Cadmium(II) complexes with 3,5-di(1H-imidazol-1-yl)benzoate: topological and structural diversity tuned by counteranions†
Zhi
Su
,
Kai
Cai
,
Jian
Fan
,
Shui-Sheng
Chen
,
Man-Sheng
Chen
and
Wei-Yin
Sun
*
Coordination Chemistry Institute, State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing National Laboratory of Microstructures, Nanjing University, Nanjing, 210093, China. E-mail: sunwy@nju.edu.cn; Fax: +86 25 8331 4502
First published on 15th September 2009
Abstract
Four new coordination polymers [Cd(L)(OAc)]·H2O (1), [Cd(L)(µ2-OH)]·H2O (2), [Cd2(L)2(µ3-SO4)(H2O)2]·5H2O (3) and [Cd(L)2]·H2O (4) with different structures and topologies were obtained by solvo/hydrothermal reactions of varied Cd(II) salts with 3,5-di(1H-imidazol-1-yl)benzoic acid (HL) and 1,3-bis(4-pyridyl)propane (bpp). The structures of 1–4 were characterized by single-crystal X-ray diffraction analysis and the results showed that no bpp ligands were incorporated in the resulting complexes, and more interestingly, complexes 1–3 were obtained by reactions of metal to ligand ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, however, when the reactions were carried out with a metal to ligand ratio of 1:2, the resulting complexes had the same structure, namely [Cd(L)2]·H2O (4). In contrast to the 2D network with 63-hcb topology of complex 1 with acetate as terminal ligand, complex 2 is a 3D (4,6)-connected net with µ2-hydroxyl bridging group. Complex 3 is an unprecedented 3D self-penetrating (3,4,5)-connected net with each sulfate as a µ3-bridge to link three Cd(II) atoms, and complex 4 is a 3D self-penetrating (3,5)-connected net formed by two alternate chiral helical chains linking 2-fold interpenetrating 2D (4,82) nets. The results showed that the L− ligand has varied coordination modes and the counteranions have a great impact on the structure of the complexes.
:1, however, when the reactions were carried out with a metal to ligand ratio of 1:2, the resulting complexes had the same structure, namely [Cd(L)2]·H2O (4). In contrast to the 2D network with 63-hcb topology of complex 1 with acetate as terminal ligand, complex 2 is a 3D (4,6)-connected net with µ2-hydroxyl bridging group. Complex 3 is an unprecedented 3D self-penetrating (3,4,5)-connected net with each sulfate as a µ3-bridge to link three Cd(II) atoms, and complex 4 is a 3D self-penetrating (3,5)-connected net formed by two alternate chiral helical chains linking 2-fold interpenetrating 2D (4,82) nets. The results showed that the L− ligand has varied coordination modes and the counteranions have a great impact on the structure of the complexes.
Introduction
Metal–organic frameworks (MOFs) have attracted much attentions from chemists not only due to their intriguing structures and diverse topologies, but also due to their potential applications in many fields.1,2 Topological analysis was found to be an efficient approach to access and simplify the MOFs' structures, especially for the complicated two- (2D) and three-dimensional (3D) structures.3 Wells, O'Keeffe and Yaghi, et al. have performed important and interesting work in this field.4 The self-penetration (also refers to the self-catenation or polyknotting), as one type of entanglement in polymeric architectures, is of great interest owing to its intriguing aesthetic structure as well as the promising application as functional materials.3a,5 Self-penetrated structure is a single net having the feature that the smallest topological rings are catenated by other rings belonging to the same net.6 However, limited MOFs with the self-penetrating phenomenon have been observed.3a,5d,7On the other hand, the imidazole-containing ligands, especially for the imidazole-containing flexible ligand as the auxiliary ligands, have proven popular in recent years in the construction of MOFs with diverse topology.8 However, the ligands with rigid imidazole and carboxylate groups have not been studied thoroughly up to now,9 and in this paper we designed and synthesized a rigid ligand with one carboxylate and two imidazole groups, namely 3,5-di(1H-imidazol-1-yl)benzoic acid (HL), as a continued investigation extended from our systematic research of imidazole-containing tripodal ligand 1,3,5-tris(1-imidazolyl)benzene, which has remarkable features in the construction of MOFs.10 First, its rigid skeleton may favor the construction of a high-dimensional structure with high thermal stability; second, the variable coordination modes of the carboxylate group can satisfy different geometric requirement of the metal centers; third, weak interactions, such as hydrogen bonding, π–π interactions, can be expected in the structural construction.11
Herein, four Cd(II) complexes with different structures and topologies, namely [Cd(L)(OAc)]·H2O (1) (OAc = acetate), [Cd(L)(µ2-OH)]·H2O (2), [Cd2(L)2(µ3-SO4)(H2O)2]·5H2O (3), [Cd(L)2]·H2O (4), were obtained by reactions of HL with distinct Cd(II) salts in the presence of 1,3-bis(4-pyridyl)propane (bpp). The results revealed that ligand L− can have varied coordination modes and is an efficient building block in the construction of MOFs. The influence of the counteranions and reaction conditions on the structures was discussed and thermogravimetric analyses and photoluminescence properties of the complexes were investigated.
