Aryl azo imidazoles assisted assembly of anion/anion–water through salt formation†‡
Avijit
Pramanik
,
Subhojit
Majumdar
and
Gopal
Das
*
Department of Chemistry, Indian Institute of Technology Guwahati, Assam 781 03, India. E-mail: gdas@iitg.ernet.in; Fax: +91-361-258-2349; Tel: +91-361-258-2313
First published on 7th September 2009
Abstract
Aryl azo dyes containing imidazole group can act as an acid or base. They are also potential hydrogen bond donor and acceptor. The imidazolium form of these dyes can form salts with various inorganic anions. The effect of the anion shape (linear, planner, spherical) on higher-dimensional supramolecular assembles of these salts has been analyzed and rationalized. Ligand assisted anion or anion–water assembly formation has been discussed. The presence of water in the crystal structure plays a vital role in the formation of higher dimensional networks. All supramolecular networks of salt 1–5 are guided by various noncovalent interactions, especially directional hydrogen bonds, halogen bonds and π-stacking interactions. The interaction of anions with the ligands in the solution phase was also investigated by absorption and fluorescent spectra.
Introduction
Supramolecular architectures assembled from various delicate noncovalent interactions such as hydrogen bonds, π⋯π stacking, C–H⋯π and electrostatic interactions, etc., have attracted intense interest in recent years because of their wide applications for catalysis, material and life sciences.1,2 Especially, the application of intermolecular hydrogen bonds is a well-known and efficient tool in the field of organic crystal design because of its strength and directional properties.3 Strong classical hydrogen bonds, such as O–H⋯N and O–H⋯O, are ideal for rationalizing and systemizing the relationship between hydrogen bond acceptor and donor molecules. Currently, H-bonding interactions have been widely used as one of the principal means to control such molecular assemblies during crystallization and thereby to engineer the crystal structures.4 Over the last several decades, although considerable effort has been devoted to the investigation of the relationships between a component structure and the resulting H-bonded crystal structure, it still remains difficult to predict how a particular molecule will pack in the crystal by such interactions.5 Thus more studies are still required to further understand such systems. The hydrogen bond between different anions and heterocyclic nitrogen atoms has been proven to be a useful and powerful organizing force and was utilized for the formation of supramolecules. Imidazole and its derivatives are ubiquitous in the biological and biochemical structures and functions and thus deserve special attention in the construction of some interesting salts and their dimentional frameworks in the recent years.6 On the other hand, the role of the anions in the supramolecular chemistry is of great interest because of its application in ion pair recognition and especially in anion exchange.7 It is also conceivable that great efforts have been directed towards the development of organic molecular crystals containing a variety of imidazole architectures.8Our interest lies in the design azo compounds containing a heterocyclic system. The presence of –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– group can lead to stabilizing a low-valent metal oxidation state due to its π acidity and the presence of low lying azo-centered π*-molecular orbitals.9 The azo function is photochromic,10 redox acive,11 pH responsive12 and its complexes act as a molecular switch.13 Earlier we have already reported the formation of various 3D supramolecular assemblies of a series of different aryl azo dyes based on their coordination chemistry.14 In this report, we chose to investigate a series of imidazole containing diazonium compounds [X = Br (L1), X = I (L2) and L3] and their corresponding salts with different various anions (Scheme 1). We have focused on the aryl azo dye assisted formation of an anion and anion–water cluster in the solid-state. We have paid much attention to the effect of counterions like ClO4− (tetrahedral), NO3− (planner), and SCN− (linear) upon the formation of supramolecular architectures with aryl azo imidazolium as building blocks. The main reason for our interest in the role of the anions upon the topology is to be synthesizing materials with useful properties and interesting supramolecular architectures. We have also investigated the solution phase interaction between aryl azo systems in the presence of a different anion by monitoring their absorption and fluorescent spectral properties.
N– group can lead to stabilizing a low-valent metal oxidation state due to its π acidity and the presence of low lying azo-centered π*-molecular orbitals.9 The azo function is photochromic,10 redox acive,11 pH responsive12 and its complexes act as a molecular switch.13 Earlier we have already reported the formation of various 3D supramolecular assemblies of a series of different aryl azo dyes based on their coordination chemistry.14 In this report, we chose to investigate a series of imidazole containing diazonium compounds [X = Br (L1), X = I (L2) and L3] and their corresponding salts with different various anions (Scheme 1). We have focused on the aryl azo dye assisted formation of an anion and anion–water cluster in the solid-state. We have paid much attention to the effect of counterions like ClO4− (tetrahedral), NO3− (planner), and SCN− (linear) upon the formation of supramolecular architectures with aryl azo imidazolium as building blocks. The main reason for our interest in the role of the anions upon the topology is to be synthesizing materials with useful properties and interesting supramolecular architectures. We have also investigated the solution phase interaction between aryl azo systems in the presence of a different anion by monitoring their absorption and fluorescent spectral properties.
