Influence of the steric hindrance of organic amines on the supramolecular network based on polyoxovanadates†
Zhihui
Yi
b,
Xiaoyang
Yu
c,
Wujiong
Xia
a,
Liyan
Zhao
a,
Chao
Yang
a,
Qian
Chen
a,
XiaoLin
Wang
a,
Xianzhu
Xu
a and
Xiao
Zhang
*a
aState Key Lab of Urban Water Resource and Environment (SKLUWRE) & Academy of Fundamental and Interdisciplinary Science, Harbin Institute of Technology, Harbin, Heilongjiang 150080, P. R. China. E-mail: zhangx@hit.edu.cn; Fax: +086-45186403193; Tel: +086-45186403193
bState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, P. R China
cJiLin Institute of Chemical Technology, Jilin City, 132022, P. R. China
First published on 4th September 2009
Abstract
Three new supramolecular architectures based on polyoxovanadates H6V10O28·2HMTA·8H2O(1), [Ni(enMe)3]2(VIV8VV7O36Cl)·2H3O (2) and [Co(en)3]2(VIV8VV7O36Cl)·2H3O (3) (HMTA = hexamethylenetetramine, enMe = 1,2′-propanediamine and en = ethylenediamine) have been prepared and characterized by elemental analyses, IR spectra, XPS spectra and single crystal X-ray diffraction structure analyses. Compound 1 displays a 3-D (4,8)-connected supramolecular host network, which is constructed of octamer water clusters and HMTA molecules, with 1-D channels occupied by the template [H6V10O28] units. Compound 2 possesses a 2-D (3,6)-connected supramolecular layered structure formed by water molecules and [V15O36]3− units showing honeycomb cavities occupied by the transition metal complexes (TMCs). Compound 3 exhibits a 3-D (4,6)-connected supramolecular host network built by the [V15O36]3− units and the lattice water molecules with TMCs cations as the template. By a careful inspection of the structures of 1–3, it is believed that the steric hindrances of organonitrogen ligands are important for the formation of different supramolecular networks. Compound 1 exhibits intensive photoluminescence and compound 3 displays antiferromagnetic interactions.
1. Introduction
Crystal engineering of organic–inorganic hybrid materials has provoked a significant interest originating not only from their diverse structural flexibility, but also from their widely promising potential applications in catalysis, medicine, photochemistry and electromagnetism.1 In crystal engineering, a remarkable area is supramolecular chemistry which has been defined as the chemistry of the intermolecular bond, covering the structures and functions of the entities formed by association of two or more chemical species by Lehn. A challenge in the supramolecular chemistry is the design and synthesis of host molecules capable of binding various guests including cations, anions, and neutral molecules. In this regard, the synthetic and structural aspects of cavities capable of binding small guest molecules have attracted increasing attention.2Inorganic–organic hybrids have emerged as a major research area for the rational design of functional materials during the past decades due to the wide range of applications.3 Polyoxometalates (POMs), as one kind of significant metal oxide clusters with nanosizes and abundant topologies, have recently been employed as inorganic building blocks for constructing supramolecular arrays with various organic ligands.4 Supramolecular assemblies based on POMs have been intensively investigated in many important aspects of chemistry such as catalysis, non-linear optical material, and medicine.5 Polyoxovanadates (POVs) are a prominent subclass of POMs, as compared to polyoxomolybdates and polyoxotungstates, are relatively under-investigated.6 In recent years, an important advance on POVs has been the study of supramolecular chemistry.7 In most cases, the available nucleophilic surface of oxygen atoms on POVs interacts with the donor of an organic ligand to form O–H⋯O or N–H⋯O hydrogen bonds for constructing a high-dimensional supramolecular network. In contrast, less attention was paid to the regulatory effects of POVs units, TMCs and organonitrogen ligands in the supramolecular network,8 despite these having been demonstrated to play a key role in controlling the topological structure of supramolecular architectures.
As a continuous work, we report three new supramolecular networks based on POVs herein, H6V10O28·2HMTA·8H2O (1), [Ni(enMe)3]2(VIV8VV7O36Cl)·2H3O (2) and [Co(en)3]2(VIV8VV7O36Cl)·2H3O (3). Compound 1 displays a 3-D (4,8)-connected host supramolecular network with the Schläfli symbol (412·612·84)(46)2.9 1-D column channels formed by octamer water clusters adopting the ‘chair’ configuration and HMTA molecules were found, in which [H6V10O28] units acting as the template are located. Compound 2 possesses a 2-D (3,6)-connected supramolecular layered structure with the Schläfli symbol (43·612)(43)2 formed by water molecules and [V15O36] units showing honeycomb cavities which were filled by the transition metal complexes (TMCs) [Ni(enMe)3]2+ as the template.9 Compound 3 displays a 3-D (4,6)-connected host network with the Schläfli symbol (43·612)(43·63)2 constructed from [V15O36] units and lattice water molecules with [Co(en)3]2+ cations as the template.9 These results show that the lattice water molecules, POVs units and the steric hindrance of organonitrogen ligands are important for the formation of different supramolecular networks.
