1,3-Diamino-2-hydroxypropane-N,N,N′,N′-tetraacetic acid stabilized amorphous calcium carbonate: nucleation, transformation and crystal growth
Guo-Bin
Cai
,
Shao-Feng
Chen
,
Lei
Liu
,
Jun
Jiang
,
Hong-Bin
Yao
,
An-Wu
Xu
and
Shu-Hong
Yu
*
Division of Nanomaterials & Chemistry, Hefei National Laboratory for Physical Sciences at the Microscale, Department of Chemistry, University of Science and Technology of China, Hefei, 230026, China. E-mail: shyu@ustc.edu.cn
First published on 2nd September 2009
Abstract
A low molecular weight organic molecule, 1,3-diamino-2-hydroxypropane-N,N,N′,N′-tetraacetic acid, has been found that it could stabilize amorphous calcium carbonate (ACC) for at least three days in a gas diffusion reaction, also it could control the formation of hierarchical calcite crystals. The transformation process from ACC to calcite crystals has been systematically studied. Nucleation sites and intermediates were both captured by time-dependent experiments. It is found that ACC could form a close packed film on the substrate and part of the nucleation occurred on the film. After nucleation, another form of ACC conglomeration was found to be dissolved from inside. The intermediates on the substrate were found to be composed of fibres. A rod-dumbbell-sphere transformation phenomenon was observed. Selective adsorption and the mesocrystal transformation mechanism are assumed to play a key role in the formation of intermediates with different shapes and structures.
1. Introduction
Recently, amorphous calcium carbonate (ACC) has gained a lot of attention. ACC is a very special phase of calcium carbonate. Firstly, it contains very little water, only from zero to one mole of bound water have been found in ACC extracted from organisms.1,2 This makes it very convenient to store, as revealed in plants and invertebrates.3 Secondly, ACC exhibits only short-range and limited intermediate-range ordering and no long-range structure coherence, which makes it truly amorphous.4 ACC is inherently unstable,3 and it will transform into a stable phase very fast without any stabilizers. This may be one of the reasons why it has not been studied until very recently. The most fascinating feature of ACC is that it could be tailored into various moulds, then crystallization might be controlled as mould restriction.5 Some excellent work has been done based on this feature. Aizenberg and co-workers successfully used AFM tips to make a unique nucleation site to induce the patterned calcite single crystal formation.6 Qi et al. made use of packed modified polystyrene (PS) spheres as a scaffold to obtain inverse opal photonic crystals composed of calcite.7 These studies all use ACC as the precursor, thus showing how important and amazing ACC is.Until now, the preparation of ACC mainly includes four kinds of methods, i.e. direct mix calcium ions and carbonate ions quickly,8 hydrolysis of dimethyl carbonate,9 or catalyzing decomposition of urea to give homogeneous carbonate ions10 and Kitano methods.11–13 Miniemulsion,14 Langmuir15,16 and gas diffusion technology17–19 are also used in preparing ACC particles. Quick operations or stabilizers are needed to get a less crystallized ACC. For the unstable phase, stabilizers are quite few. It has been reported that magnesium ions,20,21 phytic acid,22 poly(acrylic acid),23 poly(sodium 4-styrene sulfonate),24 poly(aspartic acid)25 and their derivatives have the ability to stabilize ACC. Up until now, low molecular weight organic molecules containing carboxyl groups have been seldomly found to stablize ACC, even the monomers of polymer stabilizers can do nothing to stabilize ACC.
Except for the preparation of ACC, it is still a challenge to make the ACC transformation process clear, as it changes too fast to capture its transformation intermediates. Based on the literatures, two main transformation pathways in solution were put forward. Rieger et al. examined the first formed ACC using cryo-TEM and in situ X-ray microscopy and considered that ACC undergoes dissolution–recrystallization to form the more thermodynamically stable state of calcium cabonate.26 Sethman et al. used in situ AFM to examine the gelatinous phase formed on calcite substrate, and consider this phase goes through an overall physical change in the shape of the precursor phase as these structures emerge, but not a dissolution–recrystallization reaction.27 Gower et al. suggested that this mechanism is analogous to the mesocrystal assembly mechanisms proposed by Cölfen and co-workers.28,29 These two mechanisms seem to be quite different.
In this paper, a low molecular weight organic molecule, 1,3-diamino-2-hydroxypropane-N,N,N′,N′-tetraacetic acid (H4dhpta), was used to control the crystallization of calcium carbonate. Powell and coworkers first used this molecule in biomineralization, and hierarchical “microtrumpet” calcite crystals were obtained.30 Herein, we found that H4dhpta could stabilize ACC for a long time. By using H4dhpta to control the calcium carbonate growth in solution with a carbon dioxide gas diffusion process, the ACC phase can be formed both in solutions and on substrates. The intermediates were captured during the transition from ACC to hierarchical calcite crystals. An integrated mechanism that includes physical changes, dissolution–recrystallization processes and mesocrystal transformation has been proposed.