Experimental
Materials and methods
All reagents were obtained from commercial sources and used as received. The solvents were distilled freshly following a standard procedure. The IR spectra were recorded on a Perkin Elmer-Spectrum One FT-IR spectrometer with KBr disks in the range 4000–400 cm−1. The absorption spectra were recorded on a Perkin Elmer Lambda- 25 UV-Visible Spectrometer at 298 K. The NMR spectra were recorded on a Varian FT−400 MHz instrument. The chemical shifts were recorded in parts per million (ppm) on the scale using tetramethylsilane (TMS) as a reference. Elemental analyses were carried out on a Perkin-Elmer 2400 automatic carbon, hydrogen and nitrogen analyzer. Powder X-ray diffraction data were recorded using a Seifert powder X-ray diffractometer (XRD 3003TT) with Cu Kα source (λ = 1.54 Å) on a glass surface of an air-dried sample. The steady-state fluorescence spectra were recorded on a Varian Cary-Bio spectrofluorimeter and corrected for emission. The fluorescence quantum yield was determined in each case by comparing the corrected spectrum with that of naphthalene (ΦF = 0.23) in ethanol by taking the area under the total emission using the following equation:| ΦS = ΦR (FSAR/FRAS)(ηS/ηR)2 |
X-Ray crystallography
The intensity data were collected using a Bruker SMART APEX-II CCD diffractometer, equipped with a fine focus 1.75 kW sealed tube Mo Kα radiation (λ = 0.71073 Å) at 273(3) K, with increasing ω (width of 0.3° per frame) at a scan speed of 3 s per frame. The SMART software was used for data acquisition. Data integration and reduction were undertaken with SAINT and XPREP15 software. Multi-scan empirical absorption corrections were applied to the data using the program SADABS.16 Structures were solved by direct methods using SHELXS-97 and refined with full-matrix least squares on F2 using SHELXL-97.17 All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were located from the difference Fourier maps and refined. Structural illustrations have been drawn with ORTEP-3 for Windows.18 Crystallographic noncovalent interactions data are summarized in Table 1.| D–H⋯A | H⋯A/Å | D⋯A/Å | D–H⋯A/° |
|---|---|---|---|
| Salt 1 | |||
| C16–Br2⋯S1 | 3.535 | 165.06 | |
| C7–Br1⋯O1 | 3.301 | 167.95 | |
| C18–H18⋯N9 | 2.835(.005) | 3.742 (7) | 165.51 (33) |
| C14–H14⋯N7 | 2.483(.005) | 2.636 (7) | 88.96 (30) |
| N6–H6N⋯S1 | 2.652(.001) | 3.483 (4) | 162.84 (23) |
| C9–H9⋯N3 | 2.451(.004) | 2.642 (7) | 91.31 (33) |
| C3–H3⋯N7 | 2.953(.004) | 3.514 (6) | 120.18 (26) |
| N6–H6N⋯π | 3.55 | ||
| Salt 2 | |||
| C2–I1⋯O2 | 3.079 | 170.30 | |
| C3–H3⋯O4 | 2.724 (36) | 3.278 (5) | 118.00 (61) |
| C2–H2⋯O1 | 2.467 (42) | 3.340 (6) | 153.95 (41) |
| C9–H9⋯O3 | 2.640 (32) | 3.728 (5) | 165.01 (33) |
| C5–H5⋯N3 | 2.490 (35) | 2.751 (4) | 94.51 (16) |
| π ⋯π (azo) | 3.532 | ||
| Salt 3 | |||
| C9–H9⋯O3 | 2.648 (4) | 3.512 (5) | 154.77 (23) |
| C8–H8⋯O2 | 2.521 (4) | 3.403 (6) | 158.34 (26) |
| C4–H4⋯N3 | 2.453 (3) | 2.703 (5) | 95.34 (24) |
| C8–H8⋯O4 | 2.937 (4) | 3.270 (6) | 102.75 (26) |
| C3–H3⋯O1 | 2.834 (5) | 3.758 (6) | 172.97 (24) |
| C8–H8⋯O3 | 2.813 (4) | 3.331 (6) | 116.31 (26) |
| C1–I1⋯O5 | 3.468 | 170.61 | |
| Salt 4 | |||
| C12–H12⋯O2 | 2.638 (2) | 3.539 (3) | 163.33 (15) |
| C7–H7⋯O1 | 2.528 (2) | 3.329 (3) | 144.53 (15) |
| C2–H2⋯O3 | 2.599 (2) | 3.232 (3) | 125.74 (15) |
| C3–H3⋯O1 | 2.783 (3) | 3.420 (3) | 126.60 (15) |
| C12–H12⋯N4 | 2.525 (2) | 2.825 (3) | 99.11 (13) |
| C5–H5⋯N3 | 2.427 (2) | 2.703 (3) | 96.97 (14) |
| C10–H10⋯O2 | 2.861 (2) | 3.459 (3) | 123.19 (15) |
| C7–H7⋯N1 | 2.528 (2) | 3.329 (3) | 144.53 (15) |
| π ⋯π (azo) | 3.578 | ||
| Salt 5 | |||
| C4–H4⋯O3 | 3.564 (6) | 2.658 (4) | 164.67 (30) |
| C9–H9⋯O1 | 2.622 (4) | 3.522 (7) | 163.27 (29) |
| C22–H22⋯O4 | 2.695 (4) | 3.607 (6) | 167.03 (30) |
| C9–H9⋯N4 | 2.498 (3) | 2.809 (5) | 99.76 (26) |
| C2–H2⋯N3 | 2.430 (4) | 2.704 (6) | 96.89 (29) |
| C15–H15⋯N7 | 2.471 (4) | 2.729 (6) | 95.91 (29) |
| C22–H22⋯N8 | 2.506 (3) | 2.814 (5) | 99.54 (28) |
| C13–H13⋯O2 | 2.928 (5) | 3.278 (6) | 103.92 (29) |
| C4–H4⋯N9 | 2.898 (5) | 3.712 (7) | 147.02 (31) |
| C6–H6⋯O1 | 2.935 (5) | 3.586 (7) | 128.31 (30) |
| C4–H4⋯O3 | 2.658 (4) | 3.564 (6) | 164.67 (30) |
| C22–H22⋯O4 | 2.695 (4) | 3.607 (6) | 167.03 (30) |
| C25–H25⋯O3 | 2.827 (3) | 3.313 (6) | 113.83 (29) |
| C3–H3⋯O4 | 2.954 (4) | 3.593 (6) | 127.17 (30) |
| C19–H19⋯O4 | 2.868 (4) | 3.564 (7) | 132.58 (32) |
| C16–H16⋯O1 | 2.942 (5) | 3.489 (7) | 119.02 (35) |
| C16–H16⋯π | 4.153 | ||
| π ⋯π (azo) | 3.4405 | ||
Synthesis
p-Bromo-2-(phenylazo) imidazole ligand (L1), p-iodo-2-(phenylazo) imidazole (L2) and 2-(naphthylazo) imidazole (L3), were synthesized following a literature method.19 The synthesis of aryl azo imiazolium salts are described below.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture solution of the compound at RT for seven days. IR (KBr disk) (cm−1); ν(SCN), 2190 (s), 2140, ν(O–H), 3590 (m), 3535 (m), ν(CN), 1593 (m), ν(NN), 1398 (m), ν(CS), 685 (m). Anal. Calcd (%) for C19H17Br2N9OS: C, 39.51; H, 2.96; N, 21.84. Found: C, 39.53; H, 2.95; N, 21.86%.
:1) mixture solution of the compound at RT for 10 d. Anal. Calcd (%) for C9H10N5O4I: C, 28.49; H, 2.65; N, 18.47. Found: C, 28.48; H, 2.66; N, 21.49%. IR (KBr disk) (cm−1); ν(NO), 1497 (s), ν(N–O), 1271 (m), ν(ONO), 992, ν(O–H), 3592 (m), 3531 (m), ν(CN), 1600 (m), ν(NN), 1406 (m).
:1) mixture solution of the compound at RT for 15 d. Anal. Calcd (%) for C9H10N4O5ICl: C, 25.96; H, 2.42; N, 13.46. Found: C, 25.98; H, 2.44; N, 13.45%. IR (KBr disk) (cm−1); ν(Cl–O), 1100 (m), 625(m), ν(O–H), 3588(m), 3537 (m), ν(CN), 1600 (m); ν(NN), 1406 (m).
O), 1497 (s), ν(N–O), 1271 (m), ν(ONO), 992, ν(CN), 1600 (m), ν(NN), 1406 (m).