2. Experimental
2.1 Materials and measurements
All reagents used were of analytical grade and obtained from commercial sources without further purification. The elemental analysis (C, H and N) was performed on a Perkin–Elmer 2400 CHN Elemental Analyzer. Inductively coupled plasma (ICP) analysis was conducted on a Perkin-Elmer Potima 3300DV spectrometer. IR (KBr pellets) spectrum was recorded with Perkin–Elmer Spectrum One FTIR spectrophotometer in the 300–4000 cm−1 regions using a powdered sample on the KBr plate. XPS analysis was performed on Thermo ESCALAB 250 spectrometer with an Mg Kα (1253.6 eV) achromatic X-ray source. Thermogravimetric analysis (TGA) was carried out on a Perkin-Elmer TAG-7 instrument from rt to 750 °C with a heating rate of 10 °C min−1. Variable temperature magnetic susceptibility measurements were performed on a Quantum Design MPMS XL-5 SQUID magnetometer. The fluorescence spectra are determined with a FluoroMax-4 (JobinYvon) fluorimeter.2.2 Syntheses
2.3 X-Ray crystallography
Single crystals of complexes 1–3 were glued on glass fibers at 293 K. Data were collected on a Rigaku RAXIS–RAPID image plate area detector using graphite monochromated Mo Kα diffraction (λ = 0.71073 Å). The structures were solved by the direct method and refined by the full–matrix least-squares method on F2 using the SHELXTL 97 crystallographic software package.10 All non–hydrogen atoms were refined anisotropically. For compounds 1 and 2, the hydrogen atoms of the ligands were localized in their calculated positions and refined using a riding model. The hydrogen atoms of the ligands in 3 were not treated owing to disorder. The hydrogen atoms of the lattice water molecules were not treated. Further details of the X-ray structural analyses are given in Table 1. Selected bond lengths and angles are listed in Table S1.†| 1 | 2 | 3 | |
|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR 2 = |∑w(|Fo|2 − |Fc|2)2/∑|w(Fo2)2|1/2. | |||
| Empirical formula | C12H46N8O36V10 | C18H66ClN12Ni2O38V15 | C12H54ClCo2N12O38V15 |
| Formula weight | 1387.97 | 1975.80 | 1892.08 |
| Crystal system | Monoclinic | Hexagonal | Hexagonal |
| Space group | C2/m | P63/m | P63/mmc |
| a/Å | 14.957(3) | 12.6031(9) | 12.1200(17) |
| b/Å | 16.879(3) | 12.6031(9) | 12.1200(17) |
| c/Å | 9.0167(18) | 22.563(3) | 22.053(4) |
| β/° | 115.94(3) | ||
| V/Å3 | 2047.0(7) | 3103.7(6) | 2805.5(8) |
| Z | 2 | 2 | 2 |
| D c /Mg m−3 | 2.252 | 2.114 | 2.240 |
| µ/mm−1 | 2.295 | 2.870 | 3.090 |
| F(000) | 1384 | 1960 | 1860 |
| Reflections collected | 10113 | 13151 | 22189 |
| Independent reflections | 2418 | 2074 | 1264 |
| Goodness-of-fit on F2 | 1.084 | 1.075 | 1.055 |
| Final Ra,b indices [I > 2σ(I)] | R 1 = 0.0351, wR2 = 0.1039 | R 1 = 0.0678, wR2 = 0.2038 | R 1 = 0.0788, wR2 = 0.2300 |
| R indices (all data) | R 1 = 0.0396, wR2 = 0.1068 | R 1 = 0.0706, wR2 = 0.2070 | R 1 = 0.0877, wR2 = 0.2403 |
3. Results and discussion
3.1 Synthesis
POVs can be regarded as synthons (chemically reasonable molecules) participating in a supramolecular reaction and influencing the supramolecular interactions to dictate a particular crystal packing. However, it is very difficult to rationally design and synthesize the high-dimensional supramolecular networks based on POVs, because many factors could influence the formation of POVs, such as initial reactants, initial concentrations, pH value, reaction time and temperature. In our case, the pH values greatly influence the structures of the products. Compound 1 was obtained at ca. pH 4, compounds 2 and 3 were obtained at pH ≈ 5. The results show that the pH value is possibly one of the important factors controlling the structures of the synthesized compounds. Some examples11 have proven that while pH > 7, the dominant building blocks for the formation of POVs are tetrahedral VO4, with the pH value decreasing, square–pyramidal VO5 and octahedral VO6 will successively dominate in an aqueous vanadate solution. These building blocks should directly determine the structures of POVs. As a further proof, there is no surprise that compound 1 contains {V10} building blocks formed from octahedral VO6 units under a pH value of ca. 4, and compounds 2 and 3 include {V15} building blocks formed from pyramidal VO5 units under ca. pH 5, respectively.Additionally, it is noteworthy that the fully oxidized NH4VO3 was used as the V source, but part of the V atoms in compounds 2 and 3 are in the reduced +4 state. As we did not use other reducing agents in the reactive mixtures, it suggests that the enMe and en ligand act as effective agents to reduce partial VV into VIV. To prove this inference, Ba(NO3)2 solution was dropped into the resultant filtrate, and white precipitate could be obtained. This suggests that some of the enMe and en ligand were oxidized into CO2 under hydrothermal conditions. Organic amine ligands acting as reducing agents under hydrothermal conditions are common in the preparation of the polyoxomolybdates and polyoxotungstates, however this comparatively rare in POVs.4d,12
3.2. Crystal structures
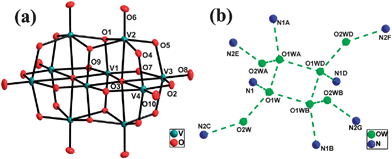 | ||
| Fig. 1 (a) Structure and labeling scheme for the poiyoxovanadate anion in 1. (b) Structure and labeling scheme for the octamer water cluster and its immediate environment in 1.[Symmetry codes: (A) x, −y, z; (B) 1 − x, y, l − z; (C) 1/2 − x, 1/2 − y, −z; (D) l − x, −y, l − z; (E) 1/2 − x, y − 1/2, −z; (F) x + 1/2, y − 1/2, l + z; (G) x + 1/2, 1/2 − y, l + z. | ||
