Hybrid polyoxometalate clusters with appended aromatic platforms†
Yu-Fei
Song
ab,
De-Liang
Long
b and
Leroy
Cronin
*b
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, BeiSanHuan East Road 15, 100029, Beijing, P. R. China
bWestCHEM, Department of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: L.Cronin@chem.gla.ac.uk; Web: http://www.croninlab.com Fax: (+44) 141 330 4888
First published on 27th August 2009
Abstract
The assembly of polyoxometalate architectures based on Mn-Anderson clusters capped with different aromatic platforms via the ‘Tris’ linker (Tris = tris(hydroxymethyl)aminomethane), allows the formation and isolation of four new hybrid polyoxometalate clusters of 2–5 with the general composition of [n-N(C4H9)4]3[MnMo6O18{(OCH2)3CN = CH-R}2]·solv., (R = –(C6H5) 2, –(C6H5O) 3, –(C11H8N) 4 and –(C13H9) 5; solv. = MeOH or H2O). These compounds were obtained simply by the reaction of compound 1, ([n–N(C4H9)4]3[MnMo6O18{(OCH2)3CNH2}2]), with the corresponding aldehyde and the aromatic platforms were chosen to explore the design principles for covalently constructed POM aromatic–organic–inorganic hybrids.
Introduction
Polyoxometalates (POMs) are a class of discrete anionic inorganic metal-oxide clusters of the form {MOx}n (where M = Mo, W, V, Nb; x = 4–7; n = 6 to 368) with unrivalled structure types,1 and with a wide range of unique physical properties and applications in areas as diverse as catalysis,2 medicine3 and nanoscale science.4 Furthermore, polyoxometalate clusters are currently the subject of many research efforts since the structures formed with POM-based building blocks can bridge multiple length scales,5 from the assembly of sub-nanoscale to protein sized molecules6 and even colloidal aggregates of clusters many hundreds of nm in size.7 Therefore, the ability to bridge such length scales, coupled with their transferable building blocks, shows that POMs are excellent building block candidates for metal-oxide-based crystal engineering.8The development of organic–inorganic hybrid POM building blocks, which comprise covalently connected cluster and organo fragments, is an important designed assembly since it allows the intimate combination of the properties of the metal-oxo and organic building blocks; this has been used to prepare polymers,9 dendrimers10 and macroporous materials.11 It is now apparent that organic components can dramatically influence the microstructures of inorganic oxides, thus providing a way for the design of novel materials.12 In this respect we are interested in the combination of highly conjugated organic molecules with POMs since this class of materials has hardly been explored, and should allow the assembly of electronically interesting systems.13–15 In addition, such organic–inorganic hybrid materials will not only combine the advantages of organic molecules such as structural fine tuning, but may also give rise to synergistic effects arising from the electronic interactions between the organic and inorganic parts.16 However, the major limitation is that the covalent functionalisation of POMs in general is not straightforward, depending critically upon the building blocks chosen.17 One route is to use a flexible synthetic strategy, in which the organic linker between the POMs and the organic site is designed to be bifunctional.18–21 Therefore in recent work we started to utilise ‘Tris’ (tris(hydroxymethyl) aminomethane (HOCH2)3NH2), in combination with a Mn-Anderson type POM21 and this was inspired by the work of Hasenknopf and Gouzerh et al., who first demonstrated the utility of this approach to construct hybrid POM-organic units.18–20