:1) mixture solution of the compound at RT for 15 d. Anal. Calcd (%) for C20H23N9O4: C, 52.95; H, 5.11; N, 27.80. Found: C, 52.94; H, 5.13; N, 27.82%. IR (KBr disk) (cm−1); ν(NO), 1497 (s), ν(N–O), 1271 (m), ν(ONO), 992, ν(O–H), 3595 (m), 3539 (m), ν(CN), 1600 (m), ν(NN), 1406 (m).
:1) mixture solution of the compound at RT for seven days. IR (KBr disk) (cm−1); ν(SCN), 2190 (s), 2140, ν(O–H), 3590 (m), 3535 (m), ν(CN), 1593 (m), ν(NN), 1398 (m), ν(CS), 685 (m). Anal. Calcd (%) for C19H17Br2N9OS: C, 39.51; H, 2.96; N, 21.84. Found: C, 39.53; H, 2.95; N, 21.86%.
:1) mixture solution of the compound at RT for 10 d. Anal. Calcd (%) for C9H10N5O4I: C, 28.49; H, 2.65; N, 18.47. Found: C, 28.48; H, 2.66; N, 21.49%. IR (KBr disk) (cm−1); ν(NO), 1497 (s), ν(N–O), 1271 (m), ν(ONO), 992, ν(O–H), 3592 (m), 3531 (m), ν(CN), 1600 (m), ν(NN), 1406 (m).
:1) mixture solution of the compound at RT for 15 d. Anal. Calcd (%) for C9H10N4O5ICl: C, 25.96; H, 2.42; N, 13.46. Found: C, 25.98; H, 2.44; N, 13.45%. IR (KBr disk) (cm−1); ν(Cl–O), 1100 (m), 625(m), ν(O–H), 3588(m), 3537 (m), ν(CN), 1600 (m); ν(NN), 1406 (m).
O), 1497 (s), ν(N–O), 1271 (m), ν(ONO), 992, ν(CN), 1600 (m), ν(NN), 1406 (m).
:1) mixture solution of the compound at RT for 15 d. Anal. Calcd (%) for C20H23N9O4: C, 52.95; H, 5.11; N, 27.80. Found: C, 52.94; H, 5.13; N, 27.82%. IR (KBr disk) (cm−1); ν(NO), 1497 (s), ν(N–O), 1271 (m), ν(ONO), 992, ν(O–H), 3595 (m), 3539 (m), ν(CN), 1600 (m), ν(NN), 1406 (m).
Results and discussion
Crystal structure studies
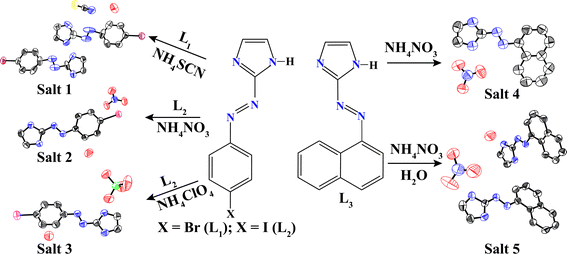
The shape of the anions of the same charge as well as the presence of solvent molecules in the crystal can reorganize the packing of the host molecule and thereby facilitate the inclusion of guest molecules.20 Various types of strong as well as weak hydrogen bonds help the host molecules to reorient themselves in order to accommodate the solvent and guest and hence facilitate the formation of different types of supramolecular interactions.21 Crystal structure analyses reveal that the five salt crystals belong to the lower-symmetry monoclinic crystal systems (Table 1). Salts 1–5 are all formed from aryl azo imidazole cations and inorganic anions with a variety of noncovalent interactions including hydrogen bonding, aryl stacking, and halogen bonding. These structures significantly differ, however, in packing modes of cation and anions in salts. Salt 1,[(L1)2H]+[SCN]−·H2O is crystallized in H2O with thiocyanate (SCN−) to yield a hydrated salt 1, which has a 3D strong H-bonded supramolecular framework. An ORTEP plot of salt 1 is shown in Fig. 1a along with the atom numbering scheme. In the asymmetric unit, one of the dye molecules is in protonated form. Protonated imidazolium cation subsequently forms a salt co-crystal with linear thiocyanate (SCN−) anion. Two of L1 are symmetrically non-equivalent and they are connected by a proton to form a homo-conjugated cationic system with a moderate, asymmetric N–H⋯N hydrogen bond with a length of 2.77(3) Å. The dihedral angle between the planes of the ring is 6.0 and 0.89°, respectively, for the protonated and unprotonated L1. However, protonation of the ligand has little effect on the azo (NN) system [L1, –NN– 1.128 Å, L1H+,-NN− 1.101 Å]. Each water molecule is linked to the two adjacent SCN− anions and one imidazole N via O–H⋯N and N–H⋯O hydrogen bonds (see ESI).†
 | ||
| Fig. 1 (a) ORTEP plot (50% probability ellipsoids) of salt 1; (b) alternating parallelogram A and B when viewed along the a axis and (c) linear water chain along the c axis. | ||
The water oxygen atom is also involved in a halogen bond with a neighboring Br1 of neutral L1. The halogen bond Br1⋯O1 distance is 3.30 Å, which is a very common halogen bond in biological processes21e and this bond is shorter than the sum of the van der Waals radii (1.85 Å for Br and 1.52 Å for O).21a The C7–Br1⋯O1 angle is 167.95°, implying a moderately strong halogen interaction. Thus, the O1 atom meets the Br1 atom in line with the C–Br bond. The nitrogen end of the SCN− anion forms a strong O–H⋯N hydrogen bond with two neighboring water molecules (see ESI).† However, the sulfur end is engaged in the formation of a halogen bond with the neighboring Br2 of cationic L1. In this case, also the halogen bond Br2⋯S1 distance is 3.535 Å, which is shorter than the sum of the van der Waals radii (1.85 Å for Br and 1.80 Å for S). The C16–Br2⋯S1 angle is 165.1°, implying that the S1 atom meets the Br2 atom in line with the C–Br bond.22 Another interesting feature is the presence of a weak N–H⋯π (N6–H6N⋯π,2.6Å) interaction between protonated imidazole N–H with the π-cloud of the CS bond of SCN− anion. The N–H bond forms an edge-to-face π-stacking interaction and is almost perpendicular to the CS bond. Anion and water molecule forms a water–thiocyanate–water–thiocyanate tetramer which has stabilized four halogen bonds and two N–H⋯O type hydrogen bonds (see ESI).† It forms a planar, zigzag arrangement of the alternating parallelogram of type A and type B when viewed along the b as well as the c axis (Fig. 1b). All water molecules are arranged linearly along the c axis (Fig. 1c). The distances between two water molecules along the c and a axes are 8.21 and 10.20 Å, respectively. The FT-IR spectra of salt 1 exhibit strong stretches at 2190, 2140, and 685 cm−1, which are in agreement with νCN and νCS.23 and there are two strong peaks 3590 (m) and 3535 (m) which may be due to ν(O–H) of H2O molecule present in the crystal system.