Experimental
Materials and methods
All commercially available chemicals are of reagent grade and used as received without further purification. Ligand HL was prepared according to procedures reported previously.12 Elemental analyses of C, H, and N were taken on a Perkin-Elmer 240C elemental analyzer at the analysis center of Nanjing University. Infrared spectra (IR) were recorded on a Bruker Vector22 FT-IR spectrophotometer by using KBr pellets. Thermogravimetric analyses (TGA) were performed on a simultaneous SDT 2960 thermal analyzer under nitrogen with a heating rate of 10 °C min−1. The luminescence spectra for the powdered solid samples were measured at room temperature on an Aminco Bowman Series2 spectrofluorometer with a xenon arc lamp as the light source. In the measurements of emission and excitation spectra the pass width is 5 nm. All measurements were carried out under the same experimental conditions.X-Ray crystallography
The crystallographic data collections for 1–4 were carried out on a Bruker Smart Apex CCD area-detector diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at 20(2) °C using the ω-scan technique. The diffraction data were integrated by using the SAINT program,13 which was also used for the intensity corrections for the Lorentz and polarization effects. Semi-empirical absorption correction was applied using the SADABS program.14 The structures were solved by direct methods and all non-hydrogen atoms were refined anisotropically on F2 by the full-matrix least-squares technique using the SHELXL-97 crystallographic software package.15 All hydrogen atoms were generated geometrically. The hydrogen atom of O2 in 2 was not found, but according to the charge balance, it must be a hydroxyl. Atom O14 in 3 disordered into two positions with site occupancy of 0.17(3) and 0.83(3), respectively. All calculations were performed on a personal computer with the SHELXL-97 crystallographic software package. Details of the crystal parameters, data collection and refinements for 1–4 are summarized in Table 1. Selected bond lengths and angles for 1–4 are listed in Table S1. Further details are provided in the ESI.†| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| a R 1 = Σ||Fo| − |Fc||/Σ|Fo|. b wR 2 = |Σw(|Fo|2 − |Fc|2)|/Σ|w(Fo)2|1/2, where w = 1/[σ2(Fo2) + (aP)2 + bP]. P = (Fo2 + 2Fc2)/3. | ||||
| Chemical formula | C15H14CdN4O5 | C13H12CdN4O4 | C26H32Cd2N8O15S | C26H20CdN8O5 |
| Formula weight | 442.69 | 400.64 | 953.34 | 636.88 |
| Temperature/K | 293(2) | 293(2) | 293(2) | 293(2) |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/c | C2/c | P-1 | C2/c |
| a/Å | 11.689(10) | 13.0295(17) | 10.7731(15) | 32.732(3) |
| b/Å | 18.209(15) | 13.8924(18) | 12.8762(18) | 8.3330(8) |
| c/Å | 8.209(7) | 7.6163(10) | 13.8455(19) | 17.4514(15) |
| α/° | 90 | 90 | 99.3870(10) | 90 |
| β/° | 110.557(2) | 90.000(2) | 111.108(2) | 91.845(2) |
| γ/° | 90 | 90 | 106.958(3) | 90 |
| V/Å3 | 1636(2) | 1378.6(3) | 1634.6(4) | 4757.5(8) |
| Z | 4 | 4 | 2 | 8 |
| D c/g cm−3 | 1.789 | 1.916 | 1.908 | 1.773 |
| F(000) | 872 | 780 | 924 | 2544 |
| θ range /° | 1.86∼25.25 | 2.14∼25.04 | 1.65∼25.10 | 2.49∼26.00 |
| Reflns. collected | 8284 | 3444 | 8291 | 12344 |
| Independent reflns. | 2941 | 1231 | 5714 | 4664 |
| Goodness-of-fit | 0.875 | 1.120 | 0.879 | 1.141 |
| R 1 (I > 2σ (I)) | 0.0454 | 0.0265 | 0.0546 | 0.0417 |
| wR 2 (I > 2σ (I)) | 0.0758 | 0.0686 | 0.0913 | 0.1033 |
Results and discussions
Synthesis and the thermal stability of the complexes
Complexes 1–4 were synthesized by the solvo/hydrothermal method in the presence of bpp. It is noteworthy that no crystals were obtained by using base NaOH or Bu4NOH to neutralize the carboxylic acid without addition of bpp. The complete deprotonation of the carboxylic group of the HL ligand to give L− in 1–4 was confirmed by IR spectral data, since no IR bands in the range of 1760–1680 cm−1 were observed in the IR spectra of 1–4 (see Experimental section), as well as by the results of crystallographic analysis (vide infra). In addition, the metal to ligand ratio is crucial for the formation of the complexes. It is interesting to find that complexes 1–3 were obtained by reactions of metal to ligand ratio of 1:1 (no single crystals were obtained in preparation of 4 with metal to ligand ratio of 1:1), however, when the metal to ligand ratio was changed to 1:2, all the reactions resulted the same complex, namely [Cd(L)2]·H2O (4).
Thermogravimetric analyses were performed to verify the thermal stability of the complexes. As shown in Fig. S1,† complex 1 shows a weight loss of 4.14% before 200 °C corresponding to the release of free water molecule (calc. 4.07%) and further decomposition occurred at 355 °C. For 2, the weight loss starts at ca. 50 °C with the liberation of the uncoordinated water molecule with a weight loss of 5.09% (calc. 4.49%), and the decomposition of the residue was observed at about 335 °C. A total weight loss of 13.25% was observed for 3 in the temperature range 40–170 °C, which is ascribed to the loss of both coordinated and uncoordinated water molecules (calc. 13.22%), and the residue is stable up to about 325 °C. Complex 4 loses weight in the temperature range 130–170 °C, and the loss is 3.29%, which is attributed to the departure of free water molecule (calc. 2.82%) and the residue is stable up to 340 °C.
