ligand L1 and the mixture stirred for ∼1 h. The solution was reduced to ∼5 mL in vacuo and the yellow precipitate collected by suction filtration and washed with MeCN (5 mL). It was recrystallized from the water/EtOH mixture (20 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v:v). The crystals, collected after ∼2 weeks, were washed with water, EtOH and Et2O, then dried under vacuum for 2 h. Yield: 359 mg (65%). 1H-NMR (300 MHz, CDCl3, 25 °C): δ = 2.84 − 3.59 (m, 12H, NHCH2CH2S and SCH2CH), 1.98 − 2.11 (septim, 2H, CH2CH(CH3)2), 1.07 − 1.10 (m, 12H, CH(CH3)2). 13C-NMR (75.48 MHz, CDCl3, 25 °C): δ = 21.0 (s, CH3), 22.0 (s, CH3), 27.9 (s, CH2CH(CH3)2), 40.2 (s, CH2CH2S), 50.0 (s, SCH2CH), 54.5 (s, NHCH2CH2). Elemental analysis calcd for dried sample C12H27Br2NPdS2 (515.71): C, 27.95; H, 5.28; N, 2.72%. Found: C, 27.40; H, 5.37; N, 2.67%. ESI-MS (m/z, %): [6 − 2H2O − Br−]+ (436, 100). IR (KBr)/cm−1νN − H: 3493.
:1, v:v). The crystals, collected after ∼2 weeks, were washed with water, EtOH and Et2O, then dried under vacuum for 2 h. Yield: 359 mg (65%). 1H-NMR (300 MHz, CDCl3, 25 °C): δ = 2.84 − 3.59 (m, 12H, NHCH2CH2S and SCH2CH), 1.98 − 2.11 (septim, 2H, CH2CH(CH3)2), 1.07 − 1.10 (m, 12H, CH(CH3)2). 13C-NMR (75.48 MHz, CDCl3, 25 °C): δ = 21.0 (s, CH3), 22.0 (s, CH3), 27.9 (s, CH2CH(CH3)2), 40.2 (s, CH2CH2S), 50.0 (s, SCH2CH), 54.5 (s, NHCH2CH2). Elemental analysis calcd for dried sample C12H27Br2NPdS2 (515.71): C, 27.95; H, 5.28; N, 2.72%. Found: C, 27.40; H, 5.37; N, 2.67%. ESI-MS (m/z, %): [6 − 2H2O − Br−]+ (436, 100). IR (KBr)/cm−1νN − H: 3493.
Synthesis of [PdClL1](NO3) (7)
Complex 1 (92 mg, 0.2 mmol) was dissolved in water (10 mL) to obtain a yellow solution, upon which AgNO3 (169 mg, 1.0 mmol) was added. The mixture was stirred at rt for 24 h shielded from light, giving a white precipitate. The precipitate was filtered off and the filtrate extracted with CH2Cl2 (3 × 10 mL). The organic layer was combined and solvent removed in vacuo to obtain a yellow powder. This yellow powder was recrystallised by dissolving in CH2Cl2, and layered with Et2O for slow diffusion to obtain yellow crystals of complex 7. Yield: 80 mg (88%). 1H-NMR (500 MHz, CDCl3, 25 °C): δ = 2.65 − 3.39 (m, 12H, NHCH2CH2S and SCH2CH), 1.90 − 1.98 (septim, 2H, CH2CH(CH3)2), 1.00 and 1.02 (d, 12H, CH(CH3)2). 13C-NMR (125.77 MHz, CDCl3, 25 °C): δ = 21.2 (s, CH3), 21.8 (s, CH3), 27.7 (s, CH2CH(CH3)2), 38.0 (s, CH2CH2S), 49.3 (s, SCH2CH), 56.1 (s, NHCH2CH2). Elemental analysis calcd C12H27ClN2PdO3S2 (453.33): C, 31.78; H, 5.96; N, 6.18%. Found: C, 32.21; H, 5.84; N, 6.04%. ESI-MS (m/z, %): [7 – NO3−]+ (392, 100). IR (KBr)/cm−1: 3426 (νN–H) and 1384 (ν3 mode of NO3−).
All measurements were conducted on a Bruker AXS SMART APEX diffractometer, equipped with a CCD area-detector using Mo Kα radiation (λ = 0.71073 Å) (Table 1). The collecting frames of data, indexing reflection and determination of lattice parameters and polarization effects were performed with the SMART suite programs.10 The integration of intensity of reflections and scaling was carried out by SAINT.10 The empirical absorption correction was performed by SADABS.11 The space group determination, structure solution and least-squares refinements on |F|2 were carried out with the SHELXS-97 and SHELXL-97.12 The structures were solved by direct methods to locate the heavy atoms, followed by difference maps for the light non-hydrogen atoms. Anisotropic thermal parameters were refined for the rest of the non-hydrogen atoms. Hydrogen atoms were placed geometrically and refined isotropically. CCDC No.: 729211 (3), 729212 (4), 729213 (5), 729214 (6), and 729215 (7).