2. Experimental
Materials
All chemicals are of analytical grade and used as received without further purification. 1,3-Diamino-2-hydroxypropane-N,N,N′,N′-tetraacetic acid (Fw = 322.27, Scheme 1) was obtained from Alfa Aesar, while ammonium carbonate and calcium chloride were purchased from Shanghai Chemical Reagent Company. All glassware (beakers and small pieces of glass substrates) were cleaned and sonicated in ethanol for 5 min. It was rinsed with deionized water and further soaked in a piranha solution H2SO4/H2O2(3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v:v), and then rinsed with deionized water, and finally dried in air with acetone.
:1, v:v), and then rinsed with deionized water, and finally dried in air with acetone.
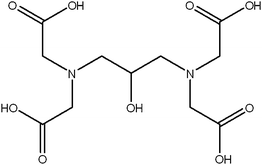 | ||
| Scheme 1 Chemical structure of 1,3-diamino-2-hydroxypropane-N,N,N′,N′-tetraacetic acid. | ||
Preparation
Stock aqueous solutions of H4dhpta (1 g L−1 ≈ 3 mM) and CaCl2 (0.25 M) was freshly prepared in deionized water separately. Various volumes of CaCl2 solution were added to this H4dhpta solution with vigorous stirring to ensure complete mixing to obtain a 1 g L−1 H4dhpta solution with different CaCl2 concentrations. The molar ratios of H4dhpta to CaCl2 were varied from 1:1 to 1:32, which makes the molar ratio of COO− ions to Ca2+ ions from 4:1 to 1:8. Equal volumes of the mixture (10 mL) were then added into 25 mL glass beakers. The beakers were covered with Parafilm punched with three needle holes and then put in a desiccator. Finally, a glass bottle (10 mL) of abrasive ammonium carbonate was placed at the bottom of the desiccator and also covered with Parafilm punched with three needle holes. After different periods of reaction time, one beaker was taken out and the glass substrate was rinsed with distilled water and acetone and allowed to dry at room temperature for characterization. Precipitates were collected by centrifugation and washed three times by water and acetone separately and then dried at room temperature.
Characterization
The collected sample was characterized on an (Philips X'Pert Pro Super) X-ray powder diffractometer with Cu Kα radiation (λ = 1.541874 Å). SEM images were obtained with a JEOL JSM-6700F scanning electron microscope operating at 10 kV and a Zeis Supra™ 40 high resolution field emission scanning electron microscope operating at 5 KV. Thermogravimetric analysis (TGA) was carried out on a Diamond TG/DTA thermal analyzer (Perkin-Elmer) with a heating rate of 10 K min−1 from room temperature to 800 °C in an air flow. FTIR spectra were measured on a Bruker Vector-22 FT-IR spectrometer from 4000 to 400 cm−1 at room temperature. Transmission electron microscopy (TEM) was performed on a JEOL-2011 high-resolution transmission electron microscope (HRTEM) operated at an acceleration voltage of 200 kV. Carbon and nitrogen were determined by standard combustion analysis with an Elementar Vario EL III elemental analyser. The film thickness was detected using an (Shimadzu XRF-1800) X-ray fluorescence spectrometer. The pH was monitored by a Delta 320 pH monitor (Mettler Toledo).3. Results and discussion
3.1 Effects of different molar ratios of H4dhpta to Ca2+ ions on crystallization of CaCO3
Mineralization of CaCO3 with a different ratio of the additive was performed. In an equal molar ratio of H4dhpta to CaCl2, no precipitates were found, indicating the strong coordination ability of H4dhpta with Ca2+ ions. When the molar ratio of CaCl2 to H4dhpta increased to 2:1, after four days, only hierarchical rods were observed (Fig. 1A). And they have two length scales, one is in 20 µm, the other is over 50 µm. No matter how long the rods are, they all show hierarchical structures similar to smaller rods. When the molar ratio of CaCl2 to H4dhpta increased to 4:1, crystals of rods, dumbbells, semi-attached spheres and spheres were found (Fig. 1b). No other morphologies existed except for bigger spheres when continuously increasing the concentration of calcium ions. In Powell's experiment, when the molar ratio of CaCl2 to H4dhpta is 25:1, ‘microtrumpets’ composed of nanocrystalline calcite were formed.30 However, in a gas diffusion process, no microtrumpet precipitated, which shows quite a big difference between slow and quick reactions.
![SEM images of precipitates prepared under different conditions. (A) [Ca2+]/[H4dhpta] = 2 : 1, (B) [Ca2+]/[H4dhpta] = 4 : 1.](/image/article/2010/CE/b911426m/b911426m-f1.gif) | ||
| Fig. 1 SEM images of precipitates prepared under different conditions. (A) [Ca2+]/[H4dhpta] = 2:1, (B) [Ca2+]/[H4dhpta] = 4:1. | ||
The phase of calcium carbonate precipitates was examined by XRD. The XRD pattern perfectly agrees with the literature (JCPDS 05-0586), as shown in Fig. 2E. This proves that these precipitates are all calcite, indicating that the organic molecule can not change the crystal polymorph, but can only influence the morphology. FT-IR spectrum is also used to verify the phase of these CaCO3 precipitates. The presence of the peaks at 713 and 876 cm−1 can be assigned as the characteristic peaks of calcite (Fig. 3B).