Crystal structure of [Cd(L)(OAc)]·H2O (1)
The results of X-ray crystallographic analysis revealed that the asymmetric unit of complex 1 contains one ligand L−, one Cd(II) cation, an acetate anion and a free water molecule. As exhibited in Fig. 1a, each Cd(II) atom is six-coordinated by two nitgrogen (N1, N3A) atoms of imidazole and two oxygen (O1B, O2B) atoms of a chelating carboxylate group from three different L− ligands, and two oxygen (O3, O4) atoms from a chelating acetate anion. The Cd–O distances are ranging from 2.300(4) to 2.406(4) Å, the Cd–N distances are 2.251(5) and 2.266(5) Å, respectively, and the coordination angles around the Cd1 are in the range of 55.36(15)–145.37(17) (Table S1). On the other hand, each ligand L− in turn links three Cd(II) atoms by its two imidazole groups and carboxylate with µ1-η1:η1-chelate fashion (Scheme 1a) to generate an infinite 2D network with typical 63-hcb topology when the centroid of each L− ligand was treated as a 3-connected node as illustrated in Fig. 1b and 1c. Three Cd(II) atoms and three L− ligands (each L− using two of its three arms connects two Cd(II) atoms) form a 28-membered macrocyclic ring through Cd–O and Cd–N bonds (Fig. 1c). In this macrometallcycle, the lengths of the edges are not equivalent, and the intermetallic separations of Cd1⋯Cd1A, Cd1⋯Cd1B and Cd1A⋯Cd1B are 10.42, 11.69 and 11.75 Å, respectively. Meanwhile, each acetate anion is alternating above and below the 2D 63-hcb plane (Fig. S2).†
 | ||
| Scheme 1 The coordination modes of L− ligands in complexes 1–4. | ||
 | ||
| Fig. 1 (a) The coordination environment of Cd(II) atom in 1 with the ellipsoids drawn at the 30% probability level. The hydrogen atoms and water molecule are omitted for clarity. Symmetry Code: A x − 1, y, z, B −x, y − 1/2, −z + 3/2. (b) The representation of 63-hcb net. Green: Cd; Red: the centroid of L− ligand. (c) The 2D network structure of 1. | ||
It can be seen clearly, the 2D layers repeat in an …ABAB… stacking sequence along the c axis, and each pair of benzene rings of the L− ligand is nearly parallel with a dihedral angle of 6.40°, and separated by a centroid-centroid distance of 4.14 Å, indicating the presence of weak π–π stacking interactions.16 Such π–π interactions linked the 2D layer to 3D framework (Fig. S3).† Furthermore, the C–H⋯O hydrogen bonds formed by the O atoms of the acetate and the H atoms of the adjacent layers further consolidate the 3D framework (Fig. S4),† and the details of the hydrogen bonds are listed in Table S2.† The O⋯O distances of 2.814 and 2.912 Å (Table S2†) indicate that there are also O–H⋯O hydrogen bonds although the hydrogen atoms of the uncoordinated water molecule could not be located.
Crystal structure of [Cd(L)(µ2-OH)]·H2O (2)
In order to evaluate the influence of the counteranion on the structure of the complex, Cd(BF4)2·6H2O was used to react with HL, instead of Cd(OAc)2·2H2O, and complex 2 was successfully isolated. The asymmetric unit of 2 contains half of a [Cd(L)(µ2-OH)]·H2O fragment, namely one Cd(II) atom sitting on the inversion center, half L− ligand lying on a twofold axis, and one µ2-hydroxyl group and one uncoordinated water molecule at special position. As exhibited in Fig. 2a, the Cd(II) atom is six-coordinated with octahedral geometry by two oxygen (O1, O1D) atoms and two nitrogen (N1B, N1C) atoms from four distinct L− ligands, and two oxygen (O2, O2E) atoms from two hydroxyl groups. The Cd1–O1, Cd1–O2 and Cd1–N1 bond lengths are 2.350(3), 2.1919(15) and 2.281(4) Å, respectively, and the angles around Cd(II) atom range from 84.39(10) to 180.0° (Table S1).† On the other hand, each L− ligand links four Cd(II) atoms using its two imidazole groups and carboxylate with µ2-η1:η1-bridging mode (Scheme 1b), which is different from the L− ligand in 1 (Scheme 1a).
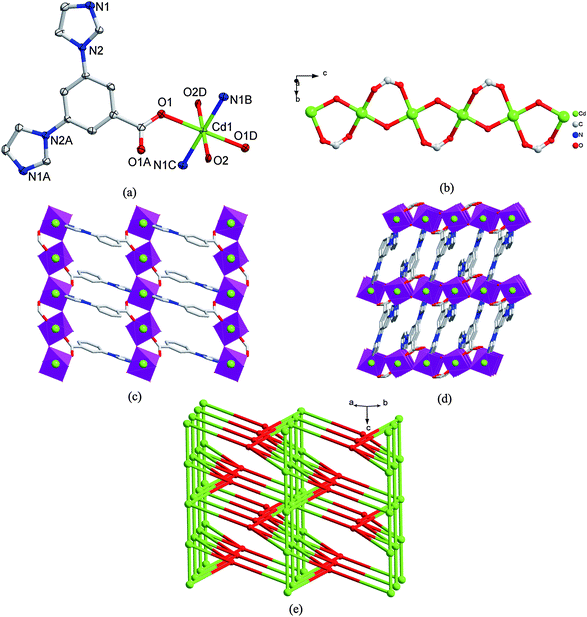 | ||
| Fig. 2 (a) The coordination environment of Cd(II) atom in 2 with the ellipsoids drawn at the 30% probability level. The hydrogen atoms and water molecule are omitted for clarity. Symmetry code: A 2 − x, y, 1/2 − z, B −x + 5/2, −y + 3/2, −z, C x − 1/2, y − 1/2, z, D −x + 2, −y + 1, −z. (b) An infinite 1D chain structure formed by µ2-O of hydroxyl and µ2-η1:η1-bridging mode of the carboxylate group in 2. (c) A view of infinite 2D network in 2. (d) 3D structure of 2. (e) Schematic illustrating topology of the binodal (4,6)-connected net of 2. Red balls: the centroid of L− ligands; Green balls: Cd(II) atoms. | ||
The most striking feature of 2 is that there are µ2-OH bridges together with the carboxylate bridges of the L− ligands linking Cd(II) atoms to form an infinite one-dimensional (1D) chain structure as illustrated in Fig. 2b. The 1D chains are linked together by the 3-imidazole group of the L− ligand via Cd1–N1 coordination bonds to give a 2D network structure as depicted in Fig. 2c. Such 2D layers are further connected by 5-imidazole group of the L− ligand to give a 3D architecture via Cd1–N1A coordination interactions (Fig. 2d). It is obvious that the structures of complexes 1 and 2 are different: 2D network of 1 with coordinated acetate counteranion, and 3D net of 2 with µ2-OH bridge. Such difference may be ascribed to the different coordination modes of L− ligand and the different size and coordination ability of the counteranions of OAc− and BF4−.