| Complex |
3
|
4
|
5
|
6
|
7
|
| Formula |
C8H21Cl2NPdOS2 |
C24H60Cl4N2Pd2O3S4 |
C16H35Cl2NPdO2S2 |
C12H31Br2NPdO2S2 |
C12H27ClN2PdO3S2 |
| MW |
388.68 |
907.58 |
514.87 |
551.72 |
453.33 |
|
T/K |
293(2) |
296(2) |
100(2) |
100(2) |
223(2) |
| Crystal size/mm3 |
0.60 × 0.12 × 0.08 |
0.20 × 0.14 × 0.08 |
0.60 × 0.08 × 0.06 |
0.60 × 0.16 × 0.08 |
0.16 × 0.10 × 0.09 |
| Cryst. syst. |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C 2/c |
C 2/c |
P |
P |
|
a/Å |
5.1868(3) |
31.988(2) |
34.835(3) |
5.4127(4) |
5.7601(4) |
|
b/Å |
11.4095(6) |
8.9087(4) |
5.1827(5) |
13.639(1) |
11.4786(7) |
|
c/Å |
12.8769(7) |
14.1037(7) |
25.391(2) |
15.232(1) |
15.235(1) |
|
α/° |
86.360(1) |
|
|
67.588(1) |
70.574(1) |
|
β/° |
89.309(1) |
97.911(1) |
107.190(2) |
85.211(2) |
85.423(1) |
|
γ/° |
77.171(1) |
|
|
80.964(2) |
77.631(1) |
|
V/Å3 |
741.52(7) |
3980.8(3) |
4379.3(7) |
1026.3(1) |
927.9(1) |
|
Z
|
2 |
8 |
8 |
2 |
2 |
|
D
calc/g cm−3 |
1.741 |
1.514 |
1.562 |
1.785 |
1.623 |
|
µ/mm−1 |
1.871 |
1.408 |
1.292 |
5.002 |
1.378 |
| Reflections collected |
9699 |
13800 |
14635 |
13428 |
8462 |
| Independent reflections [Rint] |
3401 [0.0250] |
4567 [0.0317] |
5022 [0.0505] |
4683 [0.0455] |
4208 [0.0297] |
| Parameters |
150 |
199 |
238 |
201 |
198 |
| GOF |
1.046 |
1.037 |
1.144 |
1.052 |
1.143 |
|
R
1(I > 2σ(I)) |
0.0211 |
0.0305 |
0.0556 |
0.0426 |
0.0513 |
|
wR
2(all data) |
0.0505 |
0.0776 |
0.1252 |
0.1163 |
0.1141 |
Results and discussion
Synthesis of [SNS]Pd pincers
The preparations and structures of [PdClL1]Cl (1) (L1 = bis(2-(ibutylthio)ethyl)amine) and [PdClL2]Cl (2) (L2 = bis-(2-(benzylthioethyl)amine) have been reported.8a A similar method is adopted for the preparation of [SNS]-pincer complexes [PdClL3]Cl (3), [PdClL4]Cl (4) and [PdClL5]Cl (5) from K2[PdCl4] and bis(2-(ethylthio)ethyl)amine (L3), bis(2-(tbutylthio)ethyl)amine (L4) or bis(2-(cyclohexylthio)ethyl)amine (L5) in aqueous ethanol, respectively. Addition of L1 to PdBr2(MeCN)2 gives [PdBrL1]Br (6). Ion exchange of 1 with AgNO3 in water gives [PdClL1](NO3) (7). A general structure of these pincer complexes is shown in Fig. 1.
![General structure of [PdX(SNS)]+ complexes 1–7.](/image/article/2010/CE/b917189b/b917189b-f1.gif) |
| | Fig. 1 General structure of [PdX(SNS)]+ complexes 1–7. | |
X-Ray crystal and molecular structures
All isolated complexes have been analysed by X-ray single-crystal crystallographic diffraction. They show a common tridentate [SNS] pincer motif with halide trans to the secondary amine on a mononuclear Pd(II) sq planar sphere. These cationic complexes, except for the nitrate 7 and the benzyl derivative 2 reported earlier, are invariably enriched by lattice hydration with extensive H-bonding with the halides.(Table 2)
Table 2 Hydrogen bonds parameters (in Å) in complexes 3–7. (A = acceptor; D = donor)
| D − H⋯A |
D–H/Å |
D⋯A/Å |
H⋯A/Å |
∠D–H⋯A/° |
| Complex 3 |
| O1W–H2W⋯Cl2A |
0.80 |
3.228(2) |
2.43 |
177 |
| O1W–H1W⋯Cl2 |
0.81 |
3.196(2) |
2.39 |
174 |
| N1–H⋯Cl2 |
0.79 |
3.157(2) |
2.38 |
171 |
| C2–H⋯Cl2B |
0.97 |
3.723(2) |
2.84 |
152 |
| C3–H⋯Cl1C |
0.97 |
3.745(2) |
2.83 |
158 |
| C7–H⋯Cl1C |
0.97 |
3.761(2) |
2.88 |
152 |
| C6–H⋯O1WD |
0.97 |
3.408(3) |
2.52 |
153 |
| Symmetry codes: A, 1 + x, y, z; B, −x, 1 − y, 2 − z; C, x − 1, y, z; D, 1 − x, −y, 2 − z. |
| Complex 4 |
| O1W–H1W⋯O2W |
0.86 |
2.827(4) |
1.99 |
163 |
| O1W–H2W⋯O2WA |
0.86 |
2.827(4) |
2.02 |
155 |
| O2W–H3W⋯Cl2B |
0.83 |
3.211(4) |
2.45 |
154 |
| O2W–H4W⋯Cl2 |
0.82 |
3.191(3) |
2.39 |
165 |
| N1–H⋯Cl2 |
0.78 |
3.108(2) |
2.34 |
169 |
| C1–H⋯Cl2C |
0.97 |
3.486(3) |
2.74 |
134 |
| C5–H⋯Cl1 |
0.96 |
3.553(4) |
2.68 |
152 |
| C8–H⋯O2WB |
0.97 |
3.367(4) |
2.60 |
137 |
| C11–H⋯Cl1 |
0.96 |
3.649(3) |
2.78 |
150 |
| C12–H⋯Pd1D |
0.96 |
3.773(3) |
2.81 |
177 |
| Symmetry codes: A, 1 − x, y, 1.5 − z; B, 1 − x, 1 − y, 1 − z; C, x, 1 − y, 0.5 + z; D, x, 2 − y, −0.5 + z. |
| Complex 5 |
| N1–H⋯Cl2 |
0.89 |
3.164(4) |
2.31 |
159 |
| C1–H⋯Cl2A |
0.99 |
3.657(5) |
2.81 |
144 |
| C2–H⋯Cl2B |
0.99 |
3.622(5) |
2.70 |
155 |
| Symmetry codes: A, x, 1 + y, z; B, 1 − x, 1 − y, 1 − z. |
| Complex 6 |
| N1–H⋯Br2 |
0.87 |
3.297(3) |
2.43 |
179 |
| O2W–H⋯O1W |