![XRD patterns of precipitates collected at different reaction time. (A) One day, (B) two days, (C) three days, (D) three-and-a-half days, (E) four days. [Ca2+]/[H4dhpta] = 4 : 1.](/image/article/2010/CE/b911426m/b911426m-f2.gif) | ||
| Fig. 2 XRD patterns of precipitates collected at different reaction time. (A) One day, (B) two days, (C) three days, (D) three-and-a-half days, (E) four days. [Ca2+]/[H4dhpta] = 4:1. | ||
![FT-IR spectra of calcium carbonate precipitates at different time intervals. (A) One day, (B) four days. [Ca2+]/[H4dhpta] = 4 : 1. The absorption bands at 865 cm−1 (ν2) and split bands at 1419 and 1490 cm−1(ν3) are typical of ACC. The sharp absorption bands at 713 (ν4) and 876 cm−1 (ν2) suggest the presence of a calcite phase.](/image/article/2010/CE/b911426m/b911426m-f3.gif) | ||
| Fig. 3 FT-IR spectra of calcium carbonate precipitates at different time intervals. (A) One day, (B) four days. [Ca2+]/[H4dhpta] = 4:1. The absorption bands at 865 cm−1 (ν2) and split bands at 1419 and 1490 cm−1(ν3) are typical of ACC. The sharp absorption bands at 713 (ν4) and 876 cm−1 (ν2) suggest the presence of a calcite phase. | ||
3.2 ACC stabilized by H4dhpta and its characterization
Time dependent experiments were performed systematically for studying the crystallization mechanism. In all these reactions, we found almost the same phenomenon. Firstly, the reaction solutions became milky and turbid. As the reaction time went on, the balance between gravity and buoyancy was broken and white precipitates descended to the bottom of the reaction bottles. As time went by, these precipitates seemed to be vanishing gradually. Finally, all milky precipitates disappeared and transparent crystals appeared both at the bottom of the bottle and the interface between air and solution.When the molar ratio of CaCl2 to H4dhpta was 1:2, only rods were obtained, and the transformation process was too fast to be captured. Thus, a 1:4 molar ratio of CaCl2 to H4dhpta was chosen. Precipitates collected at different time intervals were examined by X-ray diffraction, as shown in Fig. 2. Two broad peaks indicate an amorphous phase. After three days' diffusion reaction, the precipitates remained amorphous (Fig. 2A–2C), showing the capability of H4dhpta to stabilize the amorphous phase. Three-and-a-half days later, the crystals of calcite were precipitated (Fig. 2D and 2E).
The FTIR spectrum is very useful for distinguishing among different phases of calcium carbonate crystals. By detecting the absorption bands of carbonate, all crystal phases of calcium carbonate can be discriminated. Each phase of calcium carbonate has some characteristic absorption bands. Typically, the absorption bands of carbonate are divided into four parts: the symmetric stretch of the carbonate ion at about 1080 cm−1(ν1); the out-of-plane bending absorption at about 870 cm−1(ν2); the asymmetric stretch at about 1400 cm−1(ν3) and the in-plane bending at about 700 cm−1(ν4). For ACC, the in-plane bending is broadened and seems disappearing, the out-of-plane bending shifts to about 866 cm−1, and the asymmetric stretch peaks split into two parts at around 1420 and 1470 cm−1.31,32 And calcite shows two characteristic absorption bands at 876 and 712 cm−1.31–33 In the present experiments, the FT-IR spectrum of the initial precipitates clearly shows the characteristic absorption bands of ACC at 865 cm−1 (ν2) and split bands at 1419 and 1490 cm−1(ν3) (Fig. 3A). This confirms that precipitates of calcium carbonate are ACC at the initial stage. In contrast, the sharp absorption bands at 713(ν4) and 876 cm−1 (ν2) suggest the formation of calcite after four days’ reaction (Fig. 3B), which is consistent with XRD results.
Thermogravimetric analysis (TGA) is also used to analyze the water content in ACC. From the TG curve (Fig. 4), we can calculate that the lost water content is about 15.78 wt%. Then it is calculated that the possible structure of ACC is CaCO3·H2O and the result is quite similar to biogenic ACC investigated by Addadi and coworkers.34 The content of an organic molecule in ACC was also determined by detecting the content of nitrogen, hydrogen and carbon with combustion analysis. And a content of 1.095 wt% for N, 2.890 wt% for H and 13.15 wt% for C was detected. Then it can be seen that a content of ca. 12.60 wt% H4dhpta remains in the final product.
![TG curve of ACC collected after one day, [Ca2+]/[H4dhpta] = 4 : 1.](/image/article/2010/CE/b911426m/b911426m-f4.gif) | ||
| Fig. 4 TG curve of ACC collected after one day, [Ca2+]/[H4dhpta] = 4:1. | ||
The morphology of ACC particles was examined by high resolution FE-SEM and TEM. It seems that the ACC stabilized by H4dhpta has two types. One performs a block conglomeration on a scale of hundreds of nanometers (Fig. 5A, B). TEM shows that each aggregated particle is composed of nanoparticles with a diameter less than 10 nm (Fig. 5C). Thus, it is convincible that the first formed ACC is composed of nanoparticles with a size of less than 10 nm, they then agglomerate to form opalescent blocks. However, after three-and-a-half days, these ACC aggregates gradually dissolved from inside and finally disappeared, as shown in Fig. 5D.