To get a better insight into the present 3D framework structure, topological analysis was carried out for 2. Each Cd(II) atom connects four distinct L− ligands (vide supra) and two other Cd(II) atoms via µ2-OH, hence, each Cd(II) can be regarded as a 6-connected node. While each L− ligand in turn connects four Cd(II) atoms using its three arms, and thus each L− can be treated as 4-connected node. Such connectivity repeats infinitely to give the 3D framework as schematically shown in Fig. 2e. According to the simplification principle and the analysis of TOPOS,17 the resulting structure of 2 is a rare binodal (4,6)-connected net, and its Point (Schläfli) symbol is (3·42·52·6)(32·42·52·64·74·8) which can be derived from the (6,8)-connected seh net by simplifying some corresponding bonds (Fig. S5†).18 There were only a few binodal (4,6)-connected nets reported in RCSR,4b,19 such as soc, she, stp, toc, gar, ibd and so on, according to the geometry of nodes. The 4-connected nodes have square and tetrahedral geometries, and the 6-connected nodes have hexagonal, octahedral and trigonal prism geometries, respectively. It can be seen that the 4- and 6-connected node in complex 2 are tetrahedral and octahedral geometries, respectively.
Crystal structure of [Cd2(L)2(µ3-SO4)(H2O)2]·5H2O (3)
It is interesting that complex 3 with different structure was isolated as single crystals when Cd(SO4)·8/3H2O was used to react with HL. The X-ray crystallographic analysis revealed that 3 crystallized in triclinic space group P-1. In contrast to the only one unique Cd(II) atom in the asymmetric unit of 1 and 2, there are two Cd(II) atoms with different coordination environments in the asymmetric unit of 3. Cd1 atom is seven-coordinated by four carboxylate oxygen (O1, O2, O3, O4) atoms and one imidazole nitrogen (N3A) atom from three distinct L− ligands, one oxygen (O6) atom of sulfate and one terminal water (O10) molecule (Fig. 3a). The Cd1–N bond length is 2.250(6) Å and the Cd1–O bond lengths are in the range of 2.266(5)–2.563(5) Å (Table S1†). Comparably, Cd2 is six-coordinated with distorted octahedral geometry by three imidazole nitrogen (N1B, N5C, N7D) atoms from three distinct L− ligands, two oxygen (O7, O7E) atoms of two sulfates and one terminal water (O5) molecule. The bond lengths of Cd2–O7 and Cd2–O7E are 2.431(5) and 2.337(5) Å which are comparable to the other reported Cd(II) complexes.20 On the other hand, two different L− ligands in the asymmetric unit of 3 adopt the same coordination mode, each connects three Cd(II) atoms using its two imidazole groups and one carboxylate with chelate fashion, which is the same as that in complex 1 (Scheme 1a). | ||
| Fig. 3 (a) Coordination environment of Cd(II) atoms in 3 with the ellipsoids drawn at the 30% probability level. The hydrogen atoms and free water molecules are omitted for clarity. Symmetry Code: A x − 1, y, z, B −x + 2, −y + 1, −z, C −x + 2, −y, −z + 1, D −x + 1, −y, −z + 1; E −x + 2, −y + 1, −z + 1. (b) 3D framework of 3 and the Cd4(SO4)2 unit. (c) Representation of (3,4,5)-connected net (left) and a close-up view of self-penetrated rings in 3 (right). | ||
If the bridging linkages of the sulfate in 3 are ignored, the coordination of L− ligands with Cd(II) atoms forms a 2D network with 63-hcb topology which is similar to that of 1, and the adjacent layers repeat in an …ABCDA… stacking sequence along the b axis (Fig. S6).† The dihedral angles between layers A and B, layers B and C, layers C and D are 0, 8.53, and 0°, respectively, namely, the layers A and B as well as the layers C and D are parallel to each other, and the centroid-to-centroid distances are 3.73, 4.23 and 3.98 Å, respectively, indicating the presence of π–π stacking interactions (Fig. S7).† It should be noticed that the sulfate as µ3-connector links three Cd(II) atoms from the adjacent layers and two sulfate anions connect four Cd(II) atoms from adjacent four layers to form a Cd4(SO4)2 unit, which further connected the 2D networks to generate a 3D framework (Fig. 3b). Uncoordinated water molecules fill in the voids of the 3D net and occupy the 15.3% unit cell volume (250.8 Å3/1634.6 Å3) calculated by PLATON software (Fig. S8).† Therefore, the ultimate 2D network of 1 and the 3D net of 3 are caused by the different anions of OAc− and SO42− in the structural construction except for the same counteranions to balance the charge: the OAc− in 1 is just a terminal coordinating ligand, while the SO42− in 3 as bridging ligand links 2D networks to form 3D net.