0.85 |
2.792(8) |
2.01 |
152 |
| O1W–H⋯O2WA |
0.87 |
2.807(8) |
2.38 |
111 |
| O2W–H⋯Br2B |
0.86 |
3.256(4) |
2.41 |
173 |
| O1W–H⋯O2WB |
0.85 |
3.014(8) |
2.22 |
156 |
| C5–H⋯Br1C |
0.99 |
3.883(4) |
2.92 |
165 |
| C9–H⋯Br1C |
0.99 |
3.887(4) |
2.91 |
169 |
| Symmetry codes: A, 1 − x, 1 − y, 1 − z; B, 2 − x, 1 − y, 1 − z; C, x − 1, y, z. |
| Complex 7 |
| N1–H⋯O3 |
0.88 |
2.817(6) |
1.97 |
161 |
| C1–H⋯O3A |
0.98 |
3.273(6) |
2.53 |
132 |
| C2–H⋯O2B |
0.98 |
3.284(7) |
2.57 |
130 |
| C3–H⋯O1C |
0.98 |
3.256(6) |
2.30 |
164 |
| C4–H⋯O1 |
0.98 |
3.202(7) |
2.46 |
133 |
| C5–H⋯Cl1C |
0.98 |
3.773(5) |
2.80 |
171 |
| Symmetry codes: A, −x, 1 − y, 1 − z; B, 1 − x, 1 − y, 1 − z; C, x − 1, y, z. |
Each asymmetric unit of 3 (at 293 K, Fig. 2a) contains a lattice water molecule. The cationic fragments are stacked upon each other (Pd⋯Pd 5.187 Å) and supplemented by weak intramolecular inter-ligand interactions between the skeletal methylene proton of the peripheral ethyl with the coordinated chloride [C3–H⋯Cl1C 3.75 and C7–H⋯Cl1C 3.76 Å] (Fig. 2c).
![(a) A perspective view of the cationic structure of 3 ignoring the uncoordinated anion and water solvate (thermal ellipsoids at 30% level). (Pd: purple; S: yellow; N: dark blue; Cl: green; H: sky blue) Selected bond distances [Å] and angles [°]: Pd1–S1 2.3058(5); Pd1–S2 2.2954(5); Pd1–N1 2.031(2); Pd1–Cl1 2.2949(5); N1–Pd1–Cl1 179.27(5); N1–Pd1–S2 87.31(5); Cl1–Pd1–S2 91.98(2); N1–Pd1–S1 87.33(5); Cl1–Pd1–S1 93.37(2); S2–Pd1–S1 174.36(2). Pd1 is 0.0196 Å deviated from the least-squares plane of the donor ligands (S1, S2, N1, Cl1). (b) A zigzag and alternating H-bonding interactions in a water–chloride blend of 3 at 293 K. (Symmetry codes: A, 1 + x, y, z; B, x − 1, y, z) (c) A lattice view along the a direction of the supramolecular network showing the cation, anion and lattice water interconnected through H-bond.](/image/article/2010/CE/b917189b/b917189b-f2.gif) |
| | Fig. 2 (a) A perspective view of the cationic structure of 3 ignoring the uncoordinated anion and water solvate (thermal ellipsoids at 30% level). (Pd: purple; S: yellow; N: dark blue; Cl: green; H: sky blue) Selected bond distances [Å] and angles [°]: Pd1–S1 2.3058(5); Pd1–S2 2.2954(5); Pd1–N1 2.031(2); Pd1–Cl1 2.2949(5); N1–Pd1–Cl1 179.27(5); N1–Pd1–S2 87.31(5); Cl1–Pd1–S2 91.98(2); N1–Pd1–S1 87.33(5); Cl1–Pd1–S1 93.37(2); S2–Pd1–S1 174.36(2). Pd1 is 0.0196 Å deviated from the least-squares plane of the donor ligands (S1, S2, N1, Cl1). (b) A zigzag and alternating H-bonding interactions in a water–chloride blend of 3 at 293 K. (Symmetry codes: A, 1 + x, y, z; B, x − 1, y, z) (c) A lattice view along the a direction of the supramolecular network showing the cation, anion and lattice water interconnected through H-bond. | |
Absent in its butyl derivative 1 (at 223 K), the lattice of 3 shows an intercalating water–chloride supramolecular chain propagated by H-bonds (O1W–H1W⋯Cl2 3.20 and O1W–H2W⋯Cl2A 3.23 Å) in a zigzag manner (Fig. 2b). Similar structural arrangement was observed in [cis-α-CoCl2(trien)]·2H2O (trien = triethylenetetramine),7b [(dbtmen)Cl2]·3H2O (dbtmen = N,N′-dibenzyl-N,N,N′N′-tetramethylethylenediammonium),7c and [HV]Cl·H2O (V = 4′-(naphthalen-2-ylsulfanyl)-2,2′:6′,2″-terpyridine).7d The chains occupy the voids among the cationic stacks but are connected to the latter through H-bonding of chloride with the amine (N1–H⋯Cl2 3.16 Å) and methylene protons (C2–H⋯Cl2B 3.72 Å) of the SNS ligand. It is further supplemented by H-bonding between the hydrate oxygen and methylene protons (C6−H⋯O1WD 3.41 Å).(Fig. 2c)
Each asymmetric unit of 4 (296 K) comprises a cation, uncoordinated chloride and 1.5 lattice water molecules. The lattice chloride–hydrate is present in the form of an unusual polymer blend of intercalating “linear” trihydrate clusters (O1W–H1W⋯O2W 2.83 and O1W–H2W⋯O2WA 2.83 Å) and dichlorides (O2W–H3W⋯Cl2B 3.21 and O2W–H4W⋯Cl2 3.19 Å).(Fig. 3b) Ab-initio/DFT study suggested that small helical water chains may be stable.13 The bc plane shows that the cations and chlorides (Cl2) are interconnected in a rhombic-like network via weak H-bonds (N1–H⋯Cl2 3.11, C1–H⋯Cl2C 3.49 and C12–H⋯Pd1D 3.77 Å).(Fig. 3c) This network, together with the water trimer–chloride polymeric structure give a 1-D supramolecular H2O-Cl blend structure sanwiched among the cations (Fig. 3d and Table 2). Unlike the others, the complex cations do not assemble into stacks but form a 2-D supramolecular (4,4) network via weak intermolecular H-bonds among neighboring networks. The water–trimer–chloride clusters were captured between two neighboring supramolecular networks.