The most interesting structural feature of 1 is the novel 3-D supramolecular network with 1-D column channels assembled from water octamers and HMTA molecules (Fig. 2). Specifically, the O1W and its symmetry related ones are self-assembled through hydrogen bonds to form a cyclic tetramer with the distances of O1W⋯O1WA 2.813(4) Å and O1W⋯O1WB 2.783(3) Å [symmetry codes: (A) x, −y, z; (B) 1 − x, y, 1 − z]. The tetramer is decorated with O2W and its three symmetric equivalents to form an octamer with distances of O1W⋯O2W 2.859(6) Å (Fig. 1b and Table 2). The average OW⋯OW distance is 2.829 Å, which is in the range of 2.77 Å (in the ice II phase) to 2.85 Å (in liquid water).14 Each octamer links eight HMTA molecules and each HMTA molecule links four octamers via the hydrogen bonds between the water molecules and the N atoms of the HMTA molecules with the distance of O1W⋯N1 2.865(6) Å and the distance of O2W⋯N2C 2.758(5) Å [Symmetry code: (C) 1/2 − x, 1/2 − y, −z]. In this way, a 3-D (4,8)-connected supramolecular host network with the Schläfli symbol (412·612·84)(46)2 is formed (Fig. 2).9 This network possesses the 1-D column channels running parallel to the c axis, which are occupied by [H6V10O28] units as the template. In the topological structure, the [V10O28] unit is located at the 412 cavity formed by twelve rectangles (Fig. 3). To the best of our knowledge, compound 1 is the first example that [V10O28] units acting as the template fill in the 3-D supramolecular network. Additionally, the hydrogen bonds between the octamer and [H6V10O28] cluster with the distance of O5⋯O2W 2.798(6) Å are found, which further stabilized the overall 3-D supramolecular network.
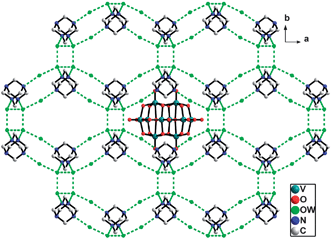 | ||
| Fig. 2 View down the c axis of the 3-D supramolecular network of compound 1, which is constructed of the octamer water clusters and HMTA molecules, with 1-D tunnels occupied by the polyanions as the template reagent. | ||
| a Symmetry codes for 1: (A) x, −y, z; (B) 1 − x, y, 1 − z; (C) 1/2 − x, 1/2 − y, −z; for 2: (A) x − 1, y − 1, z; for 3: (A) 1 − x, 1 − y, 1/2 − z, (B) x − y, −y, 1 − z. | |||
|---|---|---|---|
| Compound 1 | |||
| O1W⋯O1WA | 2.813(4) | O1W⋯O1WB | 2.783(3) |
| O1W⋯O2W | 2.859(6) | O5⋯O2W | 2.798(6) |
| N1⋯O1W | 2.865(6) | N2C⋯O2W | 2.758(5) |
| Compound 2 | |||
| O1W⋯O2A | 2.932(8) | ||
| Compound 3 | |||
| O1W⋯O5A | 2.919(22) | O1W⋯O1WB | 3.008(59) |
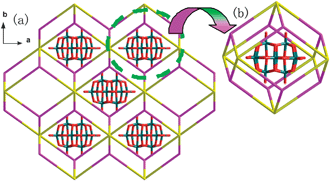 | ||
| Fig. 3 (a) Schematic representation of the (4,8)-connected supramolecular topological network with the Schläfli symbol (412·612·84)(46)2 of 1 (eight-connected nodes (the octamer water cluster) are depicted in purple and four-connected nodes (HMTA) in yellow). (b) Schematic representation of the cavity formed by the twelve rectangles filled by the polyanion. | ||
It is noteworthy that the geometry of the octamer water cluster predicted from energetic considerations is a cube with S4 or D2d symmetries.15 In 1, the octamer water cluster with a “chair” shape can be considered as a cubic arrangement destroyed by the double effects of the steric hindrance of HMTA molecules and the hydrogen bond between N2 and O2W. Specifically, N1 and N1A atoms of HMTA occupy two corners of a cube with the S4 or D2d symmetries and force the O2W and O2WA into the opposite position. At the same time, the hydrogen bonds N2C⋯O2W and N2E⋯O2WA trapped the O2W and O2WA, and stabilized the “chair” shape, therefore a “chair” shape formed. The oxygen atoms of the octamer and the nitrogen atoms trapped by the water cluster both present C2h symmetry in 1. This is noteworthy, since the C2h form of (H2O)8 is less stable than the D2d or S4 forms which have never been observed previously.16
The most interesting structural feature of 2 is the formation of a 2-D supramolecular sheet with a honeycomb cellular structure assembled by free water molecules and polyanions (Fig. 4a and Table 2). The detailed analysis shows that there is only one kind of hydrogen bonding between the lattice water molecules (O1W) and the terminal oxygen atoms (O2) of POVs [O1W⋯O2A 2.932(8) Å symmetry code: (A) x − 1, y − 1, z]. The O2 and its five symmetry related ones in a [V15O36Cl]6− anion link six water molecules and each lattice water links three adjacent [V15O36Cl]6− anions to form a 2-D (3,6)-connected supramolecular layered structure with the Schläfli symbol (43·612)(43)2 (Fig. 4).9 The angle of O2A⋯O1W⋯O2B [symmetry code: (A) x − 1, y − 1, z; (B) y –x, 1 – x, z] is 114.94(23)°, thus distorted honeycomb cavities formed from three polyanions and six lattice water were found, in which the cation complexes [Ni(enMe)3]2+ serving as the ‘guest’ units were located. It is interesting to realize that the 3-D packing view of 2, as shown in Fig. 5, exhibits pseudo channels occupied by lattice water molecules running parallel to the c-axis which was formed by spatial packing of {V15} units. The shortest distance between two adjacent sheets is about 11.28 Å (Fig. 5). To the best of our knowledge, compound 2 is the first example that the transition metal coordination complexes as guest molecules occupy the honeycomb cellular structure of the supramolecular networks formed by POVs.