In this work we aimed to assemble a range of isostructural POM clusters, capped with a number of structurally related aromatic organic substituents, building on our recent results, see Fig. 1. Here, we use the Anderson cluster, which can be directly functionalised, both top and bottom, via the grafting of tripodal tri-alkoxides (e.g. derivatised from the ‘Tris’ ligand). This is because the number of conserved building blocks in POM-based architectures promises access to the controlled growth of nanoscale clusters with predetermined structures via a bottom-up route. Additionally, the systematic manipulation of the hybrid organic–inorganic POM-based clusters could yield a deeper insight into the self-assembly process and thereby allowing the design and construction of multifunctional materials based upon POMs.22 For example, the combination of highly delocalised, pyrene-based substituents with POM-based building blocks will allow us to examine the architectural aspects with respect to self assembly and crystallisation, see Fig. 1.21
![A recent synthetic approach to the hybrid POM-aromatic architecture. LEFT: Representation of the [Mn-Anderson-(pyrene-tris)2]3−. RIGHT: Resulting structure of the compound produced showing the large void spaces. C – grey, O – red, H – white, N – blue, Mo – green (Mn and TBA cations not shown); H-bonds in purple.](/image/article/2010/CE/b910434h/b910434h-f1.gif) | ||
| Fig. 1 A recent synthetic approach to the hybrid POM-aromatic architecture. LEFT: Representation of the [Mn-Anderson-(pyrene-tris)2]3−. RIGHT: Resulting structure of the compound produced showing the large void spaces. C – grey, O – red, H – white, N – blue, Mo – green (Mn and TBA cations not shown); H-bonds in purple. | ||
In the case of the [Mn-Anderson-(pyrene-tris)2]3−, when this was crystallised with TBA+ (tetrabutyl ammonium) counter ions, a crystalline material with nanoscale void spaces was formed and it could be seen that this network was built exclusively from C–H⋯O hydrogen bonds between the pyrene groups and the Anderson cluster. Further, there is the total absence of π⋯π interactions in the structure despite the eighteen pyrene moieties present in the unit cell. This is very interesting and this observation could help develop a new architectural principle if it could be shown that C–H⋯O hydrogen bonds could dominate the assembly of the supramolecular structure of the aromatic-POM hybrids.
Herein, we report the successful extension of this synthetic and architectural design strategy in the synthesis and self assembly of four new architectures with different end-capped aromatic rings grafted onto Mn-Anderson cluster: [n-N(C4H9)4]3[MnMo6O18{(OCH2)3CN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–(C6H5)}2]·4H2O (2), [n–N(C4H9)4]3[MnMo6O18–{(OCH2)3CNCH–(C6H5O)}2]· 3CH3OH (3), [n-N(C4H9)4]3[MnMo6O18{(OCH2)3CNCH–(C11H8N)}2]·2CH3OH (4) and [n–N(C4H9)4]3-[MnMo6O18 {(OCH2)3CNCH–(C13H9)}2] (5). The results presented here show that systematic manipulation of POM assemblies could be realized by applying ‘POM-linker’ strategy via covalent functionalization approach to a [Mn-Anderson-(Tris)2]3− building block.
CH–(C6H5)}2]·4H2O (2), [n–N(C4H9)4]3[MnMo6O18–{(OCH2)3CNCH–(C6H5O)}2]· 3CH3OH (3), [n-N(C4H9)4]3[MnMo6O18{(OCH2)3CNCH–(C11H8N)}2]·2CH3OH (4) and [n–N(C4H9)4]3-[MnMo6O18 {(OCH2)3CNCH–(C13H9)}2] (5). The results presented here show that systematic manipulation of POM assemblies could be realized by applying ‘POM-linker’ strategy via covalent functionalization approach to a [Mn-Anderson-(Tris)2]3− building block.
Results and discussion
Compounds 2–5 were simply obtained by reaction of Mn-Anderson cluster of 1 with different aldehydes, respectively (see Scheme 1). The resulting clusters of 2–5, which incorporate imine functionalities, have been characterized by chemical analysis, spectroscopy, and by single-crystal X-ray crystallographic measurements. In these new clusters, the central Mn-Anderson structure consists of six edge-sharing MoO6 octahedra arranged around a central MnO6 unit and two Tris residues cap both sides of Mn-Anderson polyanion (this adoptes the δ form with approximate D3 symmetry). Additionally, the hydroxyl groups of the Tris molecule have been deprotonated and are ligated onto the surface of the Anderson assembly, whereas the pendant free amino groups of Tris on both sides of the Anderson clusters have been derivatized by different planar aromatic compounds as indicated in Scheme 1. The choice of these aromatic extensions was made to extend our initial studies, done with the pyrene system,21 and to attempt to assess the impact of the aromatic tethers on the assembly of the [Mn-Anderson-(Tris)2]3− in the crystalline state. | ||