![(a) A perspective view of the cationic structure of 4 ignoring the anion and water solvate (thermal ellipsoids at the 30% level). (Pd: purple; S: yellow; N: dark blue; Cl: green; H: sky blue.) Selected bond distances [Å] and angles [°]: Pd1–S1 2.3135(6); Pd1–S2 2.3199(6); Pd1–N1 2.035(2); Pd1–Cl1 2.2954(7); N1–Pd1–Cl1 176.44(6); N1–Pd1–S1 86.25(6); Cl1–Pd1–S1 94.54(2); N1–Pd1–S2 86.20(6); Cl1–Pd1–S2 92.77(2); S1–Pd1–S2 171.58(2). Pd1 deviates −0.0724 Å from the least-squares plane of the donor ligands (S1, S2, N1, Cl1). (b) The H-bonding interactions in a wave-like water trimer–chloride structure of 4 at 296 K. (Symmetry codes: A, 1 − x, 1 − y, 1 − z; B, 1 − x, y, 1.5 − z). (c) Rhomboid-like H-bonding network among the cations and anions. (d) A perspective view showing the water–chloride cluster-blends sandwiched by the cations.](/image/article/2010/CE/b917189b/b917189b-f3.gif) |
| | Fig. 3 (a) A perspective view of the cationic structure of 4 ignoring the anion and water solvate (thermal ellipsoids at the 30% level). (Pd: purple; S: yellow; N: dark blue; Cl: green; H: sky blue.) Selected bond distances [Å] and angles [°]: Pd1–S1 2.3135(6); Pd1–S2 2.3199(6); Pd1–N1 2.035(2); Pd1–Cl1 2.2954(7); N1–Pd1–Cl1 176.44(6); N1–Pd1–S1 86.25(6); Cl1–Pd1–S1 94.54(2); N1–Pd1–S2 86.20(6); Cl1–Pd1–S2 92.77(2); S1–Pd1–S2 171.58(2). Pd1 deviates −0.0724 Å from the least-squares plane of the donor ligands (S1, S2, N1, Cl1). (b) The H-bonding interactions in a wave-like water trimer–chloride structure of 4 at 296 K. (Symmetry codes: A, 1 − x, 1 − y, 1 − z; B, 1 − x, y, 1.5 − z). (c) Rhomboid-like H-bonding network among the cations and anions. (d) A perspective view showing the water–chloride cluster-blends sandwiched by the cations. | |
The structural analysis of 5 was carried out at 100 K to preserve the crystallinity. The pincer is flanked by two bulky chair-like cyclohexyl substituents which create large voids for water channel formation (Fig. 4). Each asymmetric unit comprises four disordered lattice water molecules with an occupancy of 0.5 and one uncoordinated Cl− ion. H-bonding interactions of the disordered lattice water molecular tape and uncoordinated chlorides are shown in Fig. 4b. Similar to 3, the cations stack upon each other to form a supramolecular network (Pd⋯Pd 5.183 Å). Interactions between chloride ion and the methylene [C1–H⋯Cl2A 3.66 and C2–H⋯Cl2B 3.62 Å] and the amine protons [N1–H⋯Cl2 3.16 Å] are observed. The water–chloride H-bonding interactions (O4S⋯Cl2A 2.94 Å) occur at the microchannels between the cations. (Fig. 4c and Table 2)
![(a) A perspective view of the cationic structure of 5 ignoring the water solvate (thermal ellipsoids at 30% level). (Pd: purple; S: yellow; N: dark blue; Cl: green; O: red; H: sky blue) Selected bond distances [Å] and angles [°]: Pd1–S1 2.317(1); Pd1–S2 2.315(1); Pd1–N1 2.040(4); Pd1–Cl1 2.299(1); N1–Pd1–Cl1 172.5(1); N1–Pd1–S1 86.4(1); Cl1–Pd1–S1 93.82(4); N1–Pd1–S2 86.6(1); Cl1–Pd1–S2 93.26(4); S1–Pd1–S2 172.93(4). Pd1 deviated 0.0645 Å from the least-squares plane of the donor ligands (S1, S2, N1, Cl1). (b) H-bonding network among the disordered water and chloride at 100 K. (c) H-bonded layers network of cation, anion and lattice hydrate.](/image/article/2010/CE/b917189b/b917189b-f4.gif) |
| | Fig. 4 (a) A perspective view of the cationic structure of 5 ignoring the water solvate (thermal ellipsoids at 30% level). (Pd: purple; S: yellow; N: dark blue; Cl: green; O: red; H: sky blue) Selected bond distances [Å] and angles [°]: Pd1–S1 2.317(1); Pd1–S2 2.315(1); Pd1–N1 2.040(4); Pd1–Cl1 2.299(1); N1–Pd1–Cl1 172.5(1); N1–Pd1–S1 86.4(1); Cl1–Pd1–S1 93.82(4); N1–Pd1–S2 86.6(1); Cl1–Pd1–S2 93.26(4); S1–Pd1–S2 172.93(4). Pd1 deviated 0.0645 Å from the least-squares plane of the donor ligands (S1, S2, N1, Cl1). (b) H-bonding network among the disordered water and chloride at 100 K. (c) H-bonded layers network of cation, anion and lattice hydrate. | |