![(a) View down the c axis of the 2-D hydrogen-bonded host supramolecular framework constructed from POVs and lattice water molecules with the honeycomb cellular structure filled by the template [Ni(enMe)3]2+ cations. (b) Schematic representation of the (3,6)-connected supramolecuar topological network with the Schläfli symbol (43·612)(43)2 of 2 (three-connected nodes (lattice water is depicted in green and six-connected nodes POVs is golden yellow.).](/image/article/2010/CE/b916793p/b916793p-f4.gif) | ||
| Fig. 4 (a) View down the c axis of the 2-D hydrogen-bonded host supramolecular framework constructed from POVs and lattice water molecules with the honeycomb cellular structure filled by the template [Ni(enMe)3]2+ cations. (b) Schematic representation of the (3,6)-connected supramolecuar topological network with the Schläfli symbol (43·612)(43)2 of 2 (three-connected nodes (lattice water is depicted in green and six-connected nodes POVs is golden yellow.). | ||
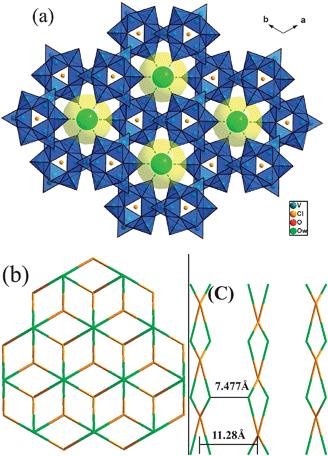 | ||
| Fig. 5 (a) Packing diagram of 2 down the c axis. The POVs are shown as polyhedrons, and the lattice water molecules as space-filling spheres. (b) The topological network of 2 in the ab plane. (c) A scheme showing the O1W⋯O1WC belonging to an adjacent sheet with a distance 7.477(3) Å in 2. | ||
It is noteworthy that compound 3 displays a 3-D supramolecular network. Detailed analysis shows that there are two kinds of hydrogen bonds between lattice water and lattice water, lattice water and O atoms (from POVs). Similar to 2, each polyanion links six lattice waters through the hydrogen bonds between O1W molecules and terminal oxygen atoms with O1W⋯ O5A 2.919(22) Å [symmetry code: (A) 1 − x, 1 − y, 1/2 − z], and the lattice water molecule links three POVs to form a 2-D (3,6)-connected supramolecular sheet (Fig. 5a, b and Table 2). However, the lattice water is a four-connected node in 3, not only linking three terminal oxygen atoms of POVs, but also linking one lattice water from an adjacent supramolecular sheet with O1W⋯O1WB 3.008(59) Å [symmetry code: (A) 1 − x, 1 − y, 1/2 − z]. Therefore, the 2-D sheet was connected by the lattice water to form a 3-D (4,6)-connected host network with the Schläfli symbol (43·612)(43·63)2 (Fig. 6).9 The 1-D tunnels along the a axis which are filled with TMCs are found in the host network.
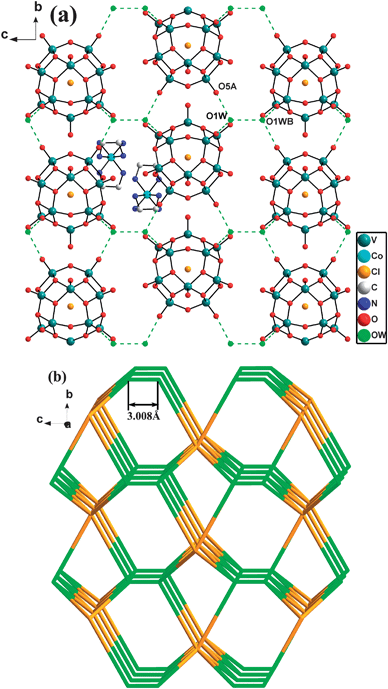 | ||
| Fig. 6 (a) View down the c axis of the 3-D supramolecular network of 3. Symmetry codes: (A) 1 − x, 1 − y, 1/2 − z, (B) x − y, −y, 1 − z. (b) Schematic representation of the (4,6)-connected supramolecular topological network with the Schläfli symbol (43·612)(43·63)2 of 3 (three-connected nodes (lattice water is depicted in green and six-connected nodes POVs is golden yellow). | ||
As can be seen in the structures of compounds 1–3, all these ones contain POVs anions, lattice water molecules and organic/TMC units, though there are no direct interactions occurring among the POVs anions, high-dimensional supramolecular networks are formed through hydrogen bonds. The host supramolecular network was constructed from hydrogen bonding interactions between water molecules and free organic ligands (in 1) or polyanions (in 2 and 3). The key role of water molecules in the supramolecular network stands out. Meanwhile, the types of polyanions and organic ligands profoundly influence the overall supramolecular network. First of all, it is noteworthy that there are two different types of POVs serving as building blocks in the three compounds, i.e. [V10O28] in 1 and [V15O36] in 2 and 3. The volume of [V10O28] is smaller than [V15O36] which makes a [V10O28] unit easier to act as a guest filling in the supramolecular network and a [V15O36] unit to serve as a six-connected nod for constructing the host supramolecular network. On the other hand, a noticeable feature of 1–3 is that the steric hindrance influences of organic amine ligands are crucial for the dimensionality of the supramolecular networks. In 1, the steric hindrance of the HMTA ligand forces the octamer water cluster to form the ‘chair’ configuration, further influence the overall supramolecular structure. Comparing the supramolecular structures of 2 and 3, we find that the longer distance between two lattice water molecules presented as OW⋯OW 7.477(26) Å in 2 (Fig. 5c) than 3.008 (59) Å (Fig. 6b) in 3 attributes to the bigger steric hindrance of the enMe ligand with terminal methyl units in 2 than en in 3. Though the components of 2 and 3 are similar, the steric hindrance difference of enMe and en makes the 2-D supramolecular sheet of 2 and the 3-D supramolecular network of 3.