![(A), (B) SEM images. (C) TEM image of ACC precipitates collected after 1 d. Insert shows the SAED pattern of the sample collected after 1 d. (D) ACC precipitates collected after three-and-a-half days. Insert shows the SAED pattern of the sample D. The structure transition from solid (C) to hollow (D) indicates dissolution from inside. [Ca2+]/[H4dhpta] = 4 : 1.](/image/article/2010/CE/b911426m/b911426m-f5.gif) | ||
| Fig. 5 (A), (B) SEM images. (C) TEM image of ACC precipitates collected after 1 d. Insert shows the SAED pattern of the sample collected after 1 d. (D) ACC precipitates collected after three-and-a-half days. Insert shows the SAED pattern of the sample D. The structure transition from solid (C) to hollow (D) indicates dissolution from inside. [Ca2+]/[H4dhpta] = 4:1. | ||
The other type of ACC is dependent on substrates and they form a densed film on substrates (Fig. 6A–D). This film might be ignored as a background in a low magnification view (Fig. 6A). However, when magnifying the flat zone (quadrate area in Fig. 6A), a rough surface composed of small particles can be observed (Fig. 6B, C). SEM image of a fracture area of this film indicates that it is a close packed film with hundreds of layers (Fig. 6D). X-Ray fluorescence spectra (XRF) demonstrate that the thickness of this film is about 166.3 nm. As these substrates were treated to be hydrophilic, there might be special interactions between substrates and the firstly formed ACC nanoparticles, which then enable these particles to form a film on the substrates. However, this could only explain a thin layer formation on substrates. For such a thick film, the interaction between particles must be considered. It can be assumed that the ACC particles here have the nature to form films before crystallization. To prove this, we chose the TEM copper grid as a substrate to see whether an ACC film can be formed on a hydrophobic surface. After 2 d, a different ACC film with some holes was obtained (Fig. 6E, F). Similar results were just reported by Chu and coworkers, who obtained similar ACC films by using maleic chitosan as an additive on TEM copper grids.35 Colloid nanoparticle self-organization was proposed to explain how these films formed. However, as differences in ACC films existed on hydrophilic and hydrophobic surfaces, much work still needs to be done to find out how these films have formed.
![(A), (B), (C), (D) SEM images of a glass substrate taken out after 1 d. (A) A low-magnification image of ACC on substrate. (B), (C) High-magnification images of a selected area in (A). (D) A fracture area of ACC films observed on the edge of a glass substrate. (E) TEM and (F) SEM images of ACC films formed on a copper grid after 2 d. Insert in (E) shows the SAED pattern of a selected area in (E). [Ca2+]/[H4dhpta] = 4 : 1.](/image/article/2010/CE/b911426m/b911426m-f6.gif) | ||
| Fig. 6 (A), (B), (C), (D) SEM images of a glass substrate taken out after 1 d. (A) A low-magnification image of ACC on substrate. (B), (C) High-magnification images of a selected area in (A). (D) A fracture area of ACC films observed on the edge of a glass substrate. (E) TEM and (F) SEM images of ACC films formed on a copper grid after 2 d. Insert in (E) shows the SAED pattern of a selected area in (E). [Ca2+]/[H4dhpta] = 4:1. | ||
3.3 Nucleation stage
The transformation of ACC to crystals was studied by taking glass substrate out of the reaction vessels at different time intervals. In about three days, ACC remained as it was before and no changes could be detected. After three-and-a-half days, white precipitates became transparent and vanished gradually, and some floaters formed at the interface between air and solutions. This suggests that the balance is broken and nucleation occurs. SEM images of precipitates on substrates at this stage are shown in Fig. 7. Separated islands with a size of about 200 nm were found on the substrates except for the aggregates showed in Fig. 5A and B. Compared with previous films, these islands are considered as the nucleation site of ACC transformation. Meanwhile, the conglomeration of the first formed ACC dissolved gradually from inside, as seen in Fig. 5D. This indirectly proves that nucleation can not form via ACC blocks, it only occurs on the interface between substrates and solutions or air and solutions, thus this is a heterogeneous nucleation process. Bigger particles with similar surface structures to that of the final crystal are also shown in Fig. 7C, D. The obvious surrounding boundaries (Fig. 7C) and the flat bottom (Fig. 7D) both indicate that these particles grew from the substrate. It is another proof demonstrating the nucleation occurs on substrates. To make sure part of the nucleation occurs on ACC films, other glass substrates were placed in a bottle one day later than ACC forms. After four days, very few crystals could be found on these substrates. Thus, some nucleations do occur on ACC films. However, as revealed by Gebauer and coworkers, a prenucleation cluster will form before nucleation occurs,36 we can't exclude the possibility that nucleation can still form in solution.![SEM images of a glass substrate in the nucleation stage that precipitated for three-and-a-half days. The islands (A), (B), surrounding boundaries (C) and the flat bottom part (Fig. 7D) indicate that nucleation occurs on substrates. [Ca2+]/[H4dhpta] = 4 : 1.](/image/article/2010/CE/b911426m/b911426m-f7.gif) | ||
| Fig. 7 SEM images of a glass substrate in the nucleation stage that precipitated for three-and-a-half days. The islands (A), (B), surrounding boundaries (C) and the flat bottom part (Fig. 7D) indicate that nucleation occurs on substrates. [Ca2+]/[H4dhpta] = 4:1. | ||
Liu et al. reported that the heterogeneous nucleation may correspond to a good structural match and synergy between biominerals and substrates at low supersaturations or a supersaturation-driven interfacial structural mismatch at high supersaturations.37 In our experiments, the substrates were treated to be hydrophilic, certain interactions must exist between substrates and ACC particles and due to the existence of ACC, supersaturation is lowered as high supersaturation will promote ACC formation, thus nucleation may occur in accordance with structural match and synergy. However, the effect of ACC itself should not be ignored. Sagi and co-workers found that biogenic ACC had a characteristic short-range order by X-ray absorption spectroscopy studies,38 so does artificial ACC revealed by Michell et al.4 Thus, we can presume that the ACC prepared here also has its own characteristic short-range order. As they closely packed on substrates, from top to bottom, a concentration gradient exists, and the gravity influence should not be neglected. On the gravity influence, the short-range order parts of ACC must be compact to attach to each other. Once the ordered part got closer, the nucleation barrier would be reduced, then nucleation occurred in accordance with the classical nucleation theory.39 So nucleation may be a result of the interactions between substrates and ACC films (that is, internal stress) or the effect of gravity or the synergetic effect of both internal stress and gravity. It is still an open question to determine which one is the key factor.