Complex 6 is the bromide derivative of 1. Their cations are isostructural but the water aggregation is different. Its structure, established at 100 K, shows an asymmetric unit comprising an uncoordinated Br− and two lattice water molecules. Particularly notable are the water steps formed from the fusion of neighboring square-like water tetramers (H2O1W⋯H2O2W⋯H2O1WA⋯H2O2WA) with similar edges (O2W–H⋯O1W 2.79 and O1W–H⋯O2WA 2.81 Å). These interactions are comparable to those in (H2O)4 (2.77Å)14a and (D2O)4 clusters (from liquid D2O) as deduced from its FIR-VRT (FIR-VRT = far-infrared vibration–rotation tunneling) spectrum (2.78 Å),14b liquid water (2.854 Å),14c,d or ice (2.77−2.84 Å).14e They are also slightly shorter than the water tetramer (O–H⋯O, 2.82 and 2.90 Å) found in the lattice of 1. The tetramers are interconnected (O1W–H⋯O2WB 3.01 Å) into a chain of water square-steps, similar to those found elsewhere.8a,14f The slope of the steps, as measured by the dihedral angle between the two mean tetramer planes (O1W⋯O2W⋯O1WA⋯O2WA and O1W⋯O2W⋯O1WB⋯O2WB) is 41.5°. Water–bromide cluster blends are formed by secondary H-bonding interactions (O2W–H⋯Br2B 3.26 Å) between water and uncoordinated bromide that are near-orthogonal to the unidirectional propagation of the steps (torsion angle of O2WA⋯O1W⋯O2WB⋯O1WD is 0°).(Fig. 5b) As the bromide ions are further linked to the stacked pincer cations (Pd⋯Pd 5.413 Å) via weak H-bonding (Br⋯H 2.91 and 2.92 Å) with the methylene and amine protons (Fig. 5c and Table 2), the entire supramolecular network is covered by these different types of H-bond interactions.
![(a) A perspective view of the cationic structure of 6 ignoring the water solvate (thermal ellipsoids at 30% level). (Pd: purple; S: yellow; N: dark blue; Br: green; O: red; H: sky blue.) Selected bond distances [Å] and angles [°]: Pd1–S1 2.302(1); Pd1–S2 2.306(1); Pd1–N1 2.040(3); Pd1–Br1 2.4208(5); N1–Pd1–Br1 179.6(1); N1–Pd1–S1 86.9(1); Br1–Pd1–S1 93.29(3); N1–Pd1–S2 86.9(1); Br1–Pd1–S2 92.87(3); S1–Pd1–S2 173.83(4). Pd1 deviated −0.0011 Å from the least-squares plane of the donor ligands (S1, S2, N1, Br1). (b) Interconnection of the water tetramer into a step-like structure supplemented by H-bonding with bromide ions at 100 K. (c) Water-bromide blends sandwiched by the cations.](/image/article/2010/CE/b917189b/b917189b-f5.gif) |
| | Fig. 5 (a) A perspective view of the cationic structure of 6 ignoring the water solvate (thermal ellipsoids at 30% level). (Pd: purple; S: yellow; N: dark blue; Br: green; O: red; H: sky blue.) Selected bond distances [Å] and angles [°]: Pd1–S1 2.302(1); Pd1–S2 2.306(1); Pd1–N1 2.040(3); Pd1–Br1 2.4208(5); N1–Pd1–Br1 179.6(1); N1–Pd1–S1 86.9(1); Br1–Pd1–S1 93.29(3); N1–Pd1–S2 86.9(1); Br1–Pd1–S2 92.87(3); S1–Pd1–S2 173.83(4). Pd1 deviated −0.0011 Å from the least-squares plane of the donor ligands (S1, S2, N1, Br1). (b) Interconnection of the water tetramer into a step-like structure supplemented by H-bonding with bromide ions at 100 K. (c) Water-bromide blends sandwiched by the cations. | |
As 7 and 1 are direct chemical derivatives differed only by the uncoordinated ions, a comparison of their crystal structures provides a model to examine the effect of H-bonding of lattice water in the absence of water–halide cluster blends. The analysis of 7 at 223 K reveals an asymmetric unit comprising a cation, free NO3− ion, and indeed no lattice water (Fig. 6). This is perhaps surprising considering that H-bonding between nitrate and water is common even in crystal lattices.14f,g Its absence suggested the promoting role played by the halide in attracting water hydration in this system. The Pd⋯Pd separation (5.760 Å) is also longer than that in 1 (5.364 Å) The NO3− ions are sandwiched by the cations through H-bonding with the amine proton [N1–H⋯O3 2.82, C1–H⋯O3A 3.27, C2–H⋯O2B 3.28, C3–H⋯O1C 3.26 and C4–H⋯O1 3.20 Å] (Fig. 6b and Table 2).