A similar topological approach was used to analyze the intricate structure of 3. According to the above depiction, each Cd1 atom was connected by three L− ligands and one sulfate anion, hence, it can be considered as a 4-connected node through omitting the terminal coordinating water molecule. Comparably, each Cd2 atom was linked by three L− ligands and two sulfate anions, thus, it can be viewed as a 5-connected node. On the other hand, each of the two different L− ligands as well as the sulfate anion in 3 connected three Cd(II) atoms, they can thus be treated as 3-connected node, respectively. The whole framework of 3 was accordingly simplified as illustrated in Fig. 3c, which is a rare penta-nodal (3,4,5)-connected net with its Point (Schläfli) symbol (4·63·85·10)(4·82)(63·83)(63)2.21 From a structural view, the ultimate topology of 3 is the addition of 2D 63-hcb topology with 3-connected SO42− nodes. A closer examination of a portion of the network (Fig. 3c) reveals that the 6-membered circuits formed by two sulfate nodes and two Cd(II) atoms together with two L− nodes from adjacent two layers weave through the 6-membered circuits of the 2D 63-hcb without sharing a single connecting node. Thus, to the best of our knowledge, 3 can be considered as the first example of a self-penetrating MOF with (3,4,5)-connected topology.
Crystal structure of [Cd(L)2]·H2O (4)
When the HL ligand was used to react with CdCO3, which is obviously different from other Cd(II) salts used for the preparation of 1–3 since the CdCO3 can neutralize the acid by itself, and complex 4 with a different structure was isolated. It is interesting that complex 4 crystallizes in monoclinic space group C2/c without co-ligand bpp. In the asymmetric unit, there are one Cd(II) center, two L− ligands and one uncoordinated water molecule. The central Cd1 atom is penta-coordinated by four imidazole nitrogen (N1, N3A, N5, N7B) atoms and one carboxylate oxygen (O1C) atom from five different L− ligands (Fig. 4a). The Cd1–O bond distance is 2.441(3) Å, and the average Cd1–N bond length is 2.316 Å. The distances of 2.64 Å (Cd1–O2) and 2.69 Å (Cd1–O3) indicate that there are weak interactions between Cd1 and O2 as well as Cd1 and O3, but they are longer than the Cd–O bond distances in the previously reported Cd(II) complexes.20 On the other hand, two L− ligands adopt different coordination modes, one as a µ2-bridge connecting two Cd(II) atoms using its two imidazole groups and the carboxylate group is free of coordination (Scheme 1c), the other one as a µ3-bridge connecting three Cd(II) atoms by its two imidazole groups and the carboxylate with monodentate fashion (Scheme 1d). It is clear that complex 4 is different from complexes 1–3, no counteranions were presented in the framework of 4 and the charge balance is completely dependent on L− ligands, while there are OAc− in 1, OH− in 2 and SO42− in 3 as counteranions, respectively.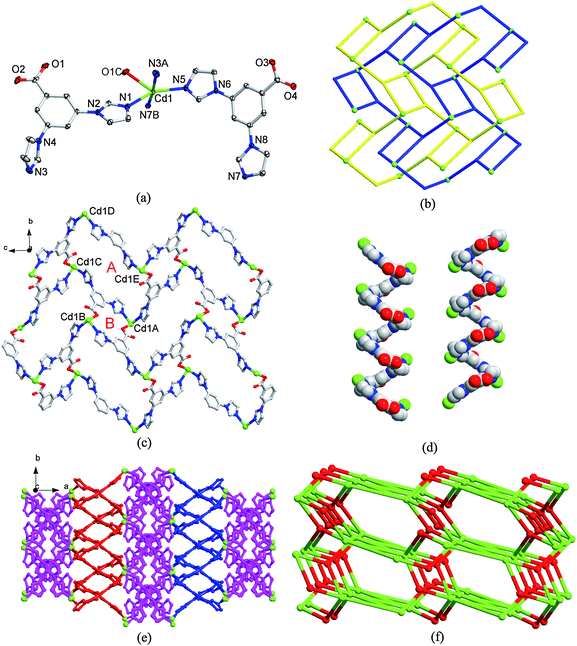 | ||
| Fig. 4 (a) Coordination environment of the Cd(II) atom in 4 with the ellipsoids drawn at the 30% probability level. The hydrogen atoms and free water molecule are omitted for clarity. Symmetry code: A x, −y, z + 1/2, B −x + 1/2, y + 1/2, C −x, −y + 1, −z + 1. (b) Representation of 2-fold interpenetrated (4·82) nets. (c) 2D network of 4 formed by µ3-bridge L− ligand and Cd(II) atoms with A and B macrocycles. (d) L- and R-handedness helical chains formed by µ2-bridge L− ligands and Cd(II) atoms. (e) 3D framework of 4. Pink: 2-fold interpenetrated (4·82) nets; blue and red: L- and R-handedness helical chains, respectively. (f) The (3,5)-connected net of 4. Green: Cd(II); Red: the centroid of L− ligand. | ||
When the µ2-bridge L− ligands are neglected, the connections between the µ3-bridge L− ligands and Cd(II) atoms form 2-fold interpenetrated 2D network with 3-connected (4,82) topology (Fig. 4b), where both the L− ligands and Cd(II) atoms act as 3-connectors, which has been rarely found in the imidazole-containing complexes and most of them are square (4,4) or hexagonal 63-hcb net like complex 1.22 There are two macrocyclic rings, A and B, in the 2D net (Fig. 4c). A, a 38-membered ring, was formed by four L− ligands (each L− using two of its three arms connects two Cd(II) atoms) and four Cd(II) atoms, with intermetallic separations of 12.71 Å of Cd1A⋯Cd1C and Cd1D⋯Cd1E, 11.59 Å of Cd1A⋯Cd1E and Cd1C⋯Cd1D, respectively. B, an 18-membered ring, was constituted by two L− ligands and two Cd(II) atoms with intermetallic separation of 9.86 Å of Cd1A⋯Cd1B (Fig. 4c). Meanwhile, the µ2-bridge L− ligands connecting Cd(II) atoms form two alternate chiral helical chains along the b axis, L- and R-handedness, respectively (Fig. 4d). The 2-fold interpenetrated layers stack in an -ABAB-sequence parallel to the (b,c) plane, and are further interconnected by the chiral helical chains to form an ultimate 3D framework (Fig. 4e).