| Scheme 1 Syntheses of new POM architectures of 2–5 by grafting various aromatic rings to the Mn-Anderson cluster of 1via covalent bonds. | ||
The reaction of 1 with benzaldehyde resulted in the formation of compound 2, which has been isolated in 63% yield as orange block crystals. Compound 2 crystallises in a tetragonal space group I41cd with Z = 16; a summary of crystal structure parameters and refinement details for this compound is given in Table 1. Detailed structural analysis (see Fig. 2a) revealed that the structure of compound 2 retains the Anderson structure of the parent compound 1 and the Mo–O bond distances and the O–Mo–O bond angles are all in the normal range for such Anderson clusters. The central Mn3+ ions are chelated by six oxygen atoms by two Tris moieties with the bond distances in the range of 1.918(9) and 2.090(8) Å. The supramolecular structure is defined by the intermolecular interactions that exist between terminal Mo–O and C–H of the benzene rings with the C⋯O bond distances ranging from 3.51(3) to 3.61(2) Å and C–H⋯O bond angles from 157.0(10) to 162.3(10)°, respectively. Further, it is these hydrogen bonds formed by the tips of the benzene rings (i.e. the hydrogen atoms in the 4 position) which connect the Anderson-based cluster spacer into a 1-D hydrogen bonded chain that is ‘coated’ with TBA cations, see Fig. 2. The TBA coating interacts with these 1-D chains via C–H⋯O hydrogen bonded interactions, which define the majority of the intermolecular interactions between the molecules in the cell (Fig. 2b). The intermolecular interactions can be observed between both bridging Mo–O–Mo and terminal Mo–O oxo ligands present in the Anderson-unit and C–H moieties of the TBA+ cations with C⋯O bond distances falling within the range of 3.50(5) to 3.60(3) Å and bond angles of the C–H⋯O interactions range 112.8(10) to 152.5(10)°, respectively.
| Compounds | 2 | 3 | 4 | 5 |
|---|---|---|---|---|
| Empirical formula | C70H140 MnMo6N5O28 | C73H144MnMo6N5O29 | C83H150MnMo6N7O27 | C84H140MnMo6N5O24 |
| M r/g mol−1 | 2130.45 | 2186.51 | 2308.68 | 2234.59 |
| a/Å | 31.8004(6) | 24.516(3) | 37.5576(15) | 14.8970(4) |
| b/Å | 31.8004(6) | 15.4509(17) | 13.7547(6) | 13.9137(4) |
| c/Å | 36.9032(16) | 24.2330(3) | 25.1345(10) | 24.7439(7) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 90 | 94.577(5) | 128.073(2) | 97.349(1) |
| γ/° | 90 | 90 | 90 | 90 |
| V/Å3 | 37318.9(19) | 9150.1(18) | 10221.6(7) | 5086.6(2) |
| Z | 16 | 4 | 4 | 4 |
| Crystal system | Tetragonal | Monoclinic | Monoclinic | Monoclinic |
| Space group | I41cd | P21/c | C2/c | P21/c |
| Crystal color | orange | orange | orange | brown |
| T (K) | 100 | 100 | 100 | 150 |
| hkl | −33 ≤ h ≤ 33, −26 ≤ k ≤ 32, −38 ≤ l ≤ 32 | −29 ≤ h ≤ 29, −18 ≤ k ≤ 18, −28 ≤ l ≤ 26 | −44 ≤ h ≤ 44, −15 ≤ k ≤ 16, −28 ≤ l ≤ 29 | −18 ≤ h ≤ 14, −16 ≤ k ≤ 15, −29 ≤ l ≤ 29 |
| No. of data collected | 54876 | 135771 | 76149 | 30648 |
| No. of unique data | 10663 | 16069 | 9053 | 9434 |
| R1 (I > 2σ(I) | 0.0570 | 0.0359 | 0.0496 | 0.0672 |
| wR2 (all data) | 0.1524 | 0.0860 | 0.1635 | 0.2066 |
 | ||
| Fig. 2 (a) Crystal structure of compound 2 showing the cluster with the appended benzene moiety. The purple arrows show the direction of the hydrogen-bonded chain. (b) Illustration of the hydrogen-bonding interaction between each Anderson cluster in the unit cell including the interactions with the TBA counterions (shown in yellow). Mn orange, Mo ink blue, C grey, N yellow. TBA shown in yellow (c) Packing projection onto the ab plane with the TBA counter ions omitted. The hydrogen-bonded interactions are shown in purple. | ||