![(a) A perspective view of the cationic structure of 7 ignoring the anion (thermal ellipsoids at 30% level). (Pd: purple; S: yellow; N: dark blue; Cl: green; O: red; H: sky blue) Selected bond distances [Å] and angles [°]: Pd1–S1 2.301(1); Pd1–S2 2.304(1); Pd1–N1 2.022(4); Pd1–Cl1 2.303(1); N1–Pd1–S1 87.1(1); N1–Pd1–Cl1 179.4(1); S1–Pd1–Cl1 92.29(5); N1–Pd1–S2 86.1(1); S1–Pd1–S2 173.26(4); Cl1–Pd1–S2 94.45(4). Pd1 deviated 0.0039 Å from the least-squares plane of the donor ligands (S1, S2, N1, Cl1). (b) A lattice view along the a direction showing H-bonding between the anions and cations without lattice hydrate.](/image/article/2010/CE/b917189b/b917189b-f6.gif) |
| | Fig. 6 (a) A perspective view of the cationic structure of 7 ignoring the anion (thermal ellipsoids at 30% level). (Pd: purple; S: yellow; N: dark blue; Cl: green; O: red; H: sky blue) Selected bond distances [Å] and angles [°]: Pd1–S1 2.301(1); Pd1–S2 2.304(1); Pd1–N1 2.022(4); Pd1–Cl1 2.303(1); N1–Pd1–S1 87.1(1); N1–Pd1–Cl1 179.4(1); S1–Pd1–Cl1 92.29(5); N1–Pd1–S2 86.1(1); S1–Pd1–S2 173.26(4); Cl1–Pd1–S2 94.45(4). Pd1 deviated 0.0039 Å from the least-squares plane of the donor ligands (S1, S2, N1, Cl1). (b) A lattice view along the a direction showing H-bonding between the anions and cations without lattice hydrate. | |
All the pincer complexes with uncoordinated halide anion (except for 2) are hydrated with the lattice water showing extensive H-bonding with the halide and the cation (Table 3). The water tetramer steps are present as microchannnels in 1 and 6. Complex 3, with only one water hydrate per cation, is the least hyrdated among the halides. Complexes 5 and 6 have the same number of lattice water, viz. 2, and in both cases, extended water channels are evident, albeit in different structural forms viz. tape-like in 5 and step-like in 6. All the complexes (1–7), except 4, show Pd⋯Pd stacks with an additional interaction between Pd and the skeletal methylene proton of the ligands. A common feature in the lattices of 1–7 is the presence of N– H⋯A (A = anion ion, Cl−, Br− or NO3−) interactions. With a smaller substituent (ethyl) in L3, complex 3 could only present a smaller lattice channel void (5.0% void volume, calculated by PLATON15) which can only accommodate an unidirectional water–choride zigzag chain. Complexes 1 and 6, both of which bear the isobutyl substituents, have the largest voids (14.5 and 13.4% respectively) to accommodate the water chains of tetramers. Among the complexes in this system with free halide (1–6), complex 2 is unique in showing no clathrating ability for water. This may be attributed to the bigger hydrophobic benzyl groups on the ligand (L2).
Table 3 Comparative structural data of 1–7
| Complex |
S-substitutent |
Anion |
Data collection temp./K |
No. of lattice water |
H2O − X cluster blend |
|
There are four disordered lattice water molecules with an occupancy of 0.5 in each asymmetric unit in 5.