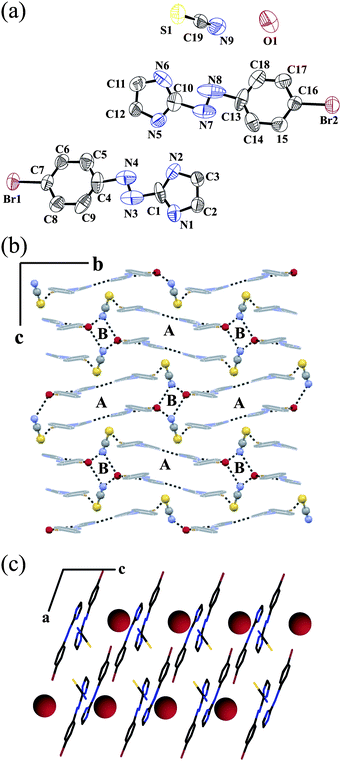
Fig. 2a shows the ORTEP plot of salt 2, [L2H]+[NO3]−·H2O along with the atom numbering scheme. The dihedral angle between the planes of the ring is 2.36°, which is less compared to the protonated ligand L1. Hence protonated L2 is more planar, which is also reflected in the increased azo bond length (–NN− 1.264 Å). Offset face-to-face π packing occurs between two adjacent benzene rings and the π cloud of NN bond 3.477 Å. It forms a staircase along the a axis (Fig. 2b).
 | ||
| Fig. 2 (a) ORTEP plot (50% probability ellipsoids) of 2; (b) staircase packing of alternating hydrophobic and hydrophilic layers along the a axis; (c) water–nitrate–water 1D chain along the a axis and (d) repeat of the hydrogen bonded ring ABA triad along the b axis and CDC triad along the c axis. | ||
It forms an alternate hydrophobic and hydrophilic layer structure. The hydrophilic layer consists of a planar NO3− anion and a water molecule. Each hydrophobic layer is connected to the hydrophilic layer by C–H⋯O type weak hydrogen bonds and halogen bonds. Each planar NO3− anion is hydrogen bonded to two water molecules and one imidazole N2–H2N via O–H⋯O and N–H⋯O type hydrogen bonds. Similar to salt 1, here also the anion forms a halogen bond with the neighboring I atom. The halogen bond distance is 3.08 Å, which is much shorter than the sum of the van der Waals radii (1.98 Å for I and 1.52 Å for O).24 The C7–I1⋯O2 angle is 170.30°, implying a moderately strong halogen interaction. Thus, the O2 atom meets the I1 atom in line with the C–I bond. However, solvent water does not form any kind of halogen bond like salt 1. Crystal water forms two O–H⋯O hydrogen bonds and one N–H⋯O type hydrogen bond with NO3− anion and imidazole N1–H1N. Water and NO3− forms an 1D infinite zigzag chain along the a axis (Fig. 2c). Each O3 of the anion forms a bifurcated hydrogen simultaneously to two water molecules. Overall, non-covalent interactions in the solid-state result in the formation of four different types of ring structures (Fig. 2d). The ABA triad ring is repeated along the b axis where the CDC type triad is propagated along the c axis. Salt 2 shows a strong sharp peak at 1497, 1271, 992 cm−1 which can be defined as the νNO stretching frequency of the unidentate bonding of the nitrate anion.23
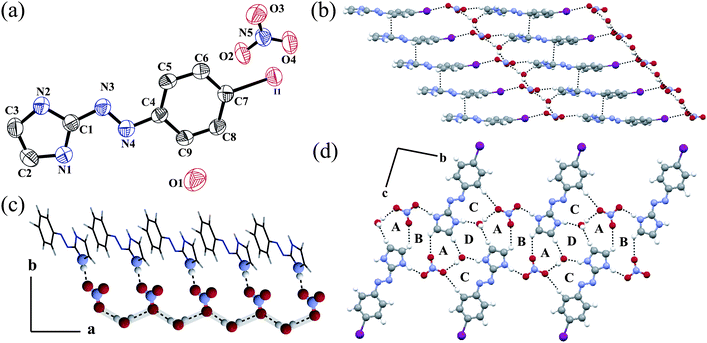
However when we changed the plane NO3− anion to a tetrahedral ClO4− anion spherical, there is a change in the overall packing and non-bonding interactions. An ORTEP plot of salt 3, [L2H]+[ClO4]−·H2O is shown in Fig. 3a along with the atom numbering scheme. The dihedral angle between the plane through the ring is 1.41° with a NN distance of 1.256 Å, which is a little shorter than the previous one. To accommodate the larger ClO4− anion in the solid state in between the two ligands, there is no stacking interaction between the aromatic planes unlike salt 2. Moreover, four ligands form a kind of elliptical pocket to accommodate both the anion and the crystal water molecule (see ESI).† Each water molecule forms two O–H⋯O type hydrogen bonds with two nearby ClO4− anions and one N–H⋯O hydrogen bond with LH+ (see ESI†) similar to that of salt 2. The relative bond lengths and angles are given in the ESI.† Similar to salt 1, here also water forms a halogen bond with the neighboring I atom. The halogen bond distance is 3.468 Å, which is a little shorter than the sum of the van der Waals radii (1.98 Å for I and 1.52 Å for O).21b,c,d The C1–I1⋯O5 angle is 171.61°, implying a moderately strong halogen interaction. However, unlike in salt 2 the anion in salt 3 does not form any halogen bonds. The anion forms O–H⋯O and N–H⋯O type hydrogen bond with water and imidazole N4–H4N.
 | ||
| Fig. 3 (a) ORTEP plot (50% probability ellipsoids) of salt 3; (b) the annulus structure of 3 formed via ionic hydrogen bonds; (c) repeat of the hydrogen bonded ring DABA tetrad along the a axis and BC diad along the b axis and (d) water–ClO4− -LH+ one-dimensional chain along the b axis. | ||
All bond lengths and angles are given in the ESI.† In salt 1 both anion and water form halogen bonds, whereas in salt 2 only the anion and in salt 3 only water form halogen bonds. The existence of a water molecule in the crystal lattice helps to form strong N–H⋯O and O–H⋯O hydrogen bonds in salt 3 which is favourable for stabilizing the annulus (Fig. 3b). The annulus structure of 3 formed via ionic hydrogen bonds with a dimension of the cavity 9.7 × 5.6 Å. The combined effect of O–H⋯O, N–H⋯O and C–H⋯O interactions in the solid state results in the formation of four different types of ring structures (Fig. 3c). Among them, ring D has the largest size. The DABA tetrad is repeated along the a axis and the BC diad is repeated in the solid state along the b axis. Along the b axis water–ClO4−−LH+ forms an infinite 1D hydrogen bonded chain (Fig. 3d).