Compound 3 was synthesized by reaction of 1 and salicyaldehyde and this compound was obtained as orange block crystals in a yield of 67%. Structural analysis of 3 by single-crystal X-ray diffraction shows that the phenol groups have been successfully capped on the Anderson cluster and it crystallizes in a monoclinic space group P21/c with a Z = 4. Compound 3, like compound 2, also comprises the tris-Anderson building block and can be formulated as [n-N(C4H9)4]3[MnMo6O18{(OCH2)3CNCH (C6H5O)}2]·3CH3OH. Similar to compound 2, extensive hydrogen bonded interactions can be seen in the structure however there are two different Anderson clusters in the asymmetric unit. One cluster-hybrid forms hydrogen bonded 1-D chains (Fig. 3c-red), via phenyl C–H interactions with the adjacent cluster unit whilst the other accepts C–H bonds into the metal oxide framework (Fig 2c-blue). The 1-D chains form between the C–H moieties of the appended phenyl groups and two terminal Mo = O groups with C⋯O distances ranging 3.338(6)–3.343(6) Å and C–H⋯O angles of 127.4(3)–166.1(3)°. The remainder of the intermolecular interactions are either solvent based or from the TBA cations; Anderson cluster interactions via C–H⋯O interactions involving both terminal and bridging oxo-ligands with the C⋯O bond distances and C–H⋯O bond angles range from 3.270(5) to 3.641(8) Å and from 133(1) to 146.1(3)° (see Fig 2c). By grafting phenol groups onto the Anderson cluster, the arrangement of the capped aromatic rings in the resulting structure of compound 3 shows differences from that in compound 2, for example, the dihedral angle of the neighbouring benzene rings in 2 is around 88(1)°, close to being perpendicular to each other; whereas the dihedral angle of two phenol rings in 3 is around 12(2)°. It should be noted that the molecular packing of 3 shows an ordered, 2-D corrugated framework whereby the TBA ‘glues’ the 1-D chains together to form a 2-D network (Fig. 3d).
 | ||
| Fig. 3 (a) X-ray single crystal structure of compound 3 and the central Mn-Anderson part with the organic appendages. (b) Shows the 1-D chain formed by three units with associated TBA cations. (c) Shows the alternating 1-D chain form by the ‘donor’ Anderson moieties (red) and acceptor groups (blue). (d) shows a representation of the molecular packing viewed from ‘c’ direction; The TBA+ cations are shown in yellow in (b) and omitted in (a), (c) and (d) for clarity (Mn orange, Mo ink blue, O red and C grey). | ||
Compound 4 was obtained as orange square crystals by reaction of 1 with 2-pyridine-benzaldehyde in a yield of 65%. This compound crystallises in a monoclinic group C2/c with a Z = 4. The crystal structure of 4 shows that the organic groups have been attached on both sides of the Mn-Anderson cluster via the grafted imine moieties. It is noted that the pyridine rings are fully co-planar with the benzene rings, which are almost perpendicular to the MnMo6 plane (Fig. 4a). The extended aromatic platforms therefore serve as multiple H-bond donors where as the Anderson cluster accepts multiple H-bonds. The packing in compound 4 reveals the formation of layers (Fig. 4b) whereby each unit can interact via C–H⋯O (C⋯O range 3.19(1)–3.39(1) Å and 127.8(6)–169.6(5)°) and π⋯π interactions (3.32(2) Å). Once again, the TBA cations are located in the ‘pores’, see Fig. 4c, and also interact via C–H⋯O interactions.
 | ||
| Fig. 4 (a) Crystal structure of compound 4 showing the central Anderson cluster and the appended organic groups. (Mn orange, Mo ink blue, O red and C grey). (b) Illustration of intermolecular interactions in compound 4. (c) Crystal packing shows the pores; solvents and TBA+ are omitted for clarity. The H-bonds are shown in purple. | ||
Compound 5 was obtained with a planar, aromatic fluorene rings capped at both sides of Mn-Anderson cluster (Fig 5a). The crystal structure of 5 crystallises in a monoclinic space group P21/c. Structural analysis of 5 shows that intermolecular interactions between the TBA+ cations and the terminal Mo–O atoms with the bond distances and bond angles, but more importantly the fluorene group is able to hydrogen bond to two terminal MoO units present in the Anderson cluster and these C⋯O interactions range 3.28(1) to 3.45(1) Å with C–H⋯O bond angles that range 141.0(6) to 173.5(6)°, respectively. As in compound 4, the extended aromatic fluorene moiety multiple (in this case 2 2) C–H hydrogen bonds fluorene which interact with two terminal oxo ligands on the Anderson cluster framework (Fig. 3b).
