The effect of fluorinated aryl substituents on the crystal structures of 1,2,3,5-dithiadiazolyl radicals†
Caroline S.
Clarke
a,
Delia A.
Haynes‡
*a,
J. Nicholas B.
Smith
a,
Andrei S.
Batsanov
b,
Judith A. K.
Howard
b,
Sofia I.
Pascu§
a and
Jeremy M.
Rawson
a
aThe Department of Chemistry, The University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW
bThe Department of Chemistry, The University of Durham, South Road, Durham, UK DH1 3LE
First published on 2nd October 2009
Abstract
The crystal structures of six new partially fluorinated aryl 1,2,3,5-dithiadiazolyls [ArCNSSN]˙ are reported [Ar = 2,6-F2C6H3 (4); Ar = 3,4-F2C6H3 (5); Ar = 3,5-F2C6H3 (6); Ar = 2,3,6-F3C6H2 (9); Ar = 2,4,6-F3C6H2 (11); and Ar = 3,4,5-F3C6H2 (12)] and compared with three previously reported structures in this series (Ar = 2,3-F2C6H3 (1); Ar = 2,5-F2C6H3 (3); Ar = 2,3,4-F3C6H2 (7)]. Radical 4 is shown to be polymorphic. Molecular electrostatic potential maps have been used to rationalise these structures. These reveal that whilst single F atoms appear to have little structure-directing influence, combinations of an ortho-F on the phenyl ring and a heterocyclic ring N, or several F atoms located in mutually ortho positions with respect to each other, generate significant regions of negative charge which have a substantial structure-directing influence.
Introduction
For some time, our group has been interested in the properties of 1,2,3,5-dithiadiazolyl (DTDA) radicals (Scheme 1). These neutral 7π-electron radicals are of great interest due to their potential as magnetic1 or conducting2 materials. The physical properties of these radicals are intimately linked to their solid state structures, and progress in the development of these radicals as molecular materials is strongly dependent upon the ability to control the structure at both the molecular and supramolecular levels.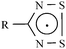 | ||
| Scheme 1 The 1,2,3,5-dithiadiazolyl radical. | ||
At the molecular level EPR studies have revealed that the spin density distribution in these radicals appears insensitive to substituent R,3,4 and the redox behaviour of aryl derivatives has been shown to follow a Hammett-type correlation, although the coefficient indicates only very modest changes in redox behaviour over a wide range of substituents.5 Conversely, few attempts have been made to undertake a systematic analysis of the factors which affect the supramolecular structure of these materials in the solid state, with perhaps the most notable exceptions being the identification6 of the CNδ−⋯Sδ+ structure-directing functionality in cyano-substituted DTDAs, and an analysis of polymorphism in ClCNSSN˙ and HCNSSN˙, both of which exhibit heterocyclic Sδ+⋯Nδ− contacts.7
In the majority of cases DTDA derivatives adopt π*–π* dimeric structures (Fig. 1) with strong interactions between singly-occupied molecular orbitals on the monomers. The dimerisation energy in solution has been estimated at ca. 35 kJ mol−1 for a range of derivatives (R = Ph, tBu and CF3)8 and renders the majority of DTDAs diamagnetic in the solid state. This dimerisation energy is significant when placed in the context of hydrogen bonds (which lie approximately in the range 2–167 kJ mol−1)9 and other non-covalent intermolecular interactions. One group of perfluorophenyl DTDA derivatives (R = p-XC6F4) has attracted particular attention3a,6b,10,11 since a number retain their paramagnetic nature in the solid state. Of these, β-p-NCC6F4CNSSN˙ undergoes a phase transition to a canted antiferromagnetic state below 36 K,10 whereas p-O2NC6F4CNSSN˙ orders as a ferromagnet below 1.3 K.6b
 | ||
| Fig. 1 Modes of association in dithiadiazolyl radicals; (a) cis-cofacial; (b) twisted; (c) trans-antarafacial; (d) trans-cofacial. | ||
The insensitivity of the spin density distribution on the heterocyclic ring to substituent R makes DTDAs particularly attractive for a systematic structural study since modification of the R group can lead to variation in solid state structure without influencing the spin distribution.4 As part of our on-going research in this area we have investigated the structural variations in a series of di- and tri-fluorophenyl-1,2,3,5-dithiadiazolyls (Scheme 2) to probe the ways in which the fluorine atom may hinder the dimerisation process. The role of fluorine in determining crystal structure has been a matter of some debate,12 and this study offers an opportunity to clarify the effect of F, and its substitution patterns, on the observed crystal structures of DTDA molecules. These structures reveal that an examination of interactions between individual structure-directing groups is not always an appropriate methodology, and that a more holistic approach is required.