3.4 Crystal growth
Two main mechanisms were proposed to explain how crystals formed, i.e. so called classical and non-classical crystallization. In the classical mechanism, crystallizaiton is based on the atom/molecule, after nucleation, the crystal grows further via a ion-by-ion attachment and unit cell replication.40 In contrast, non-classical crystallization takes particles as mediates, and crystal growth goes through oriented attachment or mesocrystal formation.41,42 The key to distinguish these two mechanisms is to capture the intermediates in the crystallization process.By taking out substrates at different time intervals, we did obtain the intermediates in ACC transformation, thus it is helpful to elucidate the formation mechanism of the final crystals.
The intermediates were captured at the same time with nucleation sites. They have a very short life of <1 h. SEM images of the sample are shown in Fig. 8. Rods (Fig. 8A), dumbells (Fig. 8C, E), semi-attached spheres (Fig. 8G), and whole spheres (Fig. 8I) were found on same substrate. These results were also found in other systems, such as CaCO3,43 BaCO3,44 fluoroapatite,45 BaSO4,46 SrC2O4,47 Bi2S348 and so on. They all showed a growth process from rods to dumbells and finally to spheres. Thus, the intermediates here may also grow through this rods–dumbells–spheres process. The difference here is that the intermediates took the place of the final crystals. Detailed images of these intermediates were shown in Fig. 8 left. Most of them have a huge amount of fibers or analogues around the surfaces. Thus, from structure similarity, it is presumed that the intermediates’ growth process is similar to fluoroapatite growth via hexanol prismatic seeds in a gelatin gel revealed by Kniep and coworkers.49,50 That is, the crystals grow via mesocrystal mediates after nucleation. The first formed intermediates are tiny rods composed of nanoparticles and stabilized by selected adsorption of H4dhpta molecules, and the rods can serve as seeds for further growth. After the rods’ formation, spliting growth which is possibly directed by intrinsic electric fields works at both ends of the rods, then dumbell-like and finally sphere-like particles are formed.51–53
![SEM images of intermediates obtained after three-and-a-half days, [Ca2+]/[H4dhpta] = 4 : 1, the right-column images are a magnification of the left-column ones. The morphology transforms from fibres (D) to prisms (F), indicating a non-classical crystallization process.](/image/article/2010/CE/b911426m/b911426m-f8.gif) | ||
| Fig. 8 SEM images of intermediates obtained after three-and-a-half days, [Ca2+]/[H4dhpta] = 4:1, the right-column images are a magnification of the left-column ones. The morphology transforms from fibres (D) to prisms (F), indicating a non-classical crystallization process. | ||
The basic units of these intermediates, fibres, are not quite the same. Some are very thin (Fig. 8B, D, J), some are more thick and round (Fig. 8F) and some seem to begin to crystallize and clear boundaries are shown (Fig. 8H). This must be the ripening process that occurred after intermediates formed and reached their critical sizes. From the morphology similarity (Fig. 8D, F, H), it is obvious to see a clear morphology transformation process from fibres to prisms. Thus the classical crystallization process is not suitable here, as unit cell replication must be prisms at first, not last. We can presume that fibres are the basic units for the crystal ripening process, and they orient along the diagonal line of the prisms. Then, after they reach the critical size, the transformation occurs. From the unclear boundaries in Fig. 8F, it can be assumed that this ripening process is also a nanoparticles’ self-assembly process, as an ion-by-ion attachment must show a clear unit cell boundary.54 Thus, all intermediates and their transformation process come from mesocrystal formation.