3.3 Valence bond calculations and XPS characterizations
Bond valence sum (BVS) calculations for compounds 1–3 are listed in Table 3.18 For 1, the result shows that the valence states of the V atoms are +5, however they do not clearly identify the reduced O sites. This is due to the protonation withdrawing electron density not only from the protonated oxygen atom but also from unprotonated oxygen atoms in the anion. Similar protonation behavior has also been observed and reported.19 In 1, the six protons belong to the whole polyanion, not special oxygen atom, so they do not show clearly bond-valence sums of O atoms to indicate them being hydroxyl O atoms. The XPS gives a peek at 517.5 eV attributed to VV2p3/2 (Fig. S4).† This result further confirms the composition of compound 1.| 1 | 2 | 3 | |||||
|---|---|---|---|---|---|---|---|
| atoms | ∑s | atoms | ∑s | atoms | ∑s | atoms | ∑s |
| V1 | 5.04 | O4 | 1.78 | V1 | 4.56 | V1 | 4.03 |
| V2 | 5.07 | O5 | 1.50 | V2 | 4.63 | V2 | 4.55 |
| V3 | 5.05 | O6 | 1.66 | V3 | 3.98 | V3 | 4.49 |
| V4 | 5.06 | O7 | 1.84 | ||||
| O1 | 1.90 | O8 | 1.68 | ||||
| O2 | 1.85 | O9 | 1.89 | ||||
| O3 | 1.96 | O4 | 1.78 | ||||
For 2 and 3, the BVS calculations show the average value calculated for 8 V(IV) and 7 V(V) is equal to 4.47 and the experimental value is equal to 4.47 for 2 and 4.42 for 3, respectively. The BVS values in 2 and 3, however, do not clearly identify the reduced V(IV) sites. This is due to the possible delocalization for the d electrons of the reduced V(IV) centers over the polyanion framework involving all V atoms as found in heteropolyblues.20 The XPS gives two peaks at 517.3 and 515.4 eV attributed to VV2p3/2 and VIV2p3/2 for 2, and 517.3 eV and 515.7 eV attributed to VV2p3/2 and VIV2p3/2 for 3, respectively (Fig. S5 and S6).† XPS of compounds 2 and 3 give peaks at 854.8 and 781.8 eV attributed to Ni2+2p3/2 and Co2+2p3/2, respectively (Fig. S7 and S8).† All these results further confirm the structure analyses.
3.4 Variable temperature magnetic susceptibility of compound 3
The variable temperature magnetic susceptibility of 3 was mesured between 4 and 300 K. Fig. 7 shows the magnetic behavior of 3 in the form of χMTvs.T and χM−1vs.T plots, where χM is the molar magnetic susceptibility. The χMT value of compound 3 at 300 K is 2.06 cm3 mol−1 K, much smaller than the expected value (6.0 cm3 mol−1 K) for the six uncoupled S = 1/2 spins of V4+ atoms and two uncoupled S = 3/2 spins of the Co2+ atoms, indicative of antiferromagnetic coupling. Initially χMT value decreases slightly on decreasing temperature, and it stabilizes at ca. 1.70 cm3 mol−1 K. Below ca. 50 K the χMT value increases and reaches a maximum of ca. 2.56 cm3 mol−1 K at 12 K. Below 12 K the χMT value decreases rapidly, reaching 1.34 cm3 mol−1 K at 4 K. At the initial decrease of the χMT value with decreasing temperature, the plateau close to 1.70 cm3 mol−1 K, and the further decrease below 12 K are a clear indication that several exchange pathways, of different efficiency operate in this cluster. The relationship of 1/χMversusT at 4–300 K can be fitted to the Curie–Weiss law, χM = Ng2µβ2s(s + 1)/(3k(T − θ)) with the Curie constant C = 2.03 cm3 K mol−1 and the Weiss constant θ = −7.01 K. The negative θ value suggests that an antiferromagnetic interaction exists in 3. We tentatively speculate such magnetic behavior may be attributed to the characteristics of the combination of zero-field splitting of Co2+ in distorted octahedral environments and strong antiferromagnetic couplings between adjacent paramagnetic centers. The detailed magnetism investigation is ongoing and will be reported later.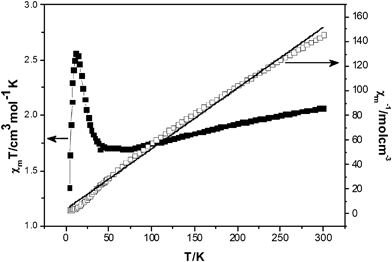 | ||
| Fig. 7 Plot of the temperature dependence of χMT and χM−1 for 3. | ||
The temperature dependence of χMT for 3 demonstrates the existence of strong antiferromagnetic coupling interactions, which are a common feature for most polyoxovanadate clusters.21 Unfortunately, it is too difficult to fit the experimental magnetic data of these extended high-nuclearity heteropolymetallic spin systems using a suitable theoretical model.22
3.5 Fluorescence properties