 | ||
| Scheme 2 The isomers of di- and trifluorophenyl-1,2,3,5-dithiadiazolyl. | ||
Experimental
All the dithiadiazolyls 1–12 were synthesized using standard literature methods4 from commercially available starting materials. All reagents {Li[N(SiMe3)2], SCl2, Zn/Cu (Aldrich); benzonitriles (Apollo, Avocado, Fluorochem, Lancaster)} were used as received. All reactions were carried out using standard double-manifold techniques and a glovebox (Saffron Scientific) which typically provided an atmosphere containing less than 10 ppm H2O. Et2O was freshly distilled off Na prior to use and THF freshly distilled off CaH2. SO2 (Aldrich) was stored over P4O10 before use. The in-house supply of nitrogen, dried by passing through a P4O10 column, was used in experimentation. The preparation of 1 is described as a typical protocol, though reductions were undertaken in either THF or liquid SO2, and the chloride salt of 11 was synthesized in hexane.Synthesis of 1
Li[N(SiMe3)2] (1.39 g, 8.3 mmol) and 2,3-difluorobenzonitrile (1.30 g, 8.3 mmol) were stirred in Et2O (40 ml) overnight to yield a pale yellow solution. The solution was cooled to 0 °C and SCl2 (1.5 ml, 24.45 mmol) added slowly with stirring. The resultant orange precipitate was stirred (3 h), filtered and pumped to dryness in vacuo. Zn/Cu couple (0.50 g, 7.69 mmol) was added and the mixture stirred in dry THF (20 ml) overnight and the resultant black suspension dried in vacuo. The solid residue was heated to 90 °C and sublimed to yield black/green blocks of the product on the cold finger. The crystals were removed from the cold finger and the process repeated until no new product could be sublimed. Yield 0.54 g, 28%. Found: C 37.79, H 1.34, N 12.51%. Required for C7H3F2N2S2: C 38.71, H 1.38, N 12.90%. +EI-MS: 216.9706 [(C6F2H3)CNSSN]+, 171 [(C6F2H3)CNS]+, 139 [(C6F2H3)CN]+, 112 [C6F2H3]+, 78 [S2N]+.Yields were not optimized for this structural study, but typically fell in the range 10–35%. Recovered yields, microanalytical data, mass spectral data and sublimation conditions for 1–12 (excluding 1, 8 and 10) are available as ESI.†
X-Ray diffraction
Crystals of 1, 3, 4, 5, 6, 9, 11 and 12 for single crystal X-ray diffraction studies were grown by sublimationin vacuo and/or under ca. 1/3 atm of N2. The single crystal structures of 1 and 3 have been reported previously.13,14 We identified two different morphologies of 4 (4α and 4β) which were dependent upon sublimation conditions.15 Radicals 2, 7, 8 and 10 provided only polycrystalline samples despite multiple attempts at sublimation using a variety of experimental conditions (temperature and pressure). We have successfully determined the structure of one of these isomers (7) from powder diffraction using synchrotron radiation.16Single crystals were mounted on the end of glass fibres using perfluorinated polyether oil.17XRD data for 4α, 9 and 12 were obtained using a Siemens SMART 1K