After 4 d of precipitation, crystals with prismatic boundaries were obtained. In a molar ratio of [Ca]/[H4dhpta] = 4:1, rods, dumbells, semi-attached spheres and spheres with a similar size of the intermediates were also found (Fig. 9), indicating a ripening and in situ crystallization process of the intermediates. The fracture of one-half of a dumbell broken perpendicularly (Fig. 9D) shows that the center represents particles rather than rods, thus indicating a particle’s self-assembly process again. As in the molar ratio of [Ca]/[H4dhpta] = 2:1, rods were the only shape that existed (Fig. 1A), then it is convincible that the coordination effect of H4dhpta with calcium ions is the key factor to control crystal growth and this must be the origin of selective adsorption.
![SEM images of crystals collected after 4 d, [Ca2+]/[H4dhpta] = 4 : 1.](/image/article/2010/CE/b911426m/b911426m-f9.gif) | ||
| Fig. 9 SEM images of crystals collected after 4 d, [Ca2+]/[H4dhpta] = 4:1. | ||
However, after two weeks’ overgrowth, typical Ostwald ripen phenomena were observed.55 That is, at a molar ratio of [Ca]/[H4dhpta] = 4:1, spheres were the dominate morphology, and other morphologies seemed to be reduced or vanished (Fig. 10A). At a molar ratio of [Ca]/[H4dhpta] = 2:1, the dominant rods were on the scale of more than 50 µm, and the small rods about 20 µm long seemed to have dissolved from the inside gradually and only a framework was left (Fig. 10B, C, D).
![SEM images of crystals collected after two weeks, (A) [Ca2+]/[H4dhpta] = 4 : 1, (B), (C), (D) [Ca2+]/[H4dhpta] = 2 : 1.](/image/article/2010/CE/b911426m/b911426m-f10.gif) | ||
| Fig. 10 SEM images of crystals collected after two weeks, (A) [Ca2+]/[H4dhpta] = 4:1, (B), (C), (D) [Ca2+]/[H4dhpta] = 2:1. | ||
3.5 Mechanism
The pH of the reaction solution was monitored during experiments. At a molar ratio of [Ca]/[H4dhpta] = 4:1, the initial pH of the reaction solution was around 2.50. One day later, the pH of the reaction solution was 9.14 ± 0.03 after the precipitation of ACC. Then the pH value of the reaction solution increased slightly as time went by. After 2 d, the pH of the solution was 9.25 ± 0.04. When crystallization began after three-and-a-half days, the pH value was 9.33 ± 0.02. After 4 d, the pH value was 9.48 ± 0.01. According to the results by Gebauer et al,36 a more stable type of ACC (ACCI) precipitated at a high binding strength in the cluster (the range of the pH values is from 9.00 to 9.50). Thus, in our experiments, the ACC formed here belongs to ACCI. The present results just prove that this type of ACC does exhibit a calcite short-range order and transforms finally into calcite.
It has been reported that the alkaline earth metal complexes with H4dhpta as well as other EDTA-type alkaline earth metal complexes have a 1:1 mononuclear structure in solution.56 The stability constant for the complex of H4dhpta with Ca2+ is around 10−6,57,58 which means the complex is more stable than CaCO3. Thus, it is reasonable that no crystals can be formed at a 1:1 molar ratio of H4dhpta to Ca2+ due to the strong coordination ability of H4dhpta with the Ca2+ ions. Then, in a solution with excess Ca2+ ions, H4dhpta can only coordinate part of the Ca2+ ions, and in the participation of this complex, ACC is stabilized. This complex is possibly adsorbed on the surface of ACC and the final calcite crystals, as is clear from the results that ACC dissolves from the inside (Fig. 5D) and unstable calcite crystals dissolve from the inside too (Fig. 10D). Thus, the crystallization process may be controlled by selective adsorption besides the mesocrystal transformation.
The schematic mechanism is shown in Fig. 11. First, amorphous calcium carbonate nanoparticles are formed homogeneously in the solution (Fig. 11A), and crystallization is based on these ACC nanoparticles. These ACC nanoparticles have a great affinity towards each other. In solution, without any boundaries, these particles will aggregate together into sphere-like agglomerates (Fig. 11B, upper). Also, they have a very strong affinity towards hydrophilic substrates and form a close packed film on the substrate (Fig. 11B, down). Under gravity control and/or internal stress, some nucleations occur on the film (Fig. 11D). These nuclei are more stable than ACC, and ACC aggregates dissolve from inside to release calcium and carbonate ions (Fig. 11C). After nucleation, organic molecules play an important role in the crystal growth stage. By coordination, organic molecules selectively adsorbed on some coaxis faces, which slowed down the interfacial energy of these faces. Then, to eliminate the high energy face, those nanoparticles self-assembled along the same axis and a basic rod unit formed (Fig. 11E). After the rod unit formed, a possible splitting growth occurs under the control of intrinsic electric fields.51–53,59
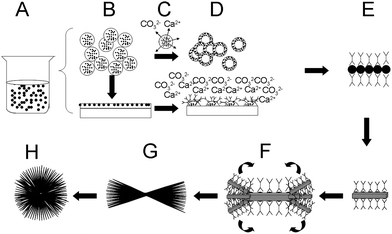 | ||
| Fig. 11 Schematic illustrations of the transition process from ACC to hierarchical crystals. | ||
Then dumbells and spheres with a fibre surface could be formed (Fig. 11F–H). However, the stabilization ability of H4dhpta is limited and these faces stabilized by H4dhpta are very unstable. When these fibres reach their critical sizes, other more stable faces are displayed as a result of particle assembly. Though crystallized well, these are still not the final morphology. Overgrowth occurs with time, and the Ostwald rules become the dominant control factor. That is, bigger spheres or rods are retained and grow even bigger, unstable morphologies like little rods and dumbells are dissolved to give ions for a more stable crystal growth and the dissolution process begins from the inside, too.