The emission spectrum of compound 1 in DMF solution at rt is depicted in Fig. 8. Compound 1 exhibits intense photoluminescence with an emission maximum at ca. 400 nm upon excitation at 268 nm. In order to understand the nature of the emission band of compound 1, we also investigated the luminescence of the free HMTA ligand used in DMF solution at rt. The HMTA ligand exhibits an emission maximum at ca. 395 nm (Fig. S9).† Compared to the emission spectrum of the HMTA ligand, the emission band of compound 1 might be assigned to the emission of the ligand.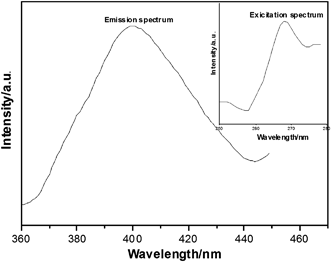 | ||
| Fig. 8 Emission spectra of compound 1 in DMF solution at rt. | ||
3.6 Thermogravimetric analyses
Thermogravimetric (TG) data for compounds 1–3 under flowing air are shown in Fig. S10–S12.† For 1, the TG curve only exhibits one step of weight loss as 34.80% in the temperature range of 180–390 °C, corresponding to the loss of crystal water and HMTA molecules (Fig. S10).† The weight of the residue is 65.20% which is in good accordance with the V2O5 as residue (calculated value: 65.52%).For 2, the TG curve (Fig. S11)† shows the first weight loss of 1.78% (calculated, 1.83%) from 100–300 °C which is ascribed to the release of two lattice waters. The second weight loss is 23.68% (calculated, 24.33%) in the temperature range 300–390 °C, corresponding to the decomposition of chloride anions and enMe molecules. The maximum mass loss of up to 660 °C is 64.77%, and the residue could be CoO and mixed-valence vanadium oxides V3O7/V6O13 based on calculations, suggesting that a part of the vanadium(V) atoms are deoxidized by NH3 generated during the thermal decomposition. Similar thermal decomposition behavior has also been observed and reported in other polyoxovanadate compounds.23 Upon further heating, a continuous weight increase corresponds to an uptake of oxygen accompanied by a valence state change of the vanadium ions. Compound 3 exhibits thermal gravimetric behavior (Fig. S12)† similar to 2. The first weight loss of 2.27% (calculated, 1.91%) from 100–250 °C is ascribed to the release of two lattice waters. The second weight loss is 18.92% (calculated, 20.94%) in the temperature range 250–400 °C, corresponding to the decomposition of chloride anions and en molecules. The maximum mass loss up to 660 °C is 70.77%, and the residue could be NiO and mixed-valence vanadium oxides V3O7/V6O13 based on calculations. It is noteworthy that the crystal water was lost from 180–390 °C for 1, 100–300 °C for 2 and 100–250 °C for 3, respectively. These temperatures are obviously higher than those in other POMs.5b,23b, 24 This behaviour indicates that the interactions of hydrogen bonds in 1–3 are very strong, which further stabilized the overall supramolecular network.
Conclusions
In summary, three new supramolecular compounds based on POVs have been synthesized. Compound 1 displays a 3-D (4,8)-connected host network with the Schläfli symbol (412·612·84)(46)2 formed by octamer water clusters and HMTA molecules. The steric hindrance of the HMTA ligand forces the octamer water cluster to form a ‘chair’ configuration, and the [H6V10O28] units acting as the template are located in the 3-D supramolecular network. Though 2 and 3 are isomorphic, they show different supramolecular frameworks. Compound 2 possesses a 2-D (3,6)-connected supramolecular layered structure with the Schläfli symbol (43·612)(43)2 with honeycomb cavities formed by the water molecules and the [V15O36] units, which were filled by the templates (TMCs). Compound 3 displays a 3-D (4,6)-connected host network with the Schläfli symbol (43·612)(43·63)2, which was constructed from the [V15O36] units and the lattice water molecules. The structure difference of 2 and 3 is due to the steric hindrance difference of organonitrogen ligands. In conclusion, the successful syntheses of 1–3 show that organic ligands show a profound influence on the supramolecular network, though the water molecules play a key role.Acknowledgements
The project was supported by the Natural Scientific Research Innovation Foundation at the Harbin Institute of Technology (HIT. NSRIF. 2008.08), the Program of Excellent Team At the Harbin Institute of Technology, the New Century Talent Program of the Chinese Ministry of Education (NECT-06-0341, 07-0242) and the China Postdoctoral Science Foundation (20070420859).Notes and references
- (a) V. Soghomonian, Q. Chen, R. C. Haushalter, J. Zubieta and C. J. O'Connor, Science, 1993, 259, 1596; (b) P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38, 2638 CrossRef; (c) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS.