CCD three-circle diffractometer. Data for 5 and 6 were collected on a RIGAKU AFC7-R four circle diffractometer, whilst data for 4β and 11 were obtained on a Nonius KappaCCD diffractometer. All diffractometers utilised monochromatic Mo Kα radiation (λ = 0.71073 Å) and the temperature was controlled using an Oxford Cryosystems cryostream device. Structures were solved and refined using SHELX97.18 The esds on intermolecular contacts were calculated using SHELXL9718 implemented through WinGX.19 Graphics were prepared using Mercury.20
Selected crystal data for 1–12 are presented in Table 1, and selected structural parameters are collected in Table 2. Intramolecular bond lengths and angles are supplied as ESI.†
| 1 | 3 | 4α | 4β | 5 | 6 | 7 | 9 | 11 | 12 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Chemical formula | C7H3F2N2S2 | C7H3F2N2S2 | C7H3F2N2S2 | C7H3F2N2S2 | C7H3F2N2S2 | C7H3F2N2S2 | C7H2F3N2S2 | C7H2F3N2S2 | C7H2F3N2S2 | C7H2F3N2S2 |
| Colour | Black/green | Black | Red | Red | Red | Red | Green-black | Green-black | Brown | Brown |
| Appearance | Blocks | Needles | Blocks | Needles | Blocks | Blocks | Polycrystalline bundles | Prisms | Needles | Blocks |
| Formula weight | 217.2 | 217.2 | 217.23 | 217.23 | 217.23 | 217.23 | 234.96 | 235.23 | 235.23 | 235.23 |
| Crystal system | Triclinic | Tetragonal | Monoclinic | Tetragonal | Monoclinic | Triclinic | Monoclinic | Tetragonal | Monoclinic | Triclinic |
| Space group | P-1 | I41/a | P21/c | I41/a | P21/n | P-1 | P21/n | I41/a | P21/c | P-1 |
| a/Å | 6.637(6) | 30.214(2) | 16.885(4) | 30.168(1) | 7.543(2) | 7.058(3) | 11.543(3) | 30.483(2) | 7.1195(1) | 5.9462(1) |
| b/Å | 8.768(11) | 30.214(2) | 11.989(4) | 30.168(1) | 11.193(2) | 15.045(5) | 20.6570(5) | 30.483(2) | 27.2091(3) | 10.2733(2) |
| c/Å | 13.546(5) | 7.1829(1) | 8.207(4) | 7.1749(2) | 18.688(4) | 16.258(6) | 7.0510(1) | 7.117(1) | 25.1504(4) | 14.3236(2) |
| α/° | 88.79(6) | 90 | 90.00 | 90.00 | 90.00 | 66.79(3) | 90 | 90.00 | 90.00 | 76.255(1) |
| β/° | 86.37(5) | 90 | 95.51(3) | 90.00 | 93.87(3) | 84.17(3) | 100.367(1) | 90.00 | 92.614(1) | 79.899(1) |
| γ/° | 80.40(9) | 90 | 90.00 | 90.00 | 90.00 | 80.40(3) | 90 | 90.00 | 90.00 | 78.649(1) |
| V/Å3 | 776(1) | 6556.3(11) | 1653.7(11) | 6529.9(4) | 1574.2(6) | 1563.2(10) | 1654.14(9) | 6613.2(11) | 4866.95(12) | 825.67(2) |
| Z | 4 | 32 | 8 | 32 | 8 | 8 | 8 | 32 | 24 | 4 |
| D calcd/g cm−3 | 1.86 | 1.761 | 1.745 | 1.768 | 1.833 | 1.846 | 1.887 | 1.890 | 1.926 | 1.892 |
| Temperature/K | 150(2) | 293 | 150(2) | 180(2) | 150(2) | 200(2) | 293 | 150(2) | 180(2) | 150(2) |
| µ/mm−1 | — | — | 0.623 | 0.631 | 0.655 | 0.659 | — | 0.648 | 0.661 | 0.649 |
| R int | — | — | 0.0382 | 0.0329 | 0.0863 | 0.0496 | — | 0.0385 | 0.0899 | 0.0250 |
| R1 [I > 2σ(I)] | — | — | 0.0589 | 0.0337 | 0.0464 | 0.0727 | — | 0.0274 | 0.0488 | 0.0321 |
| Reference | 13 | 14 | This work | This work | This work | This work | 16 | This work | This work | This work |