Then in the next part of the manuscript we have studied the effect of the increased hydrophobic nature of the ligand in the 3D solid state packing. The protonated naphthyl azo imidazole ligand L3 crystallizes in the presence of the NO3− anion. An ORTEP plot of salt 4, [L3H]+[NO3]− is shown in Fig. 4a along with the atom numbering scheme. naphthyl azo imidazole ligands are much more planar due to extended conjugation than the phenyl azo imidazole ligands. Hence, the dihedral angle between the aromatic planes is only 0.29°. The NN double bond character is decreased and the bond distance is increased (1.259 Å) as compared with the literature value.25 In the crystal lattice each NO3− anion forms three N–H⋯O type intermolecular hydrogen bondings with the imidazole hydrogen present in the neighborhood (see ESI).† The cation and anion form a 1D zigzag hydrogen bonded chain along the c axis (Fig. 4b) similar to another nitrate salt, 2. In salt 2 all ligands are oriented in the same direction. However, in this case every other ligand is oriented in the same direction and contiguous ligands are disposed in the opposite direction. This is due to the inter-ligand steric repulsions of the bulky naphthyl group in ligand L3. Several N–H⋯O and C–H⋯O interactions forms a wavy packing pattern along the b axis (Fig. 4c). Anti-parallel orientation of the ligand along the a axis helps to form offset face-to-face π stacking interactions (3.416 Å) between the naphthyl and imidazole group (Fig. 4d). Each of the π stacked dimers is clipped and stabilized in the solid state in both ends via C–H⋯O interactions with the adjacent anions. The FT-IR spectra of salt 4 exhibit strong stretches at 1420, 1275, 980 cm−1 which are in agreement with NO3 moieties.23
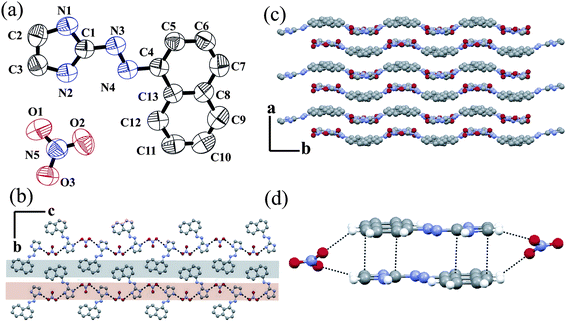 | ||
| Fig. 4 (a) ORTEP plot (50% probability ellipsoids) of salt 4; (b) zigzag H-bonded chain along the a axis; (c) wavy packing along the b axis and (d) stacking interaction in salt 4. | ||
Salt 5 is formed when a crystallization of salt 4 is carried out in the presence of water, i.e. salt 5 is the hydrated form of salt 4. An ORTEP plot of salt 5, [L3H]+[NO3]−·H2O is shown in Fig. 5a. Similar to the salt 1, it also contains one protonated and one neutral L3 in the asymmetric unit. In protonated species, the dihedral angle between the two least squares planes is only 2.31° whereas the unprotonated one is 4.75°. Both ligands form a homo-conjugate unit through the N–H⋯N bond. The protonated imidazole unit forms hydrogen bonds with water and neutral L3. On the other hand, neutral L3 forms hydrogen bonds with the anion and the protonated imidazole unit. Each anion forms two O–H⋯O bonds with the neighboring water molecules (see ESI).† Water forms two O–H⋯O bonds with the anion and one N–H⋯O bond with the imidazolium cation. All the bond noncovalent interactions are given in the ESI.† Similar to salt 2, here also exists a 1D infinite water–nitrate hydrogen bonded chain along the a axis (Fig. 5b). However, the pattern of bonding is different form salt 2. It forms an L-shape orientation of the 1D chain. The difference in the network pattern is explained by the difference in bonding of the NO3− anion with the water molecules. In salt 2, one O atom of the NO3− anion forms a bridge between two water molecules. But in salt 5, the water molecules are bridged by an O–N–O unit of the NO3− anion. Two symmetrically non-equivalent L3 ligands form a zigzag chain along the b axis (Fig. 4c). The alternate chains stack along the c axis. along the a axis it forms a rectangular 1D channel which is filled by the anion–water 1D infinite chain (see ESI).† Both L3 units are perpendicular to each other. Neutral L3 are oriented perpendicular to each other in the adjacent layer. Each perpendicular neutral unit is connected via a C–H⋯π interaction. The imidazole unit forms an offset face-to-face π–π interaction with the naphthyl moieties for both L3 units (Fig. 5d).26 The FT-IR spectra of salt 5 exhibit strong stretches at 1420, 1275, 980 cm−1 which are in agreement with coordinated O-NO2 moieties.23 and there are two extra peaks present at 3595 (m) and 3539 (m) due to ν(O–H) of the water molecule present in the crystal system.
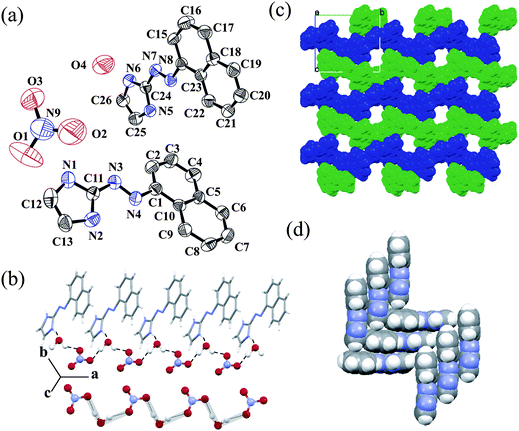 | ||
| Fig. 5 (a) ORTEP plot (50% probability ellipsoids) of salt 5; (b) 1D hydrogen bonded water–nitrate chain; (c) alternate zigzag chain of two asymmetric L3 with a 1D channel along the a axis and (d) π stacking interactions between L3. | ||
Powder X-ray diffraction (PXRD) pattern of salts 4 and 5 supports the crystalline nature of the bulk sample. Salt 5 contains a water of crystallization. Salt 5 was heated at 80 °C for 2 h in vacuum and the resulted crystalline solid was studied by PXRD. The PXRD pattern was close to that of the anhydrous salt 4 (see ESI).† However, we are unable to maintain the single crystalline nature of the heated sample and thus not being able to confirm the final structure by single crystal X-ray studies. However, the FT-IR spectrum and the PXRD pattern are to those of salt 4. Similarly, in salt 1, salt 2 and salt 3 there are also significant changes in the peak positions as well as intensities before and after water removal (see ESI).†
Absorption spectroscopy
The UV-Vis absorption spectra of ligands were recorded in MeOH at 298 K. L1 and L2 show a broad absorption band with maxima at 360 and 380 nm, respectively (Fig. 6a). However the naphthyl chromophore in L3 shows a red shifted absorption band at 392 and 410 nm with ε ∼ 104 M−1cm−1 for both peaks. These absorption bands correspond to intra-ligand (n–π*/π–π*) transitions.27 An addition of ammonium salts has a negligible effect on the absorption spectra of L1 and L2. However, the effect is more pronounced in the case of L3. In these ligands there are two sites for protonation, i.e., azo and imidazole nitrogen. L3 has more electron density than L1–2. In neutral and acid media, the absorption spectra of L1–2 do not change much, however, in a basic medium (pH∼10) a red shift occurred due to availability of electron density in the imidazole N–H group.28 The observed Δλ value is ∼33 nm (Fig. 6a). On the other hand, a bathochromic shift is observed (Δλ ∼30 nm) in acidic pH (∼3) in the case of L3 (Fig. 6b). The probable explanation for the contradictory nature of the same type of ligands is due to the electronic environment (depending on basicity) of azo nitrogen and imidazole nitrogen.29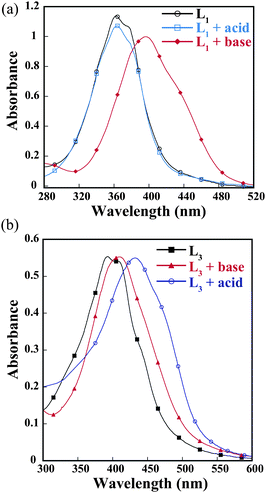 | ||
| Fig. 6 UV-Vis absorption spectra of (a) L1 and (b) L3 in the presence of acid and base. | ||