 | ||
| Fig. 5 (a) Representation of crystal structure of 5 showing the central Anderson and the two organic groups. (Mn orange, Mo ink blue, N orange and C grey). (b) Illustration of the intermolecular hydrogen bonding interactions the cluster and the C–H groups of the fluorene and the terminal Mo–O of the Anderson cluster, respectively; (c) Illustration of 5 looking down the b axis, which shows the direction of the 1-D chains. | ||
Given the weak interactions that are responsible for the assembly of the supramolecular structures of compounds 2–5, we decided to investigate the stability of the compounds using TGA and DSC. Interestingly all the compounds gave almost identical results, as shown in Fig. 6. The TGA results, done on air dried samples, show that the solvent of crystallisation is lost by the air drying and the first weight loss is at ca. 290 °C which corresponds to the decomposition and loss of three TBA+ cations in each of the samples 2–5 (see Fig. 6. and ESI†). The DSC measurements show a very sharp endothermic absorption at between 250–270 °C for compounds 2–5 (see ESI†) which is consistent with a melting process. This is interesting since despite the weak interactions that define the structures, they appear to be robust until at least 250 °C.
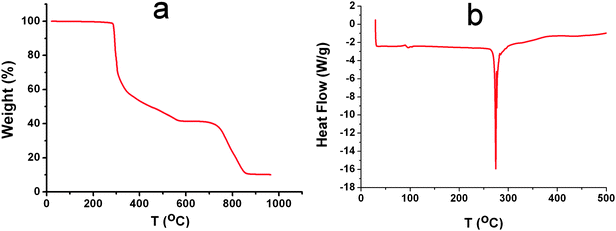 | ||
| Fig. 6 (a) Representative TGA and (b) DSC curves of 2. The TGA curve shows weight loss due to the decomposition of the TBA cations, which are observed before decomposition of the cluster unit. The DSC curve shows a very rapid melting and recrystallisation process at ca. 270 °C. | ||
Conclusions
The structural characterisation of new inorganic-organic hybrids of 2–5 clearly shows that the assembly of new POM assemblies could be achieved in Anderson-Tris system by employing ‘POM-Linker’ strategy, and this utilises the covalent functionalization of the Mn-Anderson cluster unit to give a range of building blocks, see Fig. 7. These building blocks can arrange in the solid state to give 1-D, 2-D and 3-D architectures using directed C–H⋯O interactions and the charge balancing TBA cations interact with the Anderson-units in an undirected manner. Finally, despite the fact these structures are assembled using weak interactions, it appears that these structures have some stability. In further work we will extend these initial ideas to make highly directed POM-hybrid frameworks directed by C–H⋯O interactions to produce highly functional and open frameworks. | ||
| Fig. 7 Representation of the building blocks found in compounds 2–5. The hydrogen bonded interactions are shown in purple (donor) and yellow (acceptor). | ||
Experimental
General remarks