| Twist angle | |||||||
|---|---|---|---|---|---|---|---|
| between aryl | ‘In-plane’ S⋯S (Å) | S⋯N | S⋯F | ||||
| and heterocyclic | Intradimer | Interdimer | sum of vdW radii | sum of vdW radii | sum of vdW radii | ||
| ring planes (°) | S⋯S (Å) | S⋯S (Å) (stacks) | 3.20–4.06 Å 29 | 3.20–3.63 29 | 2.90–3.41 Å 29 | Ref. | |
| a contact distances measured using Mercury, a 1 + x, y, z, b 1 + x, −1 + y, z, c x, −1 + y, z, d x, y, −1 + z, e −0.25 + y, 0.75 − x, −0.25 + z, f −0.25 + y, 0.75 − x, 0.75 + z, g 0.75 − y, 0.25 + x, 0.25 + z, h 0.75 − y, 0.25 + x, −0.75 + z, i x, 1.5 − y, 0.5 + z, j −x, −0.5 + y, 0.5 − z, k −x, 1 − y, 1 − z, l x, 1.5 − y, −0.5 + z, m 1 − x, −y, 1 − z, n − 0.5 + x, 0.5 − y, 0.5 + z, o − x, −y, 1 − z, p −1 + x, y, z, q 1 − x, 1 − y, −z, r 1 − x, 1 − y, 1 − z, s x, y, 1 + z, t 1 − x, −y, 2 − z, u 0.5 − x, −0.5 + y, 1.5 − z, v 0.5 − x, −0.5 + y, 0.5 − z, w 0.75 − y, −0.25 + x, 0.75 − z, x 0.75 − y, −0.25 + x, 1.75 − z, y 1 − x, 0.5 + y, 1.5 − z, z x, 0.5 − y, 0.5 + z, aa −x, 0.5 + y, 1.5 − z, bb −x, 1 − y, −z. | |||||||
| 1 | 5.9 | 3.020 | S2⋯S4 a…3.628 | — | — | F2⋯S4b 3.072 | 13 |
| 5.7 | F2⋯S3 b 3.549 | ||||||
| F4⋯S2c 3.028 | |||||||
| F4⋯S1 c 3.382 | |||||||
| F4⋯S4 c 3.247 | |||||||
| 3 | 21.2 | 3.203 | S1⋯S3d 4.006 | S1⋯S1e 3.517 | S1⋯N1g…3.447 | F2b⋯S2e 3.062 | 14 |
| 18.3 | 3.119 | S2⋯S4d 4.084 | S1⋯S3e 3.554 | S2⋯N1g…3.322 | F2b⋯S4e 3.064 | ||
| S3⋯S3e 3.578 | S3⋯N3g…3.776 | F4b⋯S2f 3.115 | |||||
| S3⋯S1f 3.587 | S4⋯N3g…3.390 | F4b⋯S4e 3.411 | |||||
| S1⋯N3h 3.864 | |||||||
| S2⋯N3h 3.763 | |||||||
| S3⋯N1g 3.777 | |||||||
| S4⋯N1g 3.639 | |||||||
| 4α | 48.72(27) | 3.069(3) | — | S12⋯S11i 3.361(3) | S11⋯N11i 2.974(6) | F12⋯S11j 3.376(5) | This work |
| 47.69(28) | 3.129(3) | S22⋯S21i 3.435(3) | S12⋯N11i 3.102(7) | F12⋯S12k 3.345(6) | |||
| S22 S11i 3.694(3) | S21⋯N21i 3.284(7) | F16⋯S21l 3.088(5) | |||||
| S22⋯N21i 3.498(7) | F16⋯S22d 3.234(5) | ||||||
| 4β | 29.42(4) | 3.2167(7) | S11⋯S21d 3.9939(8) | S11⋯S11g 3.4885(6) | S11⋯N11g 3.5217(17) | F12⋯S12e 3.2297(13) | This work |
| 24.93(4) | 3.0692(8) | S12⋯S22d 4.1258(8) | S11⋯S21h 3.5191(7) | S12⋯N11g 3.2139(17) | F12⋯S22e 3.1052(12) | ||
| S21⋯S11g 3.5491(7) | S11⋯N21h 3.9402(17) | F22⋯S12f 3.2407(12) | |||||
| S21⋯S21g 3.5363(6) | S12⋯N21h 3.8125(17) | F22⋯S22e 3.7534(14) | |||||
| S21⋯N11g 3.9081(17) | |||||||
| S22⋯N11f 3.6066(17) | |||||||
| S21⋯N21g 3.9046(17) | |||||||
| S22⋯N21g 3.2715(17) | |||||||
| 5 | 11.99(18) | 3.074(2) | — | — | S11⋯N21m 3.262(4) | F14⋯S11n 3.079(3) | This work |
| 8.46(17) | 3.172(2) | S21⋯N11m 3.284(4) | F14⋯S12n 2.986(3) | ||||
| F13⋯S12n 3.265(3) | |||||||
| F24⋯S21n 3.114(3) | |||||||
| F24⋯S22n 3.194(3) | |||||||
| F23⋯S22n 3.168(3) | |||||||
| S11⋯F13° 3.203(3) | |||||||
| 6 | 10.93(13) | 3.113(3) | S11⋯S21p 3.965(2) | S11⋯S32 3.666(3) | S11⋯N32 3.376(6) | F35⋯S21r 3.255(5) | This work |