From a topological view of the complicated 3D framework of 4, each µ2- and µ3-bridge L− ligand can be regarded as 2- and 3-connector, respectively, and each Cd(II) atom can be treated as 5-connector. Such connectivity repeats infinitely to give the 3D framework of 4 as schematically shown in Fig. 4f. According to the analysis of TOPOS, the resulting structure of 4 is an unprecedented binodal (3,5)-connected net, and its Point (Schläfli) symbol is (4·6·8)(4·64·85). The network of compound 4 clearly results in self-penetrating since it is generated from linking 2-fold-interpenetrated sheets (Fig. S9).† The self-penetration feature is not very common within coordination polymers, and only limited numbers of self-penetration nets have been reported.3a, 5d, 7 Similar self-penetrating (3,5)-connected net through linking two 2-fold interpenetrated (4,82) net, [Ni(oba)(bpy)]·2H2O (5) (oba = 4,4′-oxybis(benzoate); bpy = 4,4′-bipyridine) with Point (Schläfli) symbol (4·82)(4·64·84·10), has been reported by Wang et al.22c Obviously, the difference between these two complexes is that 5 uses rigid linear bpy ligand to link the 2-fold layers, while complex 4 uses chiral helical chains. On the other hand, the structure of 5 was constructed by mixed ligands, one rigid and one flexible, while the one of complex 4 was constructed by one rigid ligand through adopting different coordination modes.
Comparison and the effect of the counteranions
It is well known that the carboxylate-containing ligands are good candidates for the construction of coordination frameworks with an interesting structure and specific topology due to their variable coordination modes.23 Therefore, comparison and comprehension of the coordination modes of the ligand L− in complexes 1–4 will be a good path to understanding the complicated structures. There are four different kinds of coordination modes (Scheme 1) of the L− ligand in complexes 1–4 connecting two, three and four Cd(II) atoms, respectively. In addition, bpp was added in the preparation reactions of 1–4, however, no bpp appeared in the resulting complexes, even when a double amount of bpp was used. In the case of 1–3, the bpp may simply serve as a base to neutralizing the carboxylic acid of HL since no additional base was added, while in the case of 4, the CdCO3 can neutralize the carboxylic acid of HL and bpp can act as co-ligand, however, no complex with bpp was isolated.Furthermore, the final structures of the complexes are not only determined by the coordination modes of the ligand, but are also subtly influenced by the counteranions regarding their different sizes, shapes and coordination abilities. The obvious examples are complexes 1 and 3. The coordination of L− and Cd(II) atoms formed the 2D 63-hcb nets in both 1 and 3, and the 2D layers packed together through the π–π stacking interactions. However, the final structure of 1 is a 2D network because the acetate anions just act as the teminal ligands coordinating to the Cd(II) atoms with chelate fashion, while 3 is 3D net because the sulfate anion acts as a µ3-bridge connected to three Cd(II) atoms and further linking the 2D to 3D framework. In complex 2, OH− appearing as a µ2-bridge may probably be due to the big size and weak coordination ability of BF4−. In 4, the CO32− may be neutralized by the carboxylic acid of HL and the main framework is just constructed by the L− and Cd(II) atoms.
Photoluminescence of complexes 1–4
The mixed inorganic–organic hybrid coordination polymers have been investigated for fluorescence properties in view of potential applications. The photoluminescence of ligand HL and complexes 1–4 was studied in the solid state at room temperature under the same experimental conditions, and all measurements of emission spectra were excited at a wavelength of 397 nm. As shown in Fig. 5, emission bands were observed at 467 nm for HL, 450 nm for 1, 463 nm for 2, 466 nm for 3, 468 nm for 4, respectively. The emission bands of complexes 1–4 may be assigned to intraligand fluorescence due to their similarity to that of HL.24 In addition, it is noteworthy that complex 1 showed intense emission compared with those of other complexes, which may be attributed to the different rigidity of the crystal packing in the solid state.25 The observed blue or red shifts of the emission maximum between the complexes and the HL ligand are considered to mainly originate from the coordination interactions between the metal atoms and the ligands.24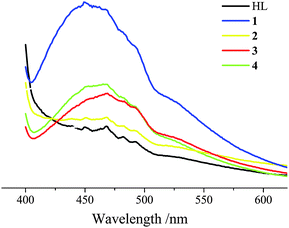 | ||
| Fig. 5 Emission spectra of HL and complexes 1–4 in the solid state at room temperature. | ||
Conclusions
Assembly reactions of Cd(II) salts with imidazole- and carboxylate-containing ligand, 3,5-di(1H-imidazol-1-yl)benzoic acid, result in the formation of four distinct coordination polymers with completely different structures and topologies. In addition, the photoluminescence properties of complexes 1–4 were investigated, and the results showed that they are candidates for photoactive materials. The present study showed that both the coordination modes of the ligand and the counteranions play an important role in determining the structure of the complexes.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant no. 20731004 and 20721002) and the National Basic Research Program of China (Grant no. 2007CB925103).References
- (a) C. H. Li, K. L. Huang, Y. N. Chi, X. Liu, Z. G. Han, L. Shen and C. W. Hu, Inorg. Chem., 2009, 48, 2010 CrossRef CAS; (b) J. Zhang and X. H. Bu, Chem. Commun., 2009, 206 RSC; (c) M. J. Prakash, Y. Zou, S. H. Hong, M. Park, M. P. N. Bui, G. H. Seong and M. S. Lah, Inorg. Chem., 2009, 48, 1281 CrossRef CAS; (d) J. Heo, Y. M. Jeon and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 7712 CrossRef CAS; (e) R. L. Sang and L. Xu, Chem. Commun., 2008, 6143 RSC; (f) S. Z. Zhan, M. Li, J. Z. Hou, J. Ni, D. Li and X. C. Huang, Chem.–Eur. J., 2008, 14, 8916 CrossRef CAS.