4. Conclusions
In conclusion, we found that H4dhpta, a low molecule weight organic molecule with carboxyl functional groups, could stabilize amorphous calcium carbonate. By interrupting the reaction at different time intervals, a whole crystallization process was successfully observed. It is found that the first formed ACC has two forms. One exists in solution and conglomerates into sphere-like precipitates. The other forms a close packed film on the substrate. The film is easily ignored as it is so dense and flat that it could be recognized as a background of the precipitate just like substrates, also for its disappearance after the crystallization finishes. However, this film is the key to inducing heterogeneous nucleation, as precipitated ACC conglomerates dissolve during the crystallization process. As various ACC films have been found, we suggest that the nucleation on ACC films may be the common behavior for heterogeneous nucleation. Thus, it is possible to combine dissolution–recrystallization26 and the substrate physical change theory27 into one mineralization system.After the nucleation, the intermediates at different crystallization stages were also captured. These intermediates were found to be composed of fibres. A rods–dumbells–spheres transformation phenomenon was observed. It is believed that these are frameworks for the final crystal formation. In situ transformation of each fibre is brought forward to explain the formation of the final similar calcite hierarchical structures. As H4dhpta here could both serve as ACC stabilizer and crystallization controller, it is expected to act as the ideal molecule for studying the way to stabilize ACC, and the crystallization and transformation process of ACC.
Acknowledgements
S.-H.Y. acknowledges funding support from the National Science Foundation of China (NSFC) (Grants Nos. 50732006, 20621061, 20671085), the National Basic Research Program of China (2010CB934700), and the Partner-Group of the Chinese Academy of Sciences—the Max Planck Society.References
- S. Raz, P. C. Hamilton, F. H. Wilt, S. Weiner and L. Addadi, Adv. Funct. Mater., 2003, 13, 480 CrossRef CAS.
- Y. Levi-Kalisman, S. Raz, S. Weiner, L. Addadi and I. Sagi, Adv. Funct. Mater., 2002, 12, 43 CrossRef CAS.
- S. Weiner, I. Sagi and L. Addadi, Science, 2005, 309, 1027 CrossRef CAS.
- F. M. Michel, J. MacDonald, J. Feng, B. L. Phillips, L. Ehm, C. Tarabrella, J. B. Parise and R. J. Reeder, Chem. Mater., 2008, 20, 4720 CrossRef CAS.
- H. Cölfen, Angew. Chem., Int. Ed., 2008, 47, 2351 CrossRef.
- J. Aizenberg, D. A. Muller, J. L. Grazul and D. R. Hamann, Science, 2003, 299, 1205 CrossRef CAS.
- C. Li and L. M. Qi, Angew. Chem., Int. Ed., 2008, 47, 2388 CrossRef CAS.
- N. Koga, Y. Nakagoe and H. Tanaka, Thermochim. Acta, 1998, 318, 239 CrossRef CAS.
- M. Faatz, F. Gröhn and G. Wegner, Adv. Mater., 2004, 16, 996 CrossRef CAS.
- I. Sondi and E. Matijević, J. Colloid Interface Sci., 2001, 238, 208 CrossRef CAS.
- S. E. Wolf, J. Leiterer, M. Kappl, F. Emmerling and W. Tremel, J. Am. Chem. Soc., 2008, 130, 12342 CrossRef CAS.
- Y. Yamamoto, T. Nishimura, A. Sugawara, H. Inoue, H. Nagasawa and T. Kato, Cryst. Growth Des., 2008, 8, 4062 CrossRef.
- E. M. Pouget, P. H. H. Bomans, J. A. C. M. Goos, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Science, 2009, 323, 1455 CrossRef CAS.
- R. K. Pai and S. Pillai, CrystEngComm, 2008, 10, 865 RSC.
- Y. Chen, J. Xiao, Z. Wang and S. Yang, Langmuir, 2009, 25, 1054 CrossRef CAS.
- L. Dai, X. Cheng and L. B. Gower, Chem. Mater., 2008, 20, 6917 CrossRef CAS.
- T. Y. Han and J. Aizenberg, Chem. Mater., 2008, 20, 1064 CrossRef CAS.
- J. T. Han, X. Xu, D. H. Kim and K. Cho, Chem. Mater., 2005, 17, 136 CrossRef CAS.
- Y. Kim, A. N. Kulak, Y. Li, T. Batten, M. Kuball, S. P. Armesb and F. C. Meldrum, J. Mater. Chem., 2009, 19, 387 RSC.
- R. S. K. Lam, J. M. Charnock, A. Lenniec and F. C. Meldrum, CrystEngComm, 2007, 9, 1226 RSC.
- Y. Nishino, Y. Oaki and H. Imai, Cryst. Growth Des., 2009, 9, 223 CrossRef CAS.