| 11.11(15) | 3.111(3) | S12⋯S22p 3.982(3) | S12⋯S32 3.640(3) | S11⋯N42 3.684(6) | F25⋯S31q 3.062(4) | ||
| 9.45(13) | 3.150(3) | S31⋯S41p 3.945(3) | S11⋯S42 4.008(3) | S21⋯N42 3.205(6) | F25⋯S32q 3.017(5) | ||
| 11.09(16) | 3.195(3) | S32⋯S42p 3.927(3) | S12⋯S42 3.769(3) | S21⋯N32a 3.763(6) | F15⋯S41q 2.938(4) | ||
| S21⋯S32a 3.858(3) | S12⋯N22q 3.276(6) | F15⋯S42q 2.936(5) | |||||
| S22⋯S32a 3.744(3) | S22°⋯N12 3.284(6) | F15⋯S31b 3.361(4) | |||||
| S21⋯S42 3.804(3) | |||||||
| S22⋯S42 3.461(3) | |||||||
| 7 | 11.8 | 3.295 | S1⋯S4s 3.839 | — | S1⋯N4m 3.237 | (within chains) | 16 |
| 23.6 | 3.249 | S2⋯S3s 3.780 | S4⋯N1m 3.373 | F2⋯S2u 3.017 | |||
| S4⋯N4m 3.820 | F3⋯S1u 3.248 | ||||||
| S1⋯N1t 3.742 | F3⋯S2u 3.374 | ||||||
| F5⋯S3v 3.157 | |||||||
| F6⋯S4v 3.115 | |||||||
| F6⋯S3v 3.365 | |||||||
| (between chains) | |||||||
| F1⋯S4m 3.264 | |||||||
| F4⋯S4m 3.232 | |||||||
| F4⋯S1m 3.352 | |||||||
| 9 | 30.37(3) | 3.1893(7) | S11⋯S21d 3.9832(7) | S11⋯S11e 3.5901(5) | S11⋯N21h 3.8346(12) | F12⋯S12e 3.0245(9) | This work |
| 26.57(3) | 3.0748(7) | S12⋯S22d 4.0810(8) | S11⋯S21e 3.6488(5) | S12⋯N21h 3.9361(12) | F12⋯S22e 3.1447(9) | ||
| S21⋯S21e 3.5290(4) | S11⋯N11g 3.4516(12) | F22⋯S12f 3.1729(9) | |||||
| S21⋯S11f 3.5141(5) | S12⋯N11g 3.3409(12) | F22⋯S22e | |||||
| S21⋯N11g 3.9241(12) | 3.4884(10) | ||||||
| S22⋯N11g 3.8386(12) | F13⋯S12w 3.456(3) | ||||||
| S21⋯N21g 3.7735(11) | F23⋯S22x 3.4572(11) | ||||||
| S22⋯N21g 3.2313(11) | |||||||
| 11 | 29.24(6) | 3.2020(12) | S11⋯S21a 3.9936(12) | (in pinwheels) | S11⋯N52 3.347(3) | (in pinwheels) | This work |
| 30.57(6) | 3.1455(12) | S12⋯S22a 4.0266(12) | S11⋯S52 3.3929(12) | S12⋯N52 3.567(3) | F12⋯S61r 2.9420(19) | ||
| 27.98(6) | 3.2552(11) | S31⋯S41p 3.9318(11) | S21⋯S62 3.6130(12) | S21⋯N62 3.121(3) | F12⋯S51r 3.346(2) | ||
| 30.09(5) | 3.1320(11) | S32⋯S42p 4.0626(11) | S11⋯S51r 3.7249(12) | S22⋯N62 3.895(3) | F22⋯S51r 3.3221(19) | ||
| 28.58(5) | 3.1011(12) | S51⋯S61a 4.0669(11) | S11⋯S52r 3.5469(12) | S51⋯N11r 3.097(3) | F22⋯S61k 3.2416(19) | ||
| 33.23(6) | 3.2222(12) | S52⋯S62a 3.9680(12) | S21⋯S61k 4.4280(12) | S52⋯N11r 3.871(3) | F66⋯S22 2.9208(19) | ||
| S21⋯S62k 3.7510(13) | S61⋯N11r 3.517(3) | F66⋯S12 3.2441(18) | |||||
| S61⋯N21k 3.468(3) | F56⋯S12 3.273(2) | ||||||
| (between pentamers) | S62⋯N21k 3.619(3) | F56⋯S22a | |||||
| S31⋯S41° | 3.2638(18) | ||||||
| 3.4777(12) | |||||||
| S31⋯S42° | (between pinwheels) | ||||||
| 3.8709(12) | F14⋯S41y 3.257(2) | ||||||
| S32⋯S41° | F14⋯S42z 3.079(2) | ||||||
| 3.8168(12) | F24⋯S31aa 3.046(2) | ||||||
| S31⋯S31° | F24⋯S32z 3.151(2) | ||||||
| 3.2928(15) | F54⋯S42 3.317(2) | ||||||
| S31⋯S32° | F54⋯S32a 3.253(2) | ||||||
| 3.7034(11) | F64⋯S32 3.462(2) | ||||||
| S41⋯S41m | F64⋯S42 3.053(2) | ||||||
| 3.8037(16) | |||||||
| 12 | 9.07(8) | 3.1189(6) | — | S11⋯S12a 4.2330(6) | S12⋯N11p 3.5659(15) | (within chains) | This work |
| 9.05(8) | 3.0881(7) | S21⋯S12a 4.0561(7) | S12⋯N21p 3.6184(15) | F14⋯S11b 3.0522(12) | |||