|
|
1
|
i
butyl |
Cl− |
223 |
2 |
water tetramer −Cl− |
|
2
|
benzyl |
Cl− |
223 |
0 |
none |
|
3
|
ethyl |
Cl− |
293 |
1 |
water −Cl− chain |
|
4
|
t
butyl |
Cl− |
296 |
1.5 |
water trimer −Cl− chain |
|
5
|
cyclohexyl |
Cl− |
100 |
2a |
water tape −Cl− |
|
6
|
i
butyl |
Br− |
100 |
2 |
water tetramer −Br− |
|
7
|
i
butyl |
NO3− |
223 |
0 |
none |
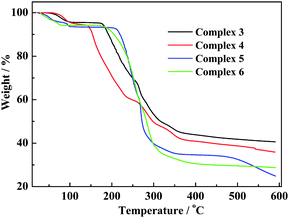
Thermal gravimetric analysis (TGA) of the fresh samples of 3–6 were carried out under N2.(Table 4 and Fig. 7) All of them show a ready weight loss attributed to dehydration from ∼30 to ∼130 °C, after which the decomposition of the anhydrous component commences. For 3 and 4, a weight-loss of 4.4% (calcd 4.5%) and 5.5% (calcd 5.9%) at 50–100 °C and 60−130 °C, respectively, corresponds to complete dehydration. For 5, the dehydration (of 6.4%, compared to calculated 7.0%) profile at 30–100 °C indicates a multistep weight-loss process. For 6, this multi-step weight-loss occurs at 30–80 °C, losing 5.7% (calcd 6.5%), corresponding to complete dehydration. Dehydration of 5 and 6 commences in ambient conditions (∼30 °C), suggesting that these forms of extended channel of water–halide blends are relatively thermo-sensitive. Facile dehydration in the form of collapse of such hydrate network would lead to rapid loss of crystallinity. This also explains why the X-ray diffraction analysis of these two compounds had to be carried out under low temperature. However, the ease of dehydration has no direct relationship with the thermal stability of the dehydrated complex. In fact, examination of the TG profiles of the four complexes (Fig. 7) showed that degradation of anhydrous 4 starts at ∼140 °C, suggesting that it is most fragile in this series. Complex 5, on the other hand, is the most robust and does not decompose until ∼200 °C.
Table 4 Selected TGA data for complexes 3–6
| Complex |
Degree of dehydration |
Dehydration temp range/° C |
Complex decomposition temp/°C |
Observed (calculated) weight-loss/% |
|
3
|
H2O |
50–100 |
∼170 |
4.4 (4.5) |
|
4
|
1.5 × H2O |
60–130 |
∼140 |
5.5 (5.9) |
|
5
|
2 × H2O |
30–100 |
∼200 |
6.4 (7.0) |
|
6
|
2 × H2O |
30–80 |
∼170 |
5.7 (6.5) |
Conclusion
This study suggested that cationic pincer complexes in the form of halide salts have a general affinity for water that blends with the halide to give different forms of water–halide clusters in the solid lattice. A common form of network organisation is to align the sq planar cations in the form of metal stacks so that the voids between and among them can accommodate the water–halide cluster blends. Both intra- and intermolecular as well as inter-ionic hydrogen-bonding interactions are prominent. The bonding network involves the lattice water, cation and anion as well as the amine proton and thioether substituent of the tridentate ligand and the coordinated halide. It lends stability to the complexes in their solid-state and promotes water miscibility and solubility in solutions. One obvious benefit is to enable the design of water-soluble pincer complexes, which are known for their catalytic applications,16 to be used as water-based catalysts in homogeneous and biphasic catalysis. Current experiments in our laboratory are directed accordingly.
Acknowledgements
This work was supported by the Ministry of Education (R-143-000-361-112), the Agency for Science, Technology & Research (R-143-000-277-305), and the National University of Singapore. We are grateful to G. K. Tan for crystallographic assistance.
Notes and references
-
(a) R. Custelcean, Chem. Commun., 2008, 295–307 RSC;
(b)
Hydrogen Bonding-New Insights, ed. S. J. Grabowski, Springer, 2006 Search PubMed;
(c)
G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond In Structural Chemistry and Biology, Oxford, 1999 Search PubMed.
- M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 CrossRef CAS.
-
(a) S. Bartoli, C. Bazzicalupi, S. Biagini, L. Borsari, A. Bencini, E. Faggi, C. Giorgi, C. Sangregorio and B. Valtancoli, Dalton Trans., 2009, 1223–1230 RSC;
(b) S. P. Gavrish, Y. D. Lampeka, P. Lightfoot and H. Pritzkow, Dalton Trans., 2007, 4708–4714 RSC;
(c) C. D. Ene, A. M. Madalan, C. Maxim, B. Jurca, N. Avarvari and M. Andruh, J. Am. Chem. Soc., 2009, 131, 4586–4587 CrossRef CAS.
-
(a) I. Bernal, C. R. Chim., 2006, 9, 1454–1466 CrossRef CAS;
(b) U. Mukhopadhyay and I. Bernal, Cryst. Growth Des., 2006, 6, 363–365 CrossRef CAS;
(c) I. Bernal, Comp. R. Chim., 2007, 10, 1209–1215 Search PubMed;
(d) A. Bialonska and I. Bernal, C. R. Chim., 2007, 10, 232–233 CrossRef CAS;
(e) S. Wallace, L. Huang, L. Massa, U. Mukhopadhyay, I. Bernal and J. Karle, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16798–16803 CrossRef CAS;
(f) I. Bernal, CrystEngComm, 2008, 10, 1265–1265 RSC;
(g) I. Bernal, C. R. Chim., 2008, 11, 942–944 CrossRef CAS.
-
(a) R. E. Babine and S. L. Bender, Chem. Rev., 1997, 97, 1359–1472 CrossRef;
(b) K. Bowman-James, Acc. Chem. Res., 2005, 38, 671–678 CrossRef CAS;
(c) X. Li, Y.-D. Wu and D. Yang, Acc. Chem. Res., 2008, 41, 1428–1438 CrossRef CAS;
(d) K. Wichmann, B. Antonioli, T. Söhnel, M. Wenzel, K. Gloe, K. Gloe, J. R. Price, L. F. Lindoy, A. J. Blake and M. Schröder, Coord. Chem. Rev., 2006, 250, 2987–3003 CrossRef CAS;
(e) S. Chakrabarti, M. F. L. Parker, C. W. Morgan, C. E. Schafmeister and D. H. Waldeck, J. Am. Chem. Soc., 2009, 131, 2044–2045 CrossRef CAS;
(f)
J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, The Royal Society of Chemistry, 2006 Search PubMed;
(g) A. M. Baruah, A. Karmakar and J. B. Baruah, Polyhedron, 2007, 26, 4479–4488 CrossRef CAS;
(h) E. C. Constable, G. Zhang, C. E. Housecroft, M. Neuburger and S. Schaffner, CrystEngComm, 2009, 11, 1014–1021 RSC.