- (a) J. P. Zhao, B. W. Hu, E. C. Sanudo, Q. Yang, Y. F. Zeng and X. H. Bu, Inorg. Chem., 2009, 48, 2482 CrossRef CAS; (b) H. Y. Ye, D. W. Fu, Y. Zhang, W. Zhang, R. G. Xiong and S. P. D. Huang, J. Am. Chem. Soc., 2009, 131, 42 CrossRef CAS; (c) C. Zhang, Y. Cao, J. F. Zhang, S. M. T. Matsumoto, Y. L. Song, J. Ma, Z. X. Chen, K. Tatsumi and M. G. Humphrey, Adv. Mater., 2008, 20, 1870 CrossRef CAS; (d) H. J. Choi, M. Dinca and J. R. Long, J. Am. Chem. Soc., 2008, 130, 7848 CrossRef CAS; (e) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keefee and O. M. Yaghi, Science, 2002, 295, 469 CrossRef; (f) M. Dinca, W. S. Han, Y. Liu, A. Dailly, C. M. Brown and J. R. Long, Angew. Chem., Int. Ed., 2007, 46, 1419 CrossRef CAS; (g) C. D. Wu and W. Lin, Angew. Chem., Int. Ed., 2007, 46, 1075 CrossRef CAS.
- (a) Y. Q. Lan, X. L. Wang, S. L. Li, Z. M. Su, K. Z. Shao and E. B. Wang, Chem. Commun., 2007, 4863 RSC; (b) L. Han, W. N. Zhao, Y. Zhou, X. Li and J. G. Pan, Cryst. Growth Des., 2008, 8, 3504 CrossRef CAS; (c) L. L. Fan, D. R. Xiao, E. B. Wang, Y. G. Li, Z. M. Su, X. L. Wang and J. Liu, Cryst. Growth Des., 2007, 7, 592 CrossRef CAS.
- (a) A. F. Wells, Three-Dimensional Nets and Polyhedra, Wiley-Interscience, New York, 1977 Search PubMed; (b) M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782 CrossRef CAS; (c) N. W. Ockwing, O. Delgado-Freedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176 CrossRef CAS.
- (a) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef; (b) X. L. Wang, C. Qin, E. B. Wang, Z. M. Su, L. Xua and S. R. Batten, Chem. Commun., 2005, 4789 RSC; (c) M. H. Bi, G. H. Li, Y. C. Zou, Z. Shi and S. H. Feng, Inorg. Chem., 2007, 46, 604 CrossRef CAS; (d) Y. Qi, Y. X. Che and J. M. Zheng, Cryst. Growth Des., 2008, 8, 3602 CrossRef CAS.
- L. Carlucci, G. Ciani and D. M. Proserpio, Coord. Chem. Rev., 2003, 246, 247 CrossRef CAS.
- (a) M. H. Bu, G. H. Li, Y. C. Zou, Z. Shi and S. H. Feng, Inorg. Chem., 2007, 46, 604 CrossRef CAS; (b) X. L. Wang, C. Qin, E. B. Wang and Z. M. Su, Chem.–Eur. J., 2006, 12, 2680 CrossRef CAS; (c) M. R. Montney, S. M. Krishnan, N. M. Patel, R. M. Supkowski and R. L. Laduca, Cryst. Growth Des., 2007, 7, 1145 CrossRef CAS.
- (a) L. Carlucci, G. Ciani, S. Maggini and D. M. Proserpio, Cryst. Growth Des., 2008, 8, 162 CrossRef CAS; (b) Y. Qi, F. Luo, S. R. Batten, Y. X. Che and J. M. Zheng, Cryst. Growth Des., 2008, 8, 2806 CrossRef CAS; (c) Y. Y. Liu, J. F. Ma, J. Yang and Z. M. Su, Inorg. Chem., 2007, 46, 3027 CrossRef CAS; (d) J. Zhang, E. Chew, S. M. Chen, J. T. H. Pham and X. H. Bu, Inorg. Chem., 2008, 47, 3495 CrossRef CAS.
- (a) J. Z. Zhang, W. R. Cao, J. X. Pan and Q. W. Chen, Inorg. Chem. Commun., 2007, 10, 1360 CrossRef CAS; (b) J. Gao, K. J. Wei, J. Ni and J. Z. Zhang, J. Coord. Chem., 2009, 62, 257 CrossRef CAS; (c) J. Xu, Q. Yuan, Z. S. Bai and W. Y. Sun, Inorg. Chem. Commun., 2009, 12, 58 CrossRef CAS.
- (a) W. Y. Sun, J. Fan, T. -a. Okamura, J. Xie, K. B. Yu and N. Ueyama, Chem.–Eur. J., 2001, 7, 2557 CrossRef CAS; (b) J. Fan, L. Gan, H. Kawaguchi, W. Y. Sun, K. B. Yu and W. X. Tang, Chem.–Eur. J., 2003, 9, 3965 CrossRef CAS; (c) S. Y. Wan, J. Fan, T. -a. Okamura, H. F. Zhu, X. M. Ouyang, W. Y. Sun and N. Ueyama, Chem. Commun., 2002, 2520 RSC; (d) J. Fan, M. H. Shu, T. -a. Okamura, Y. Z. Li, W. Y. Sun, W. X. Tang and N. Ueyama, New J. Chem., 2003, 27, 1307 RSC.