Fluorescence spectroscopy
The studied trans-2-aryl-azonaphthalene has a small fluorescence quantum yield, which is scarcely affected by the solvent (ΦF = 0.025). The fluorescence quantum yields depend weakly on the excitation wavelength, due to the presence of conformational isomers with slightly different absorption spectra in the solution.30 The emission spectra of L3 in hexane at 298 K show a locally excited (LE) broad emission of naphthalene with emission at 425 and 450 nm (λex = 400 nm), respectively. However, in other solvents a very weak emission band is observed. Dual exponential fluorescence decays confirmed the presence of two conformers cis and trans, interchanging in the solution phase31 (Fig. 7). With increasing the concentration of the ligand, the total intensity of fluorescence emission increases significantly without the formation of an excimer. Upon gradual addition of different anions to the hexane solution of L3, the intensity of the emission bands decreases. The dissociation constant32Kd was estimated from the change in fluorescence quantum yield resulted from the titration data of L3 against NH4ClO4 solution. The linear fit of the data (see ESI†) for NH4ClO4 inclusion complex was obtained by plotting log[(Φ − Φmin)/(Φmax − Φ)] as a function of the logarithm of NH4ClO4 concentration and the intercept of the linear regression determines the Kd value of 1.9 × 10−5 M mm in hexane. This value indicates the formation of a stable inclusion complex and is in consistence with good correlation coefficients (>0.99).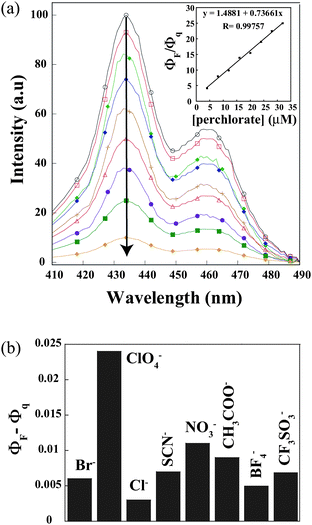 | ||
| Fig. 7 Schematic representation showing the change of fluorescence quantum yield (ΦF − Φq) of L3 upon addition of the different acids. ΦF and Φq are quantum yields of L3 in the absence and presence of a guest, respectively. Inset: Stern–Volmer plot with ammonium perchlorate. | ||
To demonstrate the selectivity of L3 towards perchlorate, we have monitored the change in fluorescence quantum yields in the presence of different anions like nitrate, thiocyanate, bromide, chloride, triflate, tetrafluoroborate and acetate. The magnitude of the quenching efficiency (ΦQ)33 follows the order of ClO4− (0.024) > NO3− (0.011) > CF3SO3− > (0.008) SCN− (0.007) > Br− (0.006) > BF4− (0.005) > Cl− (0.003) (See ESI†) which is a result of combining the steric and electronic effects through noncovalent interactions. Fig.7 clearly shows that L3 has a remarkably high selectivity toward the percholrate anion in the terms of change of the fluorescence quantum yield. The linear Stern–Volmer response of (Fig. 7, inset) ClO4− anion as a quencher is consistent with well-behaved fluorescence quenching systems.
General discussion on the observed noncovalent interactions in the solid phase and spectroscopic behaviour in the solution phase
In continuation of the anion recognition and their spectroscopic studies with variety of host molecules,33 herein we have shown that an aryl azo system containing imidazole moiety played a crucial role in forming higher-dimensional networks in the presence of a different anion. The imidazolium cation directed the anion assembly formation through a strong hydrogen bonding interaction along with weaker noncovalent interactions. We have varied the shape of the anion to evaluate the effect of higher dimension ordering in the solid-state. In salt 1 we have seen the linear SCN− anion is assembled via water to form H2O-SCN−−H2O-SCN− unique tetrameric moieties, which is not the case in other non-linear anions. However, in salt 2, the planer NO3− ion is assembled through a water molecule to form a 1D infinite zigzag chain formed by a single oxygen atom bridge of nitrate. On the other hand, in the case of salt 5, the NO3− ion is assembled with a water molecule to form a 1D infinite water-nitrate hydrogen bonded chain constituting two oxygen atoms of the nitrate ion bridged with two water molecule. In salt 4, there is no water in the crystal lattice, which drastically changes the linear chain structure. In salt 4, the naphthyl azo imidazolium ion is stacked with a nitrate ion to form a wave like structure. Salt 3 contains a spherical ClO4− ion which is assembled with water to form a 1D zigzag chain network bearing an oxygen atom of perchlorate bridged with two water molecules. In solution, the UV-visible studies shows a bathochromic and hypsochromic shift with pH, which has been explained on the basis of the electron density of the aryl group (benzyl to naphthyl) and the halogen substitution in the ligands. Fluorescence quenching studies also revel the presence of various noncovalent interactions34 between the ions which have been observed in the solid-state.Conclusions
To rationalize the influence of the noncovalent interactions of the anion on the formation of supramolecular architectures with aryl-azodiazonium salts, we have shown that the size of the anions has a marked influence upon the supramolecular assemblies. For complexes with a different type of anions (linear, plane, spherical), one dimensional layered network with identical topologies has been found. As expected, the N atom of the imidazole ring always acts as an excellent hydrogen-bonding acceptor by the formation of a strong O–H⋯N or ionic N–H⋯O and O–H⋯O hydrogen bond due to the presence of water in the crystal systems. Additionally, this study shows that complexes 1 and 5 are a good supramolecular building module that can produce bimolecular co-crystals, and its modular nature makes it possible to select a comfortable configuration in the course of formation of a self assembly hydrogen bond (L⋯LH+). Finally UV-visible and fluorescence spectroscopy studies also confirmed the existence of a weak supramolecular interaction. These results show that the types and ratios of cations and anions play a significant role in fabricating a different solid-state structure.Acknowledgements
G.D. acknowledges DST (SR/S1/IC-01/2008) and CSIR (01-2235/08/EMR-II), New Delhi India for financial support and DST-FIST for single crystal X-ray diffraction facility. A.P. wishes to thank CSIR for JRF (09/731(0045)/2007-EMR – I).Notes and references
- (a) L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469 CrossRef CAS; (b) K. Biradha and M. J. Zaworotko, J. Am. Chem. Soc., 1998, 120, 6431 CrossRef CAS; (c) L. J. Childs, N. W. Alcock and M. Hannon, Angew. Chem., Int. Ed., 2001, 40, 1079 CrossRef CAS; (d) A. D. Bond, Chem. Commun., 2002, 1664 RSC; (e) M. Kidowaki and N. Tamaoki, Chem. Commun., 2003, 290 RSC.