| S21⋯S22a 4.0794(7) | S22⋯N21p 3.6531(15) | F14⋯S12b 3.2170(12) | |||||
| F24⋯S22b 2.9404(11) | |||||||
| (between chains) | |||||||
| F13⋯S11r 3.2878(13) | |||||||
| F15⋯S21c 3.2067(11) | |||||||
| F25⋯S22bb 3.1889(12) |
Theoretical calculations
Molecular electrostatic isopotential maps (MEPs) for 1–12 were determined using restricted Hartree–Fock semi-empirical PM5 methods within the Quantum Cache programme.21 Whilst there are certain limitations to the semi-empirical methods, previous studies on HCNSSN˙ and ClCNSSN˙inter alia indicate that this method provides a good analysis of both the spin density and charge distribution in these molecules.7 In addition, excellent correlations have been observed between the redox behaviour of phenyl-substituted DTDA radicals and the calculated HOMO/LUMO energies of the DTDA ring using semi-empirical methods.5cCalculations of the energy minima and barrier to rotation between phenyl and dithidiazolyl rings have been undertaken previously,22 with both double zeta (UB3LYP/6-31G*) and triple zeta (UB3LYP/6-311G) methods offering similar energetic barriers to rotation of the phenyl ring (25 and 26 kJ mol−1 respectively), indicating reasonable convergence at this level. In this study, energies of 4′-(2-fluorophenyl)- and 4′-(2,6-difluorophenyl)-dithiadiazolyl radicals were determined using single point calculations at 10° intervals in the range 0–90° for the torsion angles between dithiadiazolyl and aryl rings. In all cases the ring geometries were otherwise fixed at those determined from a full geometry optimization. Calculations employed identical split-valence double zeta (UB3LYP/6-31G*) methods within GAMESS-UK23 to those applied previously.22
Results
The twelve possible difluorophenyl and trifluorophenyl dithiadiazolyl radicals are shown in Scheme 2. These were prepared according to the standard synthetic methods.4Vacuum sublimation of 2, 7, 8 and 10 did not afford crystals of sufficient size or quality for single crystal diffraction despite multiple attempts. Nevertheless an analysis of the remaining eight isomers (plus polymorph) has proved instructive. A number of dithiadiazolyl radicals have been shown to exhibit polymorphism or form host–guest complexes with small molecules such as N2 or CO2.7e,24 For this reason, crystalline samples were carefully scrutinised for different polymorphs and sublimation of 1–12 was carried out in vacuo (10−1 Torr) and under ca. 1/3 atm of N2 in order to probe for the presence of additional polymorphs. In our hands, with the exception of 4, each compound appeared phase pure.15 Compound 4 produced two different phases dependent upon sublimation conditions. Sublimationin vacuo produced 4α whereas sublimation under a partial atmosphere of N2 yielded 4β.The crystal structures of 4α, 4β, 5, 6, 9, 11 and 12 are described in relation to the previously reported13–15 structures of 1, 3 and 7.