[n-N(C4H9)4][α-Mo8O26] and [n-N(C4H9)4]3[MnMo6O18{(OCH2)3CNH2}2] (compound 1) were synthesized according to literature methods.17 All other chemicals, including solvents were commercially available as reagent grade from Aldrich-Sigma and used without further purification. X-ray single crystallography measurements were carried out on Bruker APEX II, Department of Chemistry, The University of Glasgow. Elemental analysis, IR, ESI-MS and 1H NMR spectra have been measured in the Department of Chemistry, Beijing University of Chemical Technology.Preparation of [n-N(C4H9)4]3[MnMo6O18{(OCH2)3CN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CH–(C6H5)}2]·4H2O (2)
CH–(C6H5)}2]·4H2O (2)
To a methanolic solution (20 mL) of compound 1 with the molecular formula of [n-N(C4H9)4]3[MnMo6O18{(OCH2)3C-NH2}2] (500 mg, 0.26 mmol), benzaldehyde (56 mg, 0.53 mmol) in 10 mL of MeOH was added and the mixture was kept refluxing for 3 hours. The reaction mixture was filtered and the filtrate was left for slow evaporation at room temperature. Yellow block crystals of compound 2 were obtained after two weeks. Compound 2 was characterized by single X-ray crystallography. Yield (308 mg, 63%). Elemental analysis for C70H132MnMo6N5O24·4H2O (2130.45): C 39.46, H 6.62, N 3.29; Found C 39.13, H 6.55, N 3.13. ESI-MS (MeCN, negative mode): 1816.4 ([M-4H2O-TBA]−). 1H NMR (d6-DMSO; ppm, 400 MHz): 0.92 (36H, –CH3), 1.30 (24H, –CH2), 1.57 (24H, –CH2), 3.15 (24H, –CH2). 7.27 (s, 2H), 7.54 (s, 2H), 7.61 (t, 2H), 7.71 (s, 2H), 7.92 (t, 2H), 8.78 (br, 2H) IR (KBr, cm−1): 3475(br), 2959(s), 2872(m), 1694(w), 1637(s), 1579(w), 1480(s), 1381(w), 1091(m), 1072(s), 1021(s), 1000(s), 902(s), 800(w), 758(w), 654(s), 562(w).
Preparation of compound 3, 4 and 5 were carried out in a similar way except that benzaldehyde were replaced with salicylaldehyde, 4-(2-pyridyl)benzaldehyde and fluorene-2-carboxaldehyde, respectively. Characterization of these compounds is listed below.
Compound 3 with the formula of [n-N(C4H9)4]3[MnMo6 O18{(OCH2)3CNCH–(C6H5O)}2]·3CH3OH
Yield (67%). Elemental analysis for C70H132MnMo6N5O26·3CH3OH (2186.51): C 40.10, H 6.64, N 3.20; Found C 40.53, H 6.72, N 3.08. ESI-MS (MeCN, negative mode): 1848.5 ([M-3CH3OH-TBA]−). 1H NMR (d6-DMSO; ppm, 400 MHz): 0.92 (36H, –CH3), 1.31 (24H, –CH2), 1.57 (24H, –CH2), 3.16 (24H, –CH2). 6.98 (s, 2H), 7.04 (t, 2H), 7.11 (t, 2H), 7.54 (s, 2H), 8.56 (br, 2H). IR (KBr, cm−1): 3450(br), 2960(s), 2932(s), 2873(m), 1622(s), 1481(s), 1458(m), 1380(w), 1278(m), 1097(m), 1034(s), 1022(s), 921(s), 904(s), 783(w), 738(s), 595(m), 564(w), 553(w).
Compound 4 with the formula of [n-N(C4H9)4]3[MnMo6 O18{(OCH2)3CNCH–(C11H8N)}2]·3CH3OH
Yield (65%). Elemental analysis for C80H138MnMo6N7O24·3CH3OH (2308.68): C 43.14, H 6.49, N 4.24; Found C 43.51, H 6.22, N 4.37. ESI-MS (MeCN, negative mode): 1970.5 ([M-TBA-3CH3OH]−). 1H NMR (d6-DMSO; ppm, 400 MHz): 0.93 (36H, –CH3), 1.31 (24H, –CH2), 1.56 (24H, –CH2), 3.15 (24H, –CH2). 7.34 (s, 2H), 7.82 (s, 4H), 7.91 (s, 2H), 8.03 (s, 2H), 8.28(s, 2H), 8.70 (s, 2H). IR (KBr, cm−1): 3476(br), 2960(s), 2873(s), 1636(s), 1605(w), 1584(m), 1481(s), 1436(s), 1380(m), 1307(m), 1223(w), 1089(s), 1023(s), 920(s), 786(m), 689(s), 583(w), 462(w).