- (a) K. T. Honman, A. M. Pivovar, J. A. Swift and M. D. Ward, Acc. Chem. Res., 2001, 34, 107 CrossRef CAS; (b) C. D. Wu, A. G. Hu, L. Zhang and W. B. Lin, J. Am. Chem. Soc., 2005, 127, 8940 CrossRef CAS; (c) V. Percec, M. Glodde, T. K. Bera, Y. Miura, I. Shiyanovskaya, K. D. Singer, V. S. K. Balagurusamy, P. A. Heiney, I. Schnell, A. Rapp, H.-W. Spiess, S. D. Hudsonk and H. Duank, Nature, 2002, 417, 384 CrossRef; (d) A. Corna, F. Rey, J. Rius, M. J. Sabater and S. Valencla, Nature, 2004, 431, 287 CrossRef CAS.
- (a) C. B. AakerOy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397 RSC; (b) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons Ltd, Chichester, 2000 Search PubMed.
- (a) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS; (b) G. Ferey, Science, 2001, 291, 994 CrossRef CAS; (c) C. Janiak, Dalton Trans., 2003, 2781 RSC; (d) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS; (e) S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS.
- (a) G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565 CrossRef CAS; (b) J. D. Dunitz, Chem. Commun., 2003, 545 RSC.
- (a) L. Dobrzanska, G. O. Lloyd, H. G. Raubenheimer and L. J. Barbour, J. Am. Chem. Soc., 2005, 127(38), 13134 CrossRef CAS; (b) L. Dobrzanska, G. O. Lloyd, H. G. Raubenheimer and L. J. Barbour, J. Am. Chem. Soc., 2006, 128, 698 CrossRef CAS; (c) X. C. Lin, Y. Y. Huang, J. P. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557 CrossRef CAS; (d) W. G. Lu, C. Y. Su, T. B. Lu, L. Jiang and J. M. Chen, J. Am. Chem. Soc., 2006, 128, 34 CrossRef CAS; (e) R. Zou, H. Sakurai and Q. Xu, Angew. Chem., Int. Ed., 2006, 45, 2542 CrossRef CAS.
- M. O. Awakeh, A. Badia, F. Brisse and X. H. Bu, Inorg. Chem., 2006, 45, 1560 CrossRef CAS.
- (a) J. C. MacDonald, P. C. Dorrestein and M. M. Pilley, Cryst. Growth Des., 2001, 1, 29 CrossRef CAS; (b) D. R. Trivedi, A. Ballabh and P. Dastidar, CrystEngComm, 2003, 5, 358 RSC; (c) Y. Akhriff, J. ServerCarrio, J. Garcia-Lozano, J. V. Folgado, A. Sancho, E. Escriva, P. Vitoria and L. Soto, Cryst. Growth Des., 2006, 6, 1124 CrossRef CAS.
- (a) D. Datta and A. Chakravorty, Inorg. Chem., 1983, 22, 1085 CrossRef CAS; (b) S. Goswami, W. Kharmawphlang, A. K. Deb and S.-M. Peng, Polyhedron, 1996, 15, 3635 CrossRef CAS.
- (a) H. Rau H. I. Durr and H. Bouas-Laurent, Photochromism: molecules and systems.Amsterdam: Elsevier; 1990 Search PubMed; (b) N. Tamai and H. Miyasaka, Chem. Rev., 2000, 100, 1875 CrossRef CAS; (c) S. Yagai, T. Karatsu and A. Kitamura, Chem.–Eur. J., 2005, 11, 4054 CrossRef CAS.
- (a) V. Glezer. In The Chemistry of the Hydrazo, Azo and Azoxy Groups; Patai, S., Ed.;Wiley & Sons: New York, 1997; p 729 Search PubMed; (b) J. Oakes, I. P. Wilkes, R. J. Clark and P. L. Gratton, J. Chem. Soc., Perkin Trans. 2, 1998, 2569 RSC.
- (a) J. H. Hartley, T. D. James and C. J. Ward, J. Chem. Soc., Perkin Trans. 1, 2000, 3155 RSC; (b) N. DiCesare and J. R. Lakowicz, J. Phys. Chem. A, 2001, 105, 6834 CrossRef CAS.
- (a) C. Renner and L. Moroder, ChemBioChem, 2006, 7, 868 CrossRef CAS; (b) I. Willner, Acc. Chem. Res., 1997, 30, 347 CrossRef CAS.
- A. Pramanik and G. Das, Cryst. Growth Des., 2008, 8, 3107 CrossRef CAS.
- S. A. I. N. T. SMART and XPREP, Siemens AnalyticalX-ray Instruments Inc., Madison, Wisconsin, USA, 1995 Search PubMed.
- G. M. Sheldrick, SADABS: software for Empirical Absorption Correction, University of Gottingen, Institute fur Anorganische Chemieder Universitat, Tammanstrasse 4, D-3400 Gottingen, Germany, 1999–2003 Search PubMed.
- G. M. Sheldrick, SHELXS-97, University of Gottingen, Germany, 1997 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- (a) P. Byabartta, S. Pal, C. Sinha, F.-L. Liao, K. Panneerselvam and T.-H. Lu, J. Coord. Chem., 2002, 55, 479 CrossRef CAS; (b) P. Chattopadhyay, B. K. Dolui and C. Sinha, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1997, 36, 429; (c) H. K. Fun, K. Chinnakali, X. F. Chen, X. H. Zhu and X. Z. You, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, IUC9900025 CrossRef.
- P. K. Thallapally, G. O. Lloyd, T. B. Wirsig, M. W. Bredenkamp, J. L. Atwood and L. J. Barbour, Chem. Commun., 2005, 5272 RSC.