- (a) M. M. Conn and J. Rebek, Jr., Chem. Rev., 1997, 97, 1647 CrossRef CAS; (b) G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Chem. Rev., 2005, 105, 1281 CrossRef CAS; (c) H. An, J. S. Bradshaw, R. M. Izatt and Z. M. Yan, Chem. Rev., 1994, 94, 939 CrossRef CAS; (d) R. Martínez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419 CrossRef CAS; (e) R. J. Sarma and J. B. Baruah, Cryst. Growth Des., 2007, 7, 989 CrossRef CAS.
- (a) A. Dolbecq, P. Mialane, L. Lisnard, J. Marrot and F. Sécheresse, Chem.–Eur. J., 2003, 9, 2914 CrossRef CAS; (b) X. Zhang, J. Q. Xu, J. H. Yu, J. Lu, Y. Xu, Y. Chen, T. G. Wang, X. Y. Yu, Q. F. Yang and Q. Hou, J. Solid State Chem., 2007, 180, 1949 CrossRef CAS; (c) J. Hao, L. Ruhlmann, Y. Zhu, Q. Li and Y. Wei, Inorg. Chem., 2007, 46, 4960 CrossRef CAS; (d) M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer, Berlin, 1983 Search PubMed.
- (a) P. Q. Zheng, Y. P. Ren, L. S. Long, R. B. Huang and L. S. Zheng, Inorg. Chem., 2005, 44, 1190 CrossRef CAS; (b) X. L. Wang, C. Qin, E. B. Wang, Y. G. Li, N. Hao, C. W. Hu and L. Xu, Inorg. Chem., 2004, 43, 1850 CrossRef CAS; (c) C. P. Pradeep, D. L. Long, G. N. Newton, Y. F. Song and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4388 CrossRef CAS; (d) Z. H. Yi, X. B. Cui, X. Zhang, G. D. Yang, J. Q. Xu, X. Y. Yu, H.-H. Yu and W. J. Duan, J. Mol. Struct., 2008, 891, 123 CrossRef CAS; (e) T. R. Veltman, A. K. Stover, A. N. Sarjeant, K. M. Ok, P. S. Halasyamani and A. J. Norquist, Inorg. Chem., 2006, 45, 5529 CrossRef CAS; (f) R. Atencio, A. Brieño and X. Galindo, Chem. Commun., 2005, 637 RSC; (g) R. Atencio, A. Briceño, P. Silva, J. A. Rodríguez and J. C. Hanson, New J. Chem., 2007, 31, 33 RSC.
- (a) H. K. Chae, D. Y. Siberio, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523 CrossRef CAS; (b) Z. Han, Y. Zhao, J. Peng, A. Tian, Q. Liu, J. Ma, E. Wang and N. Hu, CrystEngComm, 2005, 7, 380 RSC; (c) J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982 CrossRef CAS; (d) O. Kahn and C. Martinez, Science, 1998, 279, 44 CrossRef CAS; (e) C. Janiak, Angew. Chem., Int. Ed. Engl., 1997, 36, 1431 CrossRef CAS.
- (a) J. M. Breen and W. Schmitt, Angew. Chem., Int. Ed., 2008, 47, 6904 CrossRef CAS; (b) S. T. Zheng, Y. M. Chen, J. Zhang, J. Q. Xu and G. Y. Yang, Eur. J. Inorg. Chem., 2006, 397 CrossRef CAS; (c) T. Yamase, Chem. Rev., 1998, 98, 307 CrossRef CAS; (d) G. B. Karet, Z. Sun, W. E. Streib, J. C. Bollinger, D. N. Hendrickson and G. Christou, Chem. Commun., 1999, 2249 RSC.
- (a) Z. H. Yi, X. B. Cui, X. Zhang, J. H. Yu, J. Lu, J. Q. Xu, G. D. Yang, T. G. Wang, H. H. Yu and W. J. Duan, Dalton Trans., 2007, 2115 RSC; (b) W. J. Duan, X. B. Cui, Y. Xu, J. Q. Xu, H. H. Yu, Z. H. Yi, J. W. Cui and T. G. Wang, J. Solid State Chem., 2007, 180, 1875 CrossRef CAS.
- (a) C. Streb, D. L. Long and L. Cronin, CrystEngComm, 2006, 8, 629 RSC; (b) V. Coué, R. Dessapt, M. Bujoli-Doeuff, M. Evain and S. Jobic, Inorg. Chem., 2007, 46, 2824 CrossRef CAS; (c) T. Akutagawa, D. Endo, S. I. Noro, L. Cronin and T. Nakamura, Coord. Chem. Rev., 2007, 251, 2547 CrossRef CAS; (d) X. Y. Zhao, D. D. Liang, S. X. Liu, C. Y. Sun, R. G. Cao, C. Y. Gao, Y. H. Ren and Z. M. Su, Inorg. Chem., 2008, 47, 7133 CrossRef CAS; (e) Y. Wang, J. H. Yu, M. Guo and R. R. Xu, Angew. Chem., Int. Ed., 2003, 42, 4089 CrossRef CAS.
- (a) V. A. Blatov, A. P. Shevchenko, TOPOS 4.0; Samara State University, Russia, 2008 Search PubMed; (b) M. O'Keeffe and S. T. Hyde, Zeolites, 1997, 19, 370 CrossRef CAS; (c) A. F. Well, Three-Dimensional Nets and Polyhedra, Wiley, New York, 1977 Search PubMed; (d) O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acta Crystallogr., Sect. A: Found. Crystallogr., 2002, 59, 22 CrossRef.
- G. M. Sheldrick, SHELXS97 and SHELXL97. University of Göttingen, Germany Search PubMed.