Compound 5 with the formula of [n-N(C4H9)4]3[MnMo6 O18{(OCH2)3CNCH–(C13H9)}2]
Yield (60%). Elemental analysis for C84H140MnMo6N5O24 (2334.59): C 43.18, H 6.00, N 3.00; Found C 43.25, H 6.11, N 2.78. ESI-MS (MeCN, negative mode): 2088.6 ([M-TBA]−). 1H NMR (d6-DMSO; ppm, 400 MHz): 0.92 (36H, –CH3), 1.31 (24H, –CH2), 1.57 (24H, –CH2), 3.15 (24H, –CH2). 3.90 (s, 2H), 7.31 (s, 2H), 7.42 (s, 2H), 7.62 (t, 2H), 7.74 (s, 2H), 7.92 (t, 4H), 8.07 (s, 2H), 8.91 (br, 2H). IR (KBr, cm−1): 3471(br), 2959(s), 2873(s), 1634(s), 1609(m), 1481(s), 1381(w), 1088(m), 1025(s), 901(s), 739(s), 668(s), 564(7).
X-Ray crystal structure determination
Suitable single crystals of compound 2–5 were attached to a thin glass fiber by using Fomblin YR-1800 oil and mounted on a goniometer head in a general position. All data were collected on a Bruker Noinus diffractometer equipped with an APEX II CCD detector, with graphite monochromated X-radiation (λ = 0.71073 Å), running under the Collect software. Structure solution and refinement were performed by using SHELXS-9723via APEXII software package.24 The corrections for the incident and diffracted beam absorption effects were applied using empirical or numerical methods. Refinement was with SHELXL-9725 using full-matrix least-squares on F2 and all the unique data. All calculations were carried out using the WinGX package of crystallographic programs.Acknowledgements
The authors would like to thank the EPSRC, WestCHEM and the University of Glasgow and National Science Foundation of China (20801003) for funding support.References
- (a) D.-L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105 RSC; (b) M. T. Popein Comprehensive Coordination Chemistry II, Vol. 4. (Ed: A. G. Wedd) Elsevier, Oxford, 2004, p635–679 Search PubMed; (c) C. L. Hillin Comprehensive Coordination Chemistry II, Vol 4 (Ed: A. G. Wedd) Elsevier, Oxford, 2004, p679–759 Search PubMed; (d) A. Müller, P. Kögerler and A. W. M. Dress, Coord. Chem. Rev., 2001, 222, 193 CrossRef CAS.
- M. V. Vasylyev and R. Neumann, J. Am. Chem. Soc., 2004, 126, 884 CrossRef CAS; N. Mizuno, K. Yamaguchi and K. Kamata, Coord. Chem. Rev., 2005, 249, 1944 CrossRef CAS.
- B. Hasenknopf, Front. Biosci., 2005, 10, 275 CrossRef CAS.
- C. Fleming, D. L. Long, N. Mcmillan, J. Johnston, N. Bovet, V. Dhanak, N. Gadegaard, P. Kögerler, L. Cronin and M. Kadodwala, Nat. Nanotechnol., 2008, 3, 289–233 Search PubMed.
- D. L. Long and L. Cronin, Chem.–Eur. J., 2006, 12, 3698 CrossRef CAS.
- A. Müller, E. Beckmann, H. Bögge, M. Schmidtmann and A. Dress, Angew. Chem., 2002, 114, 1210 CrossRef; A. Müller, E. Beckmann, H. Bögge, M. Schmidtmann and A. Dress, Angew. Chem., Int. Ed., 2002, 41, 1162 CrossRef CAS.
- T. B. Liu, E. Diemann, H. L. Li, A. W. M. Dress and A. Müller, Nature, 2003, 426, 59 CrossRef CAS.
- D. L. Long, H. Abbas, P. Kögerler and L. Cronin, Angew. Chem., 2005, 117, 3387 CrossRef; D. L. Long, H. Abbas, P. Kögerler and L. Cronin, Angew. Chem., Int. Ed., 2005, 44, 3415 CrossRef CAS.
- B. B. Xu, M. Lu, J. H. Kang, D. G. Wang, J. Brown and Z. H. Peng, Chem. Mater., 2005, 17, 2841 CrossRef CAS; A. R. Moore, H. Kwen, A. M. Beatty and E. A. Maatta, Chem. Commun., 2000, 1793 RSC.
- H. D. Zeng, G. R. Newkome and C. L. Hill, Angew. Chem., 2000, 112, 1841 CrossRef; H. D. Zeng, G. R. Newkome and C. L. Hill, Angew. Chem. Int. Ed., 2000, 39, 1772 CAS.
- R. C. Schroden, C. F. Blanford, B. J. Melde, B. J. S. Johnson and A. Stein, Chem. Mater., 2001, 13, 1074 CrossRef CAS.
- P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., 1999, 111, 2798 CrossRef; P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem. Int. Ed., 1999, 38, 2639 CAS.
- B. B. Xu, Y. G. Wei, C. L. Barnes and Z. H. Peng, Angew. Chem., 2001, 113, 2353 CrossRef; B. B. Xu, Y. G. Wei, C. L. Barnes and Z. H. Peng, Angew. Chem., Int. Ed., 2001, 40, 2290 CrossRef CAS; Z. H. Peng, Angew. Chem., 2004, 116, 948 CrossRef; Z. H. Peng, Angew. Chem., Int. Ed., 2004, 43, 930 CrossRef CAS.
- J. L. Stark, A. L. Rheingold and E. A. Maatta, J. Chem. Soc., Chem. Commun., 1995, 1165 RSC; J. B. Strong, G. P. A. Yap, R. Ostrander, L. M. Liable-Sands, A. L. Rheingold, R. Thouvenot, P. Gouzerrh and E. A. Maatta, J. Am. Chem. Soc., 2000, 122, 639 CrossRef CAS.
- J. L. Stark, V. G. Young and E. A. Maatta, Angew. Chem., 1995, 107, 2751 CrossRef; J. L. Stark, V. G. Young and E. A. Maatta, Angew. Chem., Int. Ed. Engl., 1995, 34, 2547 CrossRef CAS; H. Kwen, V. G. Young and E. A. Maatta, Angew. Chem., 1999, 111, 1215 CrossRef; H. Kwen, V. G. Young and E. A. Maatta, Angew. Chem., Int. Ed., 1999, 38, 1145 CrossRef CAS.
- A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837–1852 RSC.
- J. C. Duhacek and D. C. Duncan, Inorg. Chem., 2007, 46, 7253 CrossRef CAS.
- P. R. Marcoux, B. Hasenknopf, J. Vaissermann and P. Gouzerh, Eur. J. Inorg. Chem., 2003, 2406 CrossRef CAS.
- B. Hasenknopf, R. Delmont, P. Herson and P. Gouzerh, Eur. J. Inorg. Chem., 2002, 1081 CrossRef CAS.
- S. Favette, B. Hasenknopf, J. Vaissermann, P. Gouzerh and C. Roux, Chem. Commun., 2003, 2664 RSC.
- (a) J. Zhang, Y.-F. Song, L. Cronin and T. Liu, J. Am. Chem. Soc., 2008, 130, 14408 CrossRef CAS; (b) Y.-F. Song, D.-L. Long and L. Cronin, Inorg. Chem., 2008, 47, 9137 CrossRef CAS; (c) D.-L. Long, Y.-F. Song, E. F. Wilson, P. Kogerler, S. X. Guo, A. M. Bond, J. Harrgreaves and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4384 CrossRef CAS; (d) Y. F. Song, N. McMillan, D. L. Long, J. Thiel, Y. Ding, H. Chen, N. Gadegaard and L. Cronin, Chem.–Eur. J., 2008, 14, 2349 CrossRef CAS; (e) Y. F. Song, H. abbas, C. Ritchie, N. McMillan, D. L. Long, N. Gadegaard and L. Cronin, J. Mater. Chem., 2007, 17, 1903 RSC; (f) Y. F. Song, D. L. Long and L. Cronin, Angew. Chem., Int. Ed., 2007, 46, 3900 CrossRef CAS.
- C. Ritchie, E. Burkholder, P. Kögerler and L. Cronin, Dalton Trans., 2006, 1712 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- Bruker Analytical X-Ray Systems, Crystallographic Software Package APEX2 version 2 PC version, Madison, WI, USA, 2006 Search PubMed.
- G. M. Sheldrick, SHELXL-97. Program for Crystal Structure Analysis, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TGA and DSC of compounds 3–5. Structures of 2–5 as cif files are available. CCDC reference numbers 733962–733965. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b910434h |
| This journal is © The Royal Society of Chemistry 2010 |