- (a) I. Alfonso, M. Bolte, M. Bru, M. I. Burguete and S. V. Luis, CrystEngComm, 2009, 11, 735 RSC; (b) T. P. Ruiz, M. F. Gomez, J. J. L. Gonzalez and A. E. Koziol, Chem. Phys., 2006, 320, 164 CrossRef CAS; (c) J. P. M Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108 CrossRef CAS; (d) Y. Lu, T. Shi, Y. Wang, H. Yang, X. Yan, X. Luo, H. Jiang and W. Zhu, J. Med. Chem., 2009, 52, 2854 CrossRef CAS; (e) P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789 CrossRef CAS.
- J. Assmore, G. Sutherland, T. K. Whidden, P. S. White and C.–M. Wong, Can. J. Chem., 1985, 63, 1209 CAS.
- (a) R. T. Conley, Infrared Spectroscopy, Allyn & Bacon, Boston, 1966 Search PubMed; (b) K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons Inc., New York, 5th edn, 1997 Search PubMed.
- (a) A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS; (b) S. Z. Zhu, C. H. Xing, W. Xu, G. F. Jin and Z. T. Li, Cryst. Growth Des., 2004, 4, 53 CrossRef CAS; (c) Z. Han, Y. Gao, X. Zhai, J. Peng, A. Tian, Y. Zhao and C. Hu, Cryst. Growth Des., 2009, 9, 1225 CrossRef CAS.
- D. Banerjee, U. S. Ray, J. S. Wu, T. H. Lu and C. Sinha, Polyhedron, 2006, 25, 3077 CrossRef CAS.
- W.-H. Wang, P.-H. Xi, X.-Y. Su, J.-B. Lan, Z.-H. Mao, J.-S. You and R.-G. Xie, Cryst. Growth Des., 2007, 7, 741 CrossRef CAS.
- (a) D. Das, B. G. Chand, K. K. Sarker, J. Dinda and C. Sinha, Polyhedron, 2006, 25, 2333 CrossRef CAS; (b) T. K. Misra, D. Das, C. Sinha, P. K. Ghosh and C. K. Pal, Inorg. Chem., 1998, 37, 1672 CrossRef CAS.
- (a) S. A. Ibrahim, N. M. Rageh, A. A. Mohamad and Y. H. Ebead, J. Chem. Eng. Data, 1999, 44, 451 CrossRef CAS; (b) H. Esener and T. Uyar, Dyes Pigm., 2007, 72, 109 CrossRef.
- H. Mustroph, R. Haessner and J. Epperlein, J. Prakt. Chem., 1984, 326, 259 CrossRef CAS.
- G. Galiazzoa, P. Bortolusb and G. Gennaric, J. Photochem. Photobiol., A, 1999, 120, 161 CrossRef.
- J. Otsuki, K. Suwa, K. K. Sarker and C. Sinha, J. Phys. Chem. A, 2007, 111, 1403 CrossRef CAS.
- G. Grynkiewicz, M. Poeniea and R. Y. Tsien, J. Biol. Chem., 1985, 260, 3440 CAS.
- (a) A. Pramanik and G. Das, Tetrahedron, 2009, 65, 2196 CrossRef CAS; (b) A. Pramanik, M. Bhuyan and G. Das, J. Photochem. Photobiol., A, 2008, 197, 149 CrossRef CAS; (c) A. Pramanik, M. Bhuyan, R. Choudhury and G. Das, J. Mol. Struct., 2008, 879, 88 CrossRef CAS; (d) G. Das, P. K. Bharadwaj, M. B. Roy and S. Ghosh, Chem. Phys., 2002, 277, 145 CrossRef CAS.
- (a) A. I. Anzellotti, M. Sabat and N. Farrell, Inorg. Chem., 2006, 45, 1638 CrossRef CAS; (b) D. F. Acevedo, J. Balach, C. R. Rivarola, M. C. Miras and C. A. Barbero, Faraday Discuss., 2006, 131, 235 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: X-Ray crystallographic file of the structures in CIF format; bond parameters, selected non-covalent interactions, networks, powder XRD, UV-visible spectra, and fluorescence spectra. CCDC reference numbers 736670–736674. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b912454c |
| ‡ Crystal data for salt 1: Fw = C19H17N9OSBr2, M = 579.30, CCDC = 736670, T = 298(2) K, monoclinic, space group P2(1)/c, a = 12.1694(3), b = 13.9841(4), c = 14.3235(4)Å, β =105.557(2)°, V = 2348.24(11) Å3, Z = 4, µ = 3.572 mm−1, 5728 reflections, 2805 unique (Rint = 0.0379), R(F) = 0.0452 (I > 2σ(I),wR(F2) = 0.0839 (all data), salt 2: Fw = C9H10N5O4I, M = 379.12, CCDC = 736671, T = 298(2) K, monoclinic, space group P2(1)/n, a = 4.88350(10), b = 10.1720(3), c = 27.4296(8) Å, β = 91.328(2)°, V = 1362.20(6) Å3, Z = 4, µ = 2.372 mm−1, 3278 reflections, 2337 unique (Rint = 0.0402), R(F) = 0.0343 (I > 2σ(I), wR(F2) = 0.0752 (all data), salt 3: Fw = C9H10N4O5ClI, M = 416.56, CCDC = 736672, T = 298(2) K, monoclinic, space group P2(1)/n, a = 5.60440(10), b = 8.8412(2), c = 29.3023(6)Å, β = 91.328(2)°, V = 1445.60(5) Å3, Z = 4, µ = 2.426 mm−1, 3537 reflections, 2712 unique (Rint = 0.0213), R(F) = 0.0356 (I > 2σ(I), wR(F2) = 0.0485 (all data), salt 4: Fw = C9H10N4O5ClI, M = C13H11N5O3, CCDC = 736673, T = 298(2) K, monoclinic, space group P2(1)/c, a = 6.8263(2), b = 11.5549(3), c = 17.0562(4)Å, β = 96.7880(10)°, V = 1335.92(6) Å3, Z = 4, µ = 0.107 mm−1, 3238 reflections, 2780 unique (Rint = 0.0251), R(F) = R1 = 0.0473 (I > 2σ(I), wR(F2) = 0.0898 (all data), salt 5: Fw = C26H23N9O4, M = 525.53, CCDC = 736674, T = 298(2) K, monoclinic, space group P2(1), a = 5.6061(2), b = 15.8135(6), c = 14.0347(5) Å, β = 97.281(2)°, V = 1234.17(8) Å3, Z = 2, µ = 0.102−1, 3192 reflections, 2996 unique (Rint = 0.0521), R(F) = R1 = 0.0418 (I > 2σ(I), wR(F2) = 0.0730 (all data) |
| This journal is © The Royal Society of Chemistry 2010 |