Molecular structure of the dithiadiazolyl ring
The heterocyclic ring geometries (see ESI†) are similar and consistent with those previously reported in other DTDA radicals e.g.ClCNSSN˙ and HCNSSN˙.7 However, the twist angles (θ) between the DTDA ring and the phenyl ring vary considerably (5.7–48.7°) (Table 2). In cases where there are two H atoms in the ortho positions (5, 6 and 12), θ is small spanning a range between 8.5 and 11.9°. These values are reflected in other phenyl-substituted dithiadiazolyls bearing two ortho–H atoms such as p-XC6H4CNSSN˙ (X = H, Cl, I, CN, CNSSN˙) (θ in range: 5.0–11.8°).5c,6a,25 Conversely, when two F atoms are positioned in the ortho positions, large twist angles are observed (24.9–48.7°). Again similar behaviour has been observed in perfluorophenyl derivatives such as p-XC6F4CNSSN˙ (X = Br, CN, NO2 and p-NCC6F4) (θ range: 32.2–68.7°).3,10,11,26 Of the derivatives containing one ortho–H and one ortho-F the behaviour is rather less well-defined. Whilst only three of the potential six derivatives containing this functionality produced crystals of a suitable quality for diffraction studies (1, 3 and 7), θ in these three structures spans the range 5.7–23.6°. The variation in θ value within each of these groups of compounds suggests a shallow minimum on the potential energy surface for rotation about the C–C bond which links the DTDA and phenyl rings.Previous DFT (UB3LYP/6-31G*) studies on PhCNSSN˙22 reveal a shallow minimum at θ = 0° with an energy barrier of 25 kJ mol−1 for complete rotation about the C–C bond between the two rings. A distortion of ±25° from co-planarity corresponds to ca. 3 kJ mol−1. Replacement of one ortho-H by F reveals a similar energy minimum at 0° but with a reduced energy barrier for complete rotation (15 kJ mol−1) and a distortion of ±35° corresponding to ca. 3 kJ mol−1. In the case of the 2,6-difluorophenyl derivative, the two ortho fluorines destabilise the planar configuration and an energy minimum occurs at θ ∼ 50° with an even lower energy barrier to rotation (11 kJ mol−1) and a nominal 3 kJ mol−1 distortion offering a range of angles in the range 30–90°. With a non-planar energy minimum, two different mechanisms arise for a 180° rotation; either via a transition state in which the two rings become coplanar, or one in which they are orthogonal. In the current case, rotation via a transition state with mutually orthogonal rings is more favourable by ca. 9.5 kJ mol−1 than one in which the two rings are coparallel. [The angular dependences of the energies (UB3LYP/6-31G*) of PhCNSSN˙, 2-FC6H4CNSSN˙ and 2,6-F2C6H3CNSSN˙ are available as ESI†].
Both the experimental observations and theoretical studies indicate that the energy barrier to distortion is low in aryl substituted DTDAs, and decreases markedly with increasing fluorination in the 2-position. This leads to larger deviations from coplanarity in the case of the fluorinated derivatives. As a consequence the torsion angle θ is likely to be rather flexible and amenable to distortions to accommodate crystal packing forces. This is particularly emphasised in the two polymorphs of 4: in 4α both θ angles (47.7 and 48.7°) fall in the conventional range for a derivative bearing two ortho-F atoms, but these angles appear unusually small in polymorph 4β (24.9 and 29.4°).
The vast majority of dithiadiazolyl radicals associate in the solid state through a π*–π* interaction between singly occupied molecular orbitals. The local a2 symmetry of this orbital allows a number of possible bonding modes to be adopted (Fig. 1).27 Whilst the majority of DTDA radicals adopt the cis-cofacial conformation, twisted and trans conformations are not unprecedented. Previous MO calculations have indicated that while the enthalpy of dimerisation is significant, the energy difference between these conformations is small.28 Indeed amongst the five reported polymorphs of ClCNSSN˙,7a–c three adopt twisted conformations whereas two adopt cis conformations. Clearly there is little energetic preference between the conformers and the mode of dimerisation is likely to be driven by other packing factors. With the exception of 1, all the di- and tri-fluorophenyl derivatives reported here adopt cis-cofacial geometries. The intradimer S⋯S distances in the cofacial dimers fall in the range 3.069–3.295 Å. Notably those structures which form ‘isolated’ dimers exhibit slightly shorter S⋯S contacts (3.069–3.172 Å, average 3.11 Å) than those which adopt π-stacked structures (3.0692–3.295 Å, average 3.17 Å). This increase in S⋯S distance for π-stacked systems presumably arises out of a bonding interaction between dimers which leads to a concomitant weakening of the intra-dimer S⋯S interaction.
Crystal structures of 4-(difluorophenyl)-1,2,3,5-dithiadiazolyls
The importance of an intermolecular contact is typically made in comparison with the sum of the van der Waals radii. A systematic study of the Cambridge Structural Database (CSD) by Nyburg and Faerman29 revealed that the van der Waals radii need not necessarily be spherical and, particularly for heavier p-block elements, may exhibit significant anisotropy. The minor radii correspond to contacts close to the molecular plane whereas major radii contacts are formed perpendicular to the plane. Within the context of the current study it is worth noting Nyburg and Faerman's estimates of the minor and major radii of N, F and S; for both N and F the radii are essentially isotropic (1.60 Å for N and 1.30–1.38 Å for F), whilst there is significantly greater anisotropy for S (1.60–2.03Å). Relevant intermolecular close contact distances for all the structures discussed here are collected in Table 2.The dimers stack along the a-axis, with alternating short intra-dimer (3.020 Å) and long inter-dimer (3.628 Å) S⋯S contacts. Individual molecules of 1 form contacts from the meta-F atom to S. Both single (3.247 Å) and bifurcated S⋯F contacts (3.028–3.549 Å) are comparable with the sum of the van der Waals radii (2.90–3.41 Å) and link dimers together along the crystallographic b-axis (Fig. 2a).