- (a) H. L. Ferreira da Silva, M. F. Minas da Piedade and M. T. Duarte, Inorg. Chim. Acta, 2003, 356, 222 CrossRef CAS; (b) I. Correia, F. Avecilla, S. Marcão and J. C. Pessoa, Inorg. Chim. Acta, 2004, 357, 4476 CrossRef CAS; (c) S. Sharma, A. Ramanan, P. Y. Zavalij and M. S. Whittingham, CrystEngComm, 2002, 4, 601 RSC; (d) K. Pavani, S. Upreti and A. Ramanan, J. Chem. Sci., 2006, 118, 159 CrossRef CAS; (e) T. G. Chirayil, E. A. Boylan, M. Mamak, P. Y. Zavalij and M. S. Whittingham, Chem. Commun., 1997, 33 RSC.
- (a) G. M. Varga, J. E. Papaconstantinou and M. T. Pope, Inorg. Chem., 1970, 9, 662 CrossRef; (b) R. A. Prados and M. T. Pope, Inorg. Chem., 1976, 15, 2547 CrossRef CAS; (c) M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34 CrossRef; (d) N. Casan-Pastor and L. C. W. baker, J. Am. Chem. Soc., 1992, 114, 10384 CrossRef CAS and references therein; (e) M. I. Khan, S. Cevik, R. J. Doedens, Q. Chen, S. Li and C. L. O'connor, Inorg. Chim. Acta, 1998, 277, 69 CrossRef CAS.
- D. C. Crans, M. Mahroof-Tahir, O. P. Anderson and M. M. Miller, Inorg. Chem., 1994, 33, 5586 CrossRef CAS.
- (a) J. L. Atwood, L. J. Barbour, T. J. Ness, C. L. Raston and P. L. Raston, J. Am. Chem. Soc., 2001, 123, 7192 CrossRef CAS; (b) G. A. Jeffrey, An Introduction to Hydrogen bonding, Oxford University Press, Oxford, UK, 1997, 160 Search PubMed.
- (a) C. J. Gruenloh, J. R. Carney, C. A. Arrington, T. S. Zwier, S. Y. Fredericks and K. D. Jordan, Science, 1997, 276, 1678 CrossRef CAS; (b) J. K. Gregory and D. C. Clary, J. Phys. Chem., 1996, 100, 18014 CrossRef CAS.
- (a) E. Tajkhorshid, P. Nollert, L. J. W. Miercke and K. Schulten, Nature, 2002, 296, 525 CAS; (b) G. Sainz, C. J. Carrell, M. V. Ponamarev, G. M. Soriano, W. A. Cramer and J. L. Smith, Biochemistry, 2000, 39, 9164 CrossRef CAS.
- T. Drezen, O. Joubert, M. Ganne and L. Brohan, J. Solid State Chem., 1998, 136, 298 CrossRef CAS.
- I. D. Brown, in: M. O'Keeffe, A. Navrotsky (Eds.), Structure and Bonding in Crystals, vol. II, Academic Press, New York, 1981 Search PubMed.
- (a) H. T. Evans, Jr and M. T. Pope, Inorg. Chem., 1984, 23, 501 CrossRef CAS; (b) W. Wang, F.-L. Zeng, X. Wang and M.-Y. Tan, Polyhedron, 1996, 15, 265 CrossRef CAS.
- (a) C.-M. Liu, D.-Q. Zhang, M. Xiong and D.-B. Zhu, Chem. Commun., 2002, 1416 RSC; (b) A. Müller, M. Koop, P. Schiffels and H. Bögge, Chem. Commun., 1997, 1715 RSC; (c) Q. Chen and C. L. Hill, Inorg. Chem., 1996, 35, 2403 CrossRef CAS.
- (a) A. Müller, R. Sessoli, E. Krickemeyer, H. Bögge, J. Meyer, D. Gatteschi, L. Pardi, J. Westphal, K. Hovemeier, R. Rohlfing, J. Döring, F. Hellweg, C. Beugholt and M. Schmidtmann, Inorg. Chem., 1997, 36, 5239 CrossRef; (b) T. Yamase, M. Suzuki and K. Ohtaka, J. Chem. Soc., Dalton Trans., 1997, 2463 RSC; (c) D. Gatteschi, B. Tsukerblatt, A. L. Barra, L. C. Brunel, A. Müller and J. Döring, J. Am. Chem. Soc., 1992, 114, 8509 CrossRef CAS; (d) A. Müller, F. Peters, M. T. Pope and D. Gatteschi, Chem. Rev., 1998, 98, 239 CrossRef.
- O. Kahn, Molecular Magnetism, VCH, Weinheim, Germany, 1993 Search PubMed.
- (a) L. Chen, F. Jiang, M. Wu, N. Li, W. Xu, C. Yan, C. Yue and M. Hong, Cryst. Growth Des., 2008, 8, 4092 CrossRef CAS; (b) A. S. J. Wery, J. M. Gutierrez-Zorrila, A. Luque and P. Roman, Polyhedron, 1996, 15, 4555 CrossRef CAS; (c) P. P. Stander and C. P. Van-Vuuren, Thermochim. Acta, 1990, 157, 347 CrossRef CAS.
- A. Schaate, S. Klingelhöfer, P. Behrens and M. Wiebcke, Cryst. Growth Des., 2008, 8, 3200 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Structural data, XPS, emission and IR spectroscopy details TG curves bond lengths and angles for 1–3, CCDC reference numbers 729399–729401. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b916793p |
| This journal is © The Royal Society of Chemistry 2010 |