- (a) S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS; (b) K. E. Holmes, P. F. Kelly and M. R. Elsegood, Dalton Trans., 2004, 3488 RSC; (c) P. S. Wang, C. N. Moorefield, M. Panzer and G. R. NewKome, Chem. Commun., 2005, 465 RSC; (d) H. F. Zhu, J. Fan, T. -a. Okamura, Z. H. Zhang, G. X. Liu, K. B. Yu, W. Y. Sun and N. Ueyama, Inorg. Chem., 2006, 45, 3941 CrossRef CAS; (e) Z. H. Zhang, T. -a. Okamura, Y. Hasegawa, H. Kawaguchi, L. Y. Kong, W. Y. Sun and N. Ueyama, Inorg. Chem., 2005, 44, 6219 CrossRef CAS.
- Z. Su, Z. S. Bai, J. Xu, T. -a. Okamura, G. X. Liu, Q. Chu, X. F. Wang, W. Y. Sun and N. Ueyama, CrystEngComm, 2009, 11, 873 RSC.
- SAINT, version 6.2; Bruker AXS, Inc., Madison, WI, 2001 Search PubMed.
- G. M. Sheldrick. SADABS, University of Göttingen, Göttingen, Germany Search PubMed.
- G. M. Sheldrick, SHELXTL, version 6.10, Bruker Analytical X-ray Systems, Madison, WI, 2001 Search PubMed.
- S. Y. Wan, Y. Z. Li, T. -a. Okamura, J. Fan, W. Y. Sun and N. Ueyama, Eur. J. Inorg. Chem., 2003, 3783 CrossRef CAS.
- (a) V. A. Blatov, IUCr CompComm Newsletter, 2006, 7, 4–38 Search PubMed TOPOS is available at http://www.topos.ssu.samara.ru; (b) V. A. Blatov, TOPOS, A Multipurpose Crystallochemical Analysis with the Program Package; Samara State University, Russia, 2004 Search PubMed; (c) R. Hoffmann, V. I. Minkin and B. K. Carpenter, Bull. Soc. Chim. Fr., 1996, 133, 117 CAS.
- V. A. Blatov and D. M. Proserpio, Acta Crystallogr., Sect. A: Found. Crystallogr., 2009, 65, 202 CrossRef.
- Reticular Chemistry Structure Resource (RCSR) at http://rcsr.anu.edu.au/.
- (a) D. P. Martin, R. M. Supowski and R. L. LaDuca, Dalton Trans., 2009, 514 RSC; (b) Y. H. He, Y. L. Feng, Y. Z. Lan and Y. H. Wen, Cryst. Growth Des., 2008, 8, 3586 CrossRef CAS; (c) M. Du, X. J. Jiang and X. J. Zhao, Inorg. Chem., 2006, 45, 3998 CrossRef CAS; (d) M. Xue, G. S. Zhu, Y. J. Zhang, Q. R. Fang, L. J. Hewitt and S. L. Qiu, Cryst. Growth Des., 2008, 8, 427 CrossRef CAS.
- M. Xue, G. S. Zhu, H. Ding, L. Wu, X. J. Zhao, Z. Jin and S. L. Qiu, Cryst. Growth Des., 2009, 9, 1481 CrossRef CAS.
- (a) J. Fan, W. Y. Sun, T. -a. Okamura, W. X. Tang and N. Ueyama, Inorg. Chem., 2003, 42, 3168 CrossRef CAS; (b) S. A. Barnett, A. J. Blake, N. R. Champness, J. E. B. Nicolson and C. Wilson, J. Chem. Soc., Dalton Trans., 2001, 567 RSC; (c) X. L. Wang, C. Qin, E. B. Wang, Y. G. Li, Z. M. Su, L. Xu and L. Carlucci, Angew. Chem., Int. Ed., 2005, 44, 5824 CrossRef CAS.
- (a) Z. H. Zhang, Z. L. Shen, T. -a. Okamura, H. F. Zhu, W. Y. Sun and N. Ueyama, Cryst. Growth Des., 2005, 5, 1191 CrossRef CAS; (b) Z. H. Zhang, T. -a. Okamura, Y. Hasegawa, H. Kawaguchi, L. Y. Kong, W. Y. Sun and N. Ueyama, Inorg. Chem., 2005, 44, 6219 CrossRef CAS; (c) Q. Chu, G. X. Liu, Y. Q. Huang, X. F. Wang and W. Y. Sun, Dalton Trans., 2007, 4302 RSC; (d) H. F. Zhu, J. Fan, T. -a. Okamura, Z. H. Zhang, G. X. Liu, K. B. Yu, W. Y. Sun and N. Ueyama, Inorg. Chem., 2006, 45, 3941 CrossRef CAS.
- (a) G. Wu, X. F. Wang, T. -a. Okamura, W. Y. Sun and N. Ueyama, Inorg. Chem., 2006, 45, 8523 CrossRef CAS; (b) S. L. Zheng, J. H. Yang, X. L. Yu, X. M. Chen and W. T. Wong, Inorg. Chem., 2004, 43, 830 CrossRef CAS; (c) L. Y. Kong, X. H. Lu, Y. Q. Huang, H. Kawaguchi, Q. Chu, H. F. Zhu and W. Y. Sun, J. Solid State Chem., 2007, 180, 331 CrossRef CAS.
- (a) K. J. Wei, Y. S. Xie, J. Ni, M. Zhang and Q. L. Liu, Cryst. Growth Des., 2006, 6, 1341 CrossRef CAS; (b) B. Valeur. Molecular Fluorescence: Principles and Applications, Wiley-VCH, Weinheim, Germany, 2002 Search PubMed; (c) G. H. Wang, Z. G. Li, H. Q. Jia, N. H. Hu and J. W. Xu, Cryst. Growth Des., 2008, 8, 1932 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Selected bond lengths and angles, and hydrogen bonding data for complexes 1–4; thermal gravimetric curves of complexes 1–4; additional structures for 1, 3 and 4. CCDC reference numbers 727675–727678. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b913639h |
| This journal is © The Royal Society of Chemistry 2010 |