- A. W. Xu, Q. Yu, W. F. Dong, M. Antonietti and H. Cölfen, Adv. Mater., 2005, 17, 2217 CrossRef CAS.
- Y. Oaki, S. Kajiyama, T. Nishimura, H. Imai and T. Kato, Adv. Mater., 2008, 20, 3633 CrossRef CAS.
- A. Cai, X. Xu, H. Pan, J. Tao, R. Liu, R. Tang and K. Cho, J. Phys. Chem. C, 2008, 112, 11324 CrossRef CAS.
- L. Dai, E. P. Douglas and L. B. Gower, J. Non-Cryst. Solids, 2008, 354, 1845 CrossRef CAS.
- J. Rieger, T. Frechen, G. Cox, W. Heckmann, C. Schmidt and J. Thieme, Faraday Discuss., 2007, 136, 265 RSC.
- I. Sethmann, A. Putnis, O. Grassmann and P. Lobmann, Am. Mineral., 2005, 90, 1213 CrossRef CAS.
- L. B. Gower, Chem. Rev., 2008, 108, 4551 CrossRef CAS.
- M. Niederberger and H. Cölfen, Phys. Chem. Chem. Phys., 2006, 8, 3271 RSC.
- S. B. Mukkamala and A. K. Powell, Chem. Commun., 2004, 918 RSC.
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959 CrossRef CAS.
- F. A. Andersen and L. Brecevic, Acta Chem. Scand., 1991, 45, 1018 CrossRef CAS.
- H. Nebel, M. Neumann, C. Mayer and M. Epple, Inorg. Chem., 2008, 47, 7874 CrossRef CAS.
- Y. Politi, T. Arad, E. Klein, S. Weiner and L. Addadi, Science, 2004, 306, 1161 CrossRef CAS.
- C. Zhong and C. C. Chu, Langmuir, 2009, 25, 3045 CrossRef CAS.
- D. Gebauer, A. Völkel and H. Cölfen, Science, 2008, 322, 1819 CrossRef CAS.
- X. Y. Liu and S. W. Lim, J. Am. Chem. Soc., 2003, 125, 888 CrossRef CAS.
- Y. Levi-Kalisman, S. Raz, S. Weiner, L. Addadi and I. Sagi, J. Chem. Soc., Dalton Trans., 2000, 3977 RSC.
- A. Laaksonen, V. Talanquer and D. W. Oxtoby, Annu. Rev. Phys. Chem., 1995, 46, 489 CrossRef CAS.
- G. Wulff, Z. Kristallogr., 1901, 34, 449 CAS.
- M. Niederberger and H. Cölfen, Phys. Chem. Chem. Phys., 2006, 8, 3271 RSC.
- H. Cölfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350 CrossRef.
- S. H. Yu, H. Cölfen, J. Hartmann and M. Antonietti, Adv. Funct. Mater., 2002, 12, 541 CrossRef CAS.
- S. H. Yu, H. Cölfen and M. Antonietti, J. Phys. Chem. B, 2003, 107, 7396 CrossRef CAS.
- R. Kniep and S. Busch, Angew. Chem., Int. Ed. Engl., 1996, 35, 2624 CrossRef CAS.
- L. Qi, H. Cölfen and M. Antonietti, Chem. Mater., 2000, 12, 2392 CrossRef CAS.
- D. Zhang, L. Qi, J. Ma and H. Cheng, CrystEngComm, 2002, 4, 536 RSC.
- J. Tang and A. P. Alivisatos, Nano Lett., 2006, 6, 2701 CrossRef CAS.
- S. Busch, U. Schwarz and R. Kniep, Adv. Funct. Mater., 2003, 13, 189 CrossRef CAS.
- H. Tlatlik, P. Simon, A. Kawska, D. Zahn and R. Kniep, Angew. Chem., Int. Ed., 2006, 45, 1905 CrossRef CAS.
- S. Busch, H. Dolhaine, A. DuChesne, S. Heinz, O. Hochrein, F. Laeri, O. Podebrad, U. Vietze, T. Weiland and R. Kniep, Eur. J. Inorg. Chem., 1999, 1643 CrossRef.
- T. X. Wang, H. Cölfen and M. Antonietti, J. Am. Chem. Soc., 2005, 127, 3246 CrossRef.
- G. Y. Chen, B. Dneg, G. B. Cai, T. K. Zhang, W. F. Dong, W. X. Zhang and A. W. Xu, J. Phys. Chem. C, 2008, 112, 672 CrossRef CAS.
- Q. Zhang, S. J. Liu and S. H. Yu, J. Mater. Chem., 2009, 19, 191 RSC.
- W. F. Ostwald, J. Phys. Chem., 1897, 22, 289 CAS.
- G. Schwarzenbach, G. Anderegg, W. Schnmder and H. Senn, Helv. Chim. Acta, 1955, 38, 1147 CrossRef CAS.
- J. H. Grimes, A. J. Huggard and S. P. Wilford, J. Inorg. Nucl. Chem., 1963, 25, 1225 CrossRef CAS.
- L. C. Thompson and S. K. Kundra, J. Inorg. Nucl. Chem., 1966, 28, 2945 CrossRef CAS.
- T. X. Wang, M. Antonietti and H. Cölfen, Chem.–Eur. J., 2006, 12, 5722 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |