Photo- and thermally-enhanced charge separation in supramolecular viologen–hexacyanoferrate complexes†
Ahmed S.
Abouelwafa
a,
Valeriu
Mereacre
b,
Teodor S.
Balaban
a,
Christopher E.
Anson
b and
Annie K.
Powell
*ab
aInstitut für Nanotechnologie, Forschungszentrum Karlsruhe, Karlsruhe Institute of Technology, Postfach 3640, Germany, D-76021, Karlsruhe, Germany. E-mail: powell@aoc.uni-karlsruhe.de; Fax: (+49) 724 782 6368; Tel: (+49) 724 782 8722
bInstitut für Anorganische Chemie der Universität Karlsruhe, Karlsruhe Institute of Technology, D-76131, Karlsruhe, Germany. Fax: (+49) 721 608 8142; Tel: (+49) 721 608 2135
First published on 7th September 2009
Abstract
Photo- and thermally enhanced charge-separation is observed in a supramolecular complex formed between viologen as electron acceptor and ferrocyanide as donor.
Introduction
Viologens are diquaternary derivatives of 4,4′-bipyridyl that act as strong electron acceptors. They can be reversibly reduced in two consecutive steps to form first a highly coloured radical cation and further to the doubly reduced species.1These properties led to a variety of applications such as supramolecular assemblies for charge separation, photocatalytic generation of hydrogen,2 photo- and electrochromic devices.3
Due to the increasing interest in donor–acceptor systems displaying long-lived charge separation which, for example, allows access to clean energy,2 we have investigated the system where ferrocyanide, [FeII(CN)6]4−, acts as electron donor with N-aromatic-substituted viologens as acceptors. This is due to the fact that the ferrocyanide unit contains both an FeII centre which can donate one electron due to its low oxidation potential, and electron-rich cyanide nitrogens which can interact with the electron-deficient viologen. Furthermore, tracing and quantifying changes in the Fe oxidation state using Mössbauer spectroscopy can be directly correlated to the overall charge separation yield. Several reports discussed the solution electrochemistry of charge transfer complexes formed between viologens and organic donor groups4 or inorganic complexes such as tris(bipy)ruthenium(II).5 Haim et al. studied the kinetics and the mechanism of the reaction of a pre-formed methyl viologen radical, MV˙+via quenching of the excited state of tris(bipy)ruthenium(II), with an in situ formed ferricyanide ion.5a Ferraudi et al. studied the factors influencing the formation of the methylviologen radical cation and its decay kinetics in aqueous and methanolic solutions.6 These studies focused on the solution chemistry of the coloured radical monocation which disproportionates in solution to give the dicationic and the doubly reduced species. This makes the viologens difficult to crystallize in their blue coloured state containing an appreciable amount of the radical species. Previous solid-state studies have shown that alkyl viologens can form intense yellow coloured charge-transfer complexes with organic and inorganic donors, but for which the amount of the radical cationic species present was not quantified.7
Although reports by Ulstrup8a and Drickamer8b showed that factors such as the solvent polarity and pressure could affect the electron transfer rate in a viologen–ferrocyanide system in solution, no solid state structural or electronic characterization were performed on this system, and the possible influence of light or heat on this donor–acceptor system in the solid state. Furthermore, we have decided to tune the properties of this system by using N-aromatic-substituted viologen with dinitrophenyl peripheries instead of the widely used alkyl viologens. This led to the enhancement of both the electron withdrawing properties of the viologen and the stability of the formed viologen radical through the phenyl-4,4′-bipyridyl π-conjugation which can accommodate the donated electron.
We report herein the direct forward reaction of viologen dication with ferrocyanide with full characterization of the resulting complex in the solid state, and the corresponding amount of the charge separated state containing the viologen radical and the ferricyanide. Furthermore, the light and heat enhancement of the charge separation is investigated in the solid state and in solution using a variety of spectroscopic techniques.
Experimental
Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was added to the melt and the reaction mixture was refluxed for 15 h at 110 °C. After complete removal of the solvent, the dark green crude product was recrystallized twice by adding hot ethanol, a few drops of water and heated in a rotary evaporator until the entire solid dissolves. The solution was left for 24 h to give large yellow needle crystals in 98% yield.
:1) was added to the melt and the reaction mixture was refluxed for 15 h at 110 °C. After complete removal of the solvent, the dark green crude product was recrystallized twice by adding hot ethanol, a few drops of water and heated in a rotary evaporator until the entire solid dissolves. The solution was left for 24 h to give large yellow needle crystals in 98% yield.
The supramolecular complex {(DNP)2[Fe(CN)6]}·20H2O 1a was obtained by addition of solid K4[Fe(CN)6]·3H2O (0.014 g, 0.033 mmol) to a dilute solution of (DNP)Cl2 (0.021 g, 0.033 mmol) in 10 mL water. The clear green solution was left at 4 °C in the dark yielding green needle crystals of 1a after 24 h in 90% yield.
Elemental analysis
Sample dried in air atmosphere: calcd for C50H28FeN18O16·8H2O: C, 44.90; H, 3.32; N, 18.86. Found: C, 45.18; H, 3.01; N, 18.24. The complex 1c was obtained by heating the fresh complex 1a at 95 °C for 2 days: calcd for C50H28FeN18O16·H2O: C, 49.58; H, 2.50; N, 20.83. Found: C, 49.93; H, 2.22; N, 20.51.The amounts of carbon, nitrogen and hydrogen in all samples were quantitatively analyzed using an Elementar Vario EL analyzer.
EPR spectroscopy
Standard X-band EPR measurements were carried out using a Bruker ESP300E spectrometer equipped with a continuous flow cryostat from Oxford Instruments in the temperature range of 300–3 K.Mössbauer spectroscopy
Spectra were acquired with a conventional spectrometer incorporating an Oxford Instruments Mössbauer-Spectromag 4000 Cryostat, equipped with a 57Co source (3.7 GBq) in a rhodium matrix in the constant-acceleration mode. Isomer shifts are given relative to α-Fe at 300 K. Spectra were fitted using the NORMOS Mössbauer Fitting Program.DSC study
The DSC was measured on a Perkin Elmer DSC Pyris 1 instrument.UV/vis spectroscopy
UV-visible spectra were recorded in 1 cm path length quartz cell on a double beam UV/vis Varian Cary 500 spectrophotometer in the range 900–200 nm. UV-light illumination experiments were performed using 15-W mercury lamp.FTIR spectroscopy
FTIR spectra were measured on a Perkin Elmer Spectrum One spectrometer in the region 400 cm−1 to 4000 cm−1 in transmission mode using 8 scans to a resolution of 4 cm−1. The samples were prepared by mixing the sample with KBr in a ca. 1:30 ratio. A disc of the sample, 1 cm in diameter, was obtained by applying 10 tones under vacuum.
Crystallography
Datasets were measured at 100 K on a Bruker SMART Apex diffractometer using graphite-monochromated Mo-Kα radiation, and corrected for absorption. Structure solution (direct methods) and full-matrix least-squares refinement against F2 (all data) were carried out using the SHELXTL software package.9 Non-H atoms were refined anisotropically, except for three lattice water oxygens that were refined as pairs of isotropic partial atoms. The H-atoms on two of the lattice waters could be located and refined. 1a: C50H68FeN18O36, 1553.07 g mol−1, orthorhombic, Ibam, a = 26.3998(15), b = 11.3528(6), c = 22.2355(12) Å, V = 6664.2(6) Å3, T = 100 K, Z = 4, Dc = 1.548 Mg m−3, µ(Mo Kα) = 0.334 mm−1, F(000) = 3232, green block 0.42 × 0.34 × 0.18 mm. 22965 reflections measured, of which 3955 unique (Rint = 0.0232). 267 parameters, wR2 = 0.2023, S = 0.991 (all data), R1 = 0.0721 (3343 with I > 2σ(I)), the largest final difference peak/hole = +0.82/−1.48 e Å−3. 1b: C50H68FeN18O36, 1553.07 g mol−1, orthorhombic, Ibam, a = 26.442(3), b = 11.3715(12), c = 22.312(2) Å, V = 6708.8(12) Å3, T = 100 K, Z = 4, Dc = 1.538 Mg m−3, µ(Mo-Kα) = 0.336 mm−1, F(000) = 3232, blackish needle 0.24 × 0.05 × 0.03 mm. 16154 reflections measured, of which 3935 unique (Rint = 0.0222). 267 parameters, wR2 = 0.2028, S = 1.041 (all data), R1 = 0.0692 (3122 with I > 2σ(I)), largest final difference peak/hole = +0.61/−1.67 e Å−3. Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 692001 and 692002.Results and disscusion
The reaction of pale yellow 1,1′-bis-(2,4-dinitrophenyl)-4,4′-bipyridinium dichloride,1b (DNP)Cl2, with colourless potassium ferrocyanide, K4[FeII(CN)6] leads to the formation of a green crystalline material 1a. The colour suggests a partial electron transfer (ET) process from the [FeII(CN)6]4− centre to the viologen acceptor, resulting in the coexistence in the crystal of small amounts of the dark-blue viologen radical cation and larger amounts of the pale yellow dicationic species. The complex retains both its crystallinity and green colour in an air atmosphere indicating a high stability of the formed viologen radical.X-Ray crystallographic analysis of the fresh green crystalline material revealed a supramolecular complex (Fig. 1) with an overall formula {(DNP)2[Fe(CN)6]}·20H2O 1a. This crystallizes in the orthorhombic space group Ibam with Z = 4; the hexacyanoferrate unit occupies the Wyckoff site c with 2/m symmetry, while the two halves of the viologen molecule are related by a crystallographic two-fold axis.
![The supramolecular complex {(DNP)2[Fe(CN)6]}·20H2O (1a) showing the interactions between the hexacyanoferrate and four surrounding viologen moieties. Colour code: Fe, green; C, black; N, blue; O, red; H, grey (lattice water molecules omitted for clarity).](/image/article/2010/CE/b915642a/b915642a-f1.gif) | ||
| Fig. 1 The supramolecular complex {(DNP)2[Fe(CN)6]}·20H2O (1a) showing the interactions between the hexacyanoferrate and four surrounding viologen moieties. Colour code: Fe, green; C, black; N, blue; O, red; H, grey (lattice water molecules omitted for clarity). | ||
Supramolecular complex formation is mediated by strong electrostatic interactions between the cyanide nitrogens and the electron-deficient viologen rings. These involve both C–H⋯N hydrogen bonds (C⋯N distances 3.158(3), 3.328(5) and 3.492(5) Å) and CN⋯N(pyr) interactions (3.532(4) Å), (Fig. 2). The Fe-C distances are in the range 1.897(9)–1.910(4) Å, with a mean value of 1.904(6) Å, consistent with a mean iron oxidation state close to FeII.10 The dihedral angle between the pyridyl rings is 30.03(7)°.
 | ||
| Fig. 2 Electrostatic interactions between the {Fe(CN)6} unit and the two (DNP) viologen moieties (left), short distances and non-planarity of the four connected six-membered rings (right) (water solvent molecules and the nitro groups are omitted for clarity). | ||
The supramolecular complex forms channels in which four viologen moieties are arranged one dimensionally along the b-axis direction (Fig. 3). There is no π-stacking between the aromatic rings because of the separation of the viologen molecular planes by the {Fe(CN)6} moieties. The packing of the {(DNP)2-[Fe(CN)6]} supramolecular complex viewed along the c-axis is shown in Fig. 3.
![Top: View of the 1D channels running along the direction of the b-axis. Bottom: Packing diagram of the {(DNP)2-[Fe(CN)6]} supramolecular complex viewed along the c-axis. Colour codes as in Fig. 1.](/image/article/2010/CE/b915642a/b915642a-f3.gif) | ||
| Fig. 3 Top: View of the 1D channels running along the direction of the b-axis. Bottom: Packing diagram of the {(DNP)2-[Fe(CN)6]} supramolecular complex viewed along the c-axis. Colour codes as in Fig. 1. | ||
Spectroscopic investigations were also consistent with a small amount of the viologen radical cation, (DNP)˙+ in the fresh sample 1a, while (DNP)2+ and [FeII(CN)6]4− remain the major components. The EPR spectrum of 1a shows a weak but sharp signal at g ≈ 2 arising from the viologen organic radical (Fig. 4). The broader signals of the FeIII species could not be clearly identified at this stage. The FTIR spectrum of 1a shows two νCN absorptions, a strong band at 2028 cm−1 assigned to [FeII(CN)6]4− and a much weaker band at 2101 cm−1, consistent with the presence of small amounts of [FeIII(CN)6]3−,11 resulting from oxidation of the ferrocyanide. The 57Fe Mössbauer spectrum of 1a at 300 K is dominated by a single line characteristic of low-spin octahedral FeII with isomer shift (IS) = −0.008 mm s−1 (relative to α-Fe).12 However, a good fit to the data required the inclusion of a low-intensity doublet corresponding to low-spin FeIII. The spectrum at 3 K is broader due to the slight onset of an intermediate spin relaxation phenomenon.
 | ||
| Fig. 4 EPR spectra of 1c measured at 180, 150, 95, 70 and 35 K. Inset: viologen radical signal appearing at g ≈2 in the EPR spectrum of 1a measured at 8 K, (the peak intensities are shown in arbitrary units). | ||
The spectrum measured at 3 K in an applied external field of 6.5 T is easier to interpret, with the major component (88.9%) a sextet with IS of 0.009 mm s−1 (with a total effective magnetic field at the nucleus Beff = 6.76 T) and intensity ratios appropriate for diamagnetic [FeII(CN)6]4− in a perpendicular applied magnetic field. There are two minor but well-defined signals to the left and right of the sextet assigned to the second and fifth lines of a low-spin [FeIII(CN)6]3− sextet (IS = −0.069 mm s−1) corresponding to 11.1% of the overall spectrum (Fig. 5). These observations are consistent with the IR and EPR spectra of the complex confirming a small but now quantifiable degree of electron transfer, leading to the formation of the mixed-valence state {(DNP)2+2[FeII(CN)6]4}1−x{(DNP)2+(DNP)+.[FeIII(CN)6]3−}x, where x = ca. 11%.
 | ||
| Fig. 5 Mössbauer spectra of 1a (upper), and 1c (lower) measured at 3 K in applied field of 6.5 T (fitting: green line: L.S. FeII; turquoise line: L.S. FeIII). | ||
Irradiation of the crystals for 24 h by sunlight or 254 nm UV radiation results in a colour change from green to dark blue, attributed to further electron transfer and formation of additional dark blue (DNP)˙+ species compared to 1a. This irradiated material, 1b, has maintained its crystallinity, and the blue crystals have a structure isomorphous to that of 1a. The X-ray crystallographic analysis of the irradiated material showed a slight increase in the Fe–C bond lengths, which are now in the range 1.887(9)–1.918(4) Å, with a mean value of 1.914(5) Å. The slight increase in the mean Fe–C distance would be consistent with a slight increase in the average oxidation state of the iron, but this cannot be counted as significant at the 3σ level. The FTIR spectrum of the irradiated material 1b shows some increase in intensity of the 2101 cm−1 band at the expense of that at 2028 cm−1, while the Mössbauer spectra of 1b are generally rather similar to those of 1a. We conclude that a somewhat greater extent of electron transfer has taken place in 1b as compared to 1a, but that irradiation of material at or near the surface to give the dark blue species prevents deeper penetration of the radiation into the crystalline material (the Mie effect).
The electron transfer process is, however, greatly enhanced by heating the green crystals of 1a. This results in both the progressive loss of the lattice water molecules and also the largely complete oxidation of the ferrocyanide centres to ferricyanide by 95 °C, with the formation of (DNP)2+(DNP)˙+[FeIII(CN)6]3−·½H2O, 1c. A DSC study on a sample of 1a shows a stepwise loss of the lattice water over the temperature range 40–120 °C, but no indication of decomposition of either ferrocyanide or viologen below 170 °C (Fig. 6). An abrupt process at 175 °C then corresponds to the decomposition and loss of all the cyanides. A thermal analysis study of potassium hexacyanoferrate(II) by Gaffar et al. showed a similar stepwise loss of lattice water molecules over the range 45–110 °C, but in their study oxidation of FeII to FeIII only takes place above 360 °C.13
 | ||
| Fig. 6 DSC study performed on the fresh sample 1a in the temperature range 35–550 °C. | ||
Unfortunately, both single-crystal and powder X-ray diffraction indicated loss of crystallinity in 1c, and it is thus not possible to determine the crystal structure of the heated sample. However, the heated crystals retain their external shape, with the crystal faces remaining well-developed and shiny, and we conclude that the transformation from 1a to 1c has not involved gross disruption of the structure, apart from the loss of nearly all lattice waters. This is supported by the IR spectrum of 1c, in which the positions of the bands from the viologen are essentially unchanged compared to the spectrum of 1a; the only significant changes are that the νCN band from ferricyanide at 2101 cm−1 is now dominant over the ferrocyanide band at 2028 cm−1, and the loss of the broad lattice water feature at ca. 3400 cm−1. Microanalysis data are also in good agreement with the formulation (DNP)2[Fe (CN)6]·½H2O, supporting the lack of decomposition of either viologen or hexacyanoferrate. We note that the dehydration process is irreversible and it is not possible to restore crystallinity by rehydration.
The EPR spectra of the heated sample 1c measured at different temperatures show a sharp peak at g = 2.07 assigned to the viologen radical, and three components at g = 0.83, 2.4 and 2.7 assigned to the FeIII(CN)6 centre14 (Fig. 4). The Mössbauer spectrum of the complex 1c measured at 3 K in an applied external field of 6.5 T shows two magnetic sextets (Fig. 5). The major sextet with an IS of −0.097 mm s−1 (Beff = 30.5 T) is assigned to the oxidized [FeIII(CN)6]3− species corresponding to ∼72% of the overall spectrum. The minor sextet is similar to the one observed in the spectrum of 1a at 3 K in 6.5 external applied field, with an IS = 0.006 mm s−1 and QS = 0.004 mm s−1 (Beff = 6.75 T) which is attributed to the remaining low-spin [FeII(CN)6]4− which did not undergo oxidation, corresponding to about 28% of the overall sample (Fig. 5). The especially strong Fe → C back-bonding existing in hexacyanoferrates weakens the triple bond of the CN group thus making the νCN appearing in the IR spectra very sensitive to the oxidation state of the Fe centre. The increase of the FeIII component at the expense of the FeII one was confirmed by subjecting the fresh sample 1a to stepwise different irradiation times and/or different temperatures, and finally observing the maximum ET enhancement by heating up to 95 °C (see Fig. 7). Similar behavior is observed in aqueous solution. A (DNP)Cl2 viologen solution changes from colourless to green when treated with K4[FeII(CN)6], with the absorption spectrum showing only a small shoulder above 400 nm assigned to a small amount of the viologen radical cation (Fig. 8: green line).
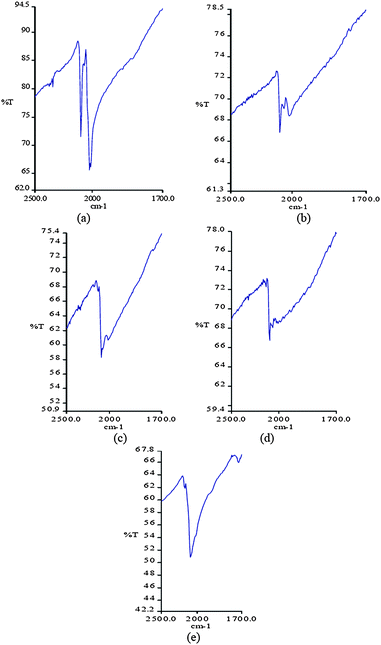 | ||
| Fig. 7 IR spectra in the range 1700–2500 cm−1 showing the shifts of the cyanide stretching vibrations depending on the sample preparation and handling (a) dried in air atmosphere (b) sunlight irradiation for 12 h (c) heating at 50 °C for 1 day (d) heated at 50 °C for 2 days and sunlight irradiation of 12 h (e) heated at 95 °C for 2 days. | ||
![Absorption spectra recorded for aqueous solutions of (i) free (DNP)Cl2 viologen (blue line, vial a), (ii) free K4[FeII(CN)6] (black line, vial b), (iii) addition of the (DNP)Cl2 solution to the K4[FeII(CN)6] one (green line, vial c), (iv) warming up the solution mixture at 50 °C for 3 min (pink line, vial d), (v) after light irradiation for 5 min (violet line), (vi) 10 minutes (brown line), (vii) 25 min (red line).](/image/article/2010/CE/b915642a/b915642a-f8.gif) | ||
| Fig. 8 Absorption spectra recorded for aqueous solutions of (i) free (DNP)Cl2 viologen (blue line, vial a), (ii) free K4[FeII(CN)6] (black line, vial b), (iii) addition of the (DNP)Cl2 solution to the K4[FeII(CN)6] one (green line, vial c), (iv) warming up the solution mixture at 50 °C for 3 min (pink line, vial d), (v) after light irradiation for 5 min (violet line), (vi) 10 minutes (brown line), (vii) 25 min (red line). | ||
Mild heating of this green solution at 50 °C for 3 min results in the colour changing to red. Although the absorption spectrum of the red solution is complicated due to the presence of a mixture of different species (e.g. viologen dication, radical cation and doubly reduced species) together with the ferricyanide peaks, two new absorption bands observed at 420 and 532 nm are assigned to the viologen radical cation monomer and dimer (formed between a doubly reduced viologen and the dication species), respectively (Fig. 8: red line).1a,3a,7b
The solution retains its colour and spectral features over time in the presence of aerial oxygen, indicating a long-lived charge separation between the generated cation, FeIII, and the (DNP)˙+ species. The increase in intensity of the broad red-shifted shoulder in the vis-NIR upon light irradiation is characteristic of viologen radical cationic species, confirming that the system is quite stable against photobleaching.2d These light and heat sensitivities of the viologen–hexacyanoferrate complexes in both the solid and solution states along with the changes in colour and oxidation states suggest that such systems are suitable candidates for the construction of smart molecular devices and switches.
Conclusions
In summary, we have characterised a new type of organic–inorganic hybrid material formed from an organic viologen acceptor with hexacyanoferrate as donor in the solid state. In contrast to previously reported viologen charge-transfer complexes, we find that both photo- and thermal enhancement of the electron transfer between donor and acceptor is possible in the solid state complex containing a well-defined quantity of the charge separated state as shown, using Mössbauer measurements.Acknowledgements
This work was supported by the DFG through the CFN and SPP 1137.Notes and references
- (a) A. Calderbank. The Dipyridilium Herbicides, Advances in Pest Control Research, 1968, 8, 127 Search PubMed; (b) J. G. Allen, German patent, 1976, DE2527638.
- (a) P. A. Brugger, P. P. Infelta, A. M. Braun and M. Grätzel, J. Am. Chem. Soc., 1981, 103, 320 CrossRef CAS; (b) N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52 CrossRef CAS; (c) L. Vermeulen and M. Thompson, Nature, 1992, 358, 656 CrossRef CAS; (d) D. L. Jiang, C. K. Choi, K. Honda, W. S. Li, T. Yuzawa and T. Aida, J. Am. Chem. Soc., 2004, 126, 12084 CrossRef CAS; (e) T. Komatsu, R. M. Wang, P. A. Zunszain, S. Curry and E. Tsuchida, J. Am. Chem. Soc., 2006, 128, 16297 CrossRef CAS; (f) M. Kirch, J. M. Lehn and J. P. Sauvage, Helv. Chim. Acta, 1979, 62, 1345 CrossRef CAS; (g) A. B. P. Lever, B. S. Ramaswamy and S. Licoccia, J. Photochem., 1982, 19, 173 CrossRef CAS.
- (a) M. Nanasawa, M. Miwa, M. Hirai and T. Kuwabara, J. Org. Chem., 2000, 65, 593 CrossRef CAS; (b) H. Kamogawa, T. Masui and M. Nanasawa, Chem. Lett., 1980, 1145 CAS; (c) M. Suzuki, M. Kimura, K. Hanabusa and H. Shirai, Chem. Commun., 1997, 2061 RSC.
- (a) C. Königstein, M. Spallart and R. Bauer, Electrochim. Acta, 1998, 43, 2435 CrossRef CAS; (b) A. Yasuda and J. Seto, J. Appl. Electrochem., 1988, 18, 333 CrossRef CAS.
- (a) L. A. Andrade de Oliveira and A. Haim, J. Am. Chem. Soc., 1982, 104, 3363 CrossRef CAS; (b) K. D. Samar and K. D. Prabir, Langmuir, 1998, 14, 5121 CrossRef.
- T. W. Ebbesen and G. Ferraudi, J. Phys. Chem., 1983, 87, 3717 CrossRef CAS.
- (a) Q. Ch. Zhang, T. Wu, X. H. Bu, T. Tran and P. Y. Feng, Chem. Mater., 2008, 20, 4170 CrossRef CAS; (b) K. B. Yoon and J. K. Kochi, J. Phys. Chem., 1991, 95, 3780 CrossRef.
- (a) M. Kjaer, I. Kristjansson and J. Ulstrup, J. Electroanal. Chem. Interfacial Electrochem., 1986, 204, 45 CrossRef; (b) W. S. Hammack and H. G. Drickamer, Chem. Phys. Lett., 1988, 151, 469 CrossRef.
- G. M. Sheldrick, SHELXTL 6.12, Bruker AXS Inc., 6300 Enterprise Lane, Madison, WI 53719-1173, USA ( 2003) Search PubMed.
- (a) B. Morosin, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3730 CrossRef; (b) J. Kuchar, J. Cernak and W. Massa, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, m418 CrossRef; (c) B. N. Figgis, B. W. Skelton and A. H. White, Aust. J. Chem., 1978, 31, 1195 CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, Wiley, New York, 5th edn, 1997, pp 105–115 Search PubMed.
- N. N. Greenwood and T. C. Gibb, Mössbauer Spectroscopy, Chapman and Hall Ltd., London, 1971 Search PubMed.
- M. A. Gaffar and M. H. Omar, J. Therm. Anal. Calorim., 2005, 81, 477 CrossRef CAS.
- (a) J. M. Baker, B. Bleaney and K. D. Bowers, Proc. Phys. Soc., London, Sect. B, 1956, 69, 1205 CrossRef; (b) H. C. Box, K. T. Lilga and E. E. Budzinski, J. Chem. Phys., 1977, 66, 2135 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR and Mössbauer spectra, microanalysis values. CCDC reference numbers 692001 and 692002. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b915642a |
| This journal is © The Royal Society of Chemistry 2010 |