-
(a) C. W. Spicer, E. G. Chapman, B. J. Finlayson-Pitts, R. A. Plastridge, J. M. Hubbe, J. D. Fast and C. M. Berkowitz, Nature, 1998, 394, 353–356 CrossRef CAS;
(b) K. W. Oum, M. J. Lakin, D. O. DeHaan, T. Brauers and B. J. Finlayson-Pitts, Science, 1998, 279, 74–77 CrossRef CAS;
(c) K. L. Foster, R. A. Plastridge, J. W. Bottenheim, P. B. Shepson, B. J. Finlayson-Pitts and C. W. Spicer, Science, 2001, 291, 471–474 CrossRef CAS.
-
(a) R. Dutzler, E. B. Campbell, M. Cadene, B. T. Chait and R. MacKinnon, Nature, 2002, 415, 287–294 CrossRef CAS;
(b) M. K. Saha and I. Bernal, Inorg. Chem. Commun., 2005, 8, 871–873 CrossRef CAS;
(c) B. R. Srinivasan, R. G. Mhalsikar, K. S. Rane, C. Näther and W. Bensch, J. Chem. Sci., 2007, 119, 21–27 CrossRef CAS;
(d) H. S. Chow, E. C. Constable, C. E. Housecroft, M. Neuburger and S. Schaffner, Dalton Trans., 2006, 2881–2890 RSC.
-
(a) S.-Q. Bai and T. S. A. Hor, Chem. Commun., 2008, 3172–3174 RSC;
(b) S.-Q. Bai, L. L. Koh and T. S. A. Hor, Inorg. Chem., 2009, 48, 1207–1213 CrossRef CAS;
(c) S. Gu and W. Chen, Organometallics, 2009, 28, 909–914 CrossRef CAS;
(d) M. Ohff, A. Ohff, M. E. van der Boom and D. Milstein, J. Am. Chem. Soc., 1997, 119, 11687–11688 CrossRef CAS;
(e) R. Huang and K. H. Shaughnessy, Organometallics, 2006, 25, 4105–4112 CrossRef;
(f) D. E. Bergbreiter, P. L. Osburn and Y.-S. Liu, J. Am. Chem. Soc., 1999, 121, 9531–9538 CrossRef CAS;
(g) P. Steenwinkel, H. Kooijman, W. J. J. Smeets, A. L. Spek, D. M. Grove and G. van Koten, Organometallics, 1998, 17, 5411–5426 CrossRef CAS.
- D. S. McGuinness, P. Wasserscheid, D. H. Morgan and J. T. Dixon, Organometallics, 2005, 24, 552–556 CrossRef CAS.
-
SMART, version 5.631, Software Reference Manuals; SAINT, version 6.63, Software Reference Manuals, Bruker AXS GmbH, Karlsruhe, Germany, 2000 Search PubMed.
-
G. M. Sheldrick, Software for Empirical Absorption Correction: SADABS, University of Göttingen, Göttingen, Germany, 2001 Search PubMed.
-
(a)
G. M. Sheldrick, Program for Crystal Structure Solution: SHELXS-97, University of Göttingen, Göttingen, Germany, 1997 Search PubMed;
(b)
G. M. Sheldrick, Program for Crystal Structures Refinement: SHELXL- 97, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- R. Parthasarathi, M. Elango, V. Subramanian and N. Sathyamurthy, J. Phys. Chem. A, 2009, 113, 3744–3749 CrossRef CAS.
-
(a) S. Supriya and S. K. Das, New J. Chem., 2003, 27, 1568–1574 RSC;
(b) J. D. Cruzan, L. B. Braly, K. Liu, M. G. Brown, J. G. Loeser and R. J. Saykally, Science, 1996, 271, 59–62 CAS;
(c) R. J. Speedy, J. D. Madura and W. L. Jorgensen, J. Phys. Chem., 1987, 91, 909–913 CrossRef CAS;
(d) A. C. Belch and S. A. Rice, J. Chem. Phys., 1987, 86, 5676–5682 CrossRef;
(e)
G. A. Jeffrey, An Introduction to Hydrogen Bonding, OUP, Oxford, 1997, pp 160–180 Search PubMed;
(f) L. R. Hanton and K. Lee, J. Chem. Soc., Dalton Trans., 2000, 1161–1166 RSC;
(g) S. Bianketti, A. J. Blake, C. Wilson, P. Hubberstey, N. R. Champness and M. Schröder, CrystEngComm, 2009, 11, 763–769 RSC.
-
(a) P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef;
(b)
A. L. Spek, PLATON.
A Multipurpose Crystallographic Tool, Utrecht University, Netherlands, 2002 Search PubMed.
-
(a) J. M. Serrano-Becerra and D. Morales-Morales, Curr. Org. Synth., 2009, 6, 169–192 Search PubMed;
(b) C. A. Kruithof, A. Berger, H. P. Dijkstra, F. Soulimani, T. Visser, M. Lutz, A. L. Spek, R. J. M. K. Gebbink and G. van Koten, Dalton Trans., 2009, 3306–3314 RSC.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.