 | ||
| Fig. 2 Packing in 1 (a) Chain-like motif parallel to the crystallographic b–axis generated from S⋯F contacts; (b) antiparallel alignment of chains generated through a bifurcated interaction between the p-H atom and the electronegative N and F atoms. The second molecule of each twisted dimer is shown in grey in order to emphasize the in-plane inter-dimer interactions (view is down the a-axis). | ||
Neighbouring chains are oriented antiparallel along the b-axis and related via an inversion centre which generates close contacts between the electronegative N and o-F atoms and the electropositive p-H on the aryl ring (N⋯H 2.544 and 2.530 Å, F⋯H 2.496 and 2.617 Å; 2.609 and 2.623 Å). This bifurcated C–H⋯N/C–H⋯F contact to DTDA derivatives bearing ortho-Fphenyl rings is a common motif in these structures and is also observed in 3, 4β and 9.
In the ab plane radicals are arranged in pinwheels (labelled A in Fig. 3). The S⋯N separations in the centre of the pinwheel range from 3.322 to 3.864 Å (cf. sum of the van der Waals radii: 3.20–3.63 Å) whereas the S⋯S separations range from 3.517–3.587 Å (cf. sum of the van der Walls radii 3.20–4.06 Å). Some structural disorder of the two fluorine positions, equivalent to a 180° flip of the phenyl ring, is observed. The major component of the disorder (Fig. 3) places the ortho-F atoms ‘exo’ to the pin-wheel and supports a bifurcated interaction between the p-H and the N and F atoms analogous to that observed in 1; the F⋯H contacts are 2.542 and 2.624 Å and N⋯H contacts are 2.584 and 2.782 Å. In the minor ‘endo’ position, there is an interaction between the N and F atoms and a heterocyclic S atom, with S⋯F contacts ranging between 3.062 and 3.411 Å (sum of the van der Waals radii 2.90–3.41 Å).
 | ||
| Fig. 3 Packing of 3 viewed down the c-axis showing the pinwheel formation (A) and channel (B) along fourfold axes. The fluorine positional disorder has been omitted for clarity and the substituents shown are the major component. | ||
Notably, the major component of the disorder also generates close F⋯F contacts of 2.410–2.814 Å between meta-F atoms and a channel at the centre of the second ‘aryl’ pin wheel (labelled B in Fig. 3). Disorder in the position of these meta-F atoms alleviates F⋯F contacts and simultaneously partially fills the void at B. This should be compared with the isostructural derivative 4β which has no meta-F atoms (Fig. 5b).
Crystal structure of α-4-(2′,6′-difluorophenyl)-1,2,3,5-dithiadiazolyl, 4α. Radical 4α crystallises in the monoclinic space groupP21/c with one cis-cofacial dimer in the asymmetric unit. The dimers form herring-bone chains, oriented approximately along the c-axis, linked via close S⋯N interactions (2.974–3.498 Å, cf. van der Waals radii 3.20–3.63 Å). These chains align coparallel to form a two-dimensional sheet (Fig. 4) in which the para-H atoms of one molecule form close contacts to the aryl ring of a symmetry-related molecule (C⋯centroid 4.126 Å, C–H-centroid angle 171°) i.e. a C–H⋯π type interaction commonly observed in aromatic molecules.30
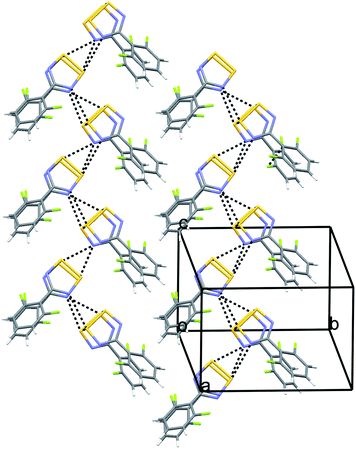 | ||
| Fig. 4 Crystal packing in 4α illustrating chain-like motif along the crystallographic c-axis. | ||
Crystal structure of β-4-(2′,6′-difluorophenyl)-1,2,3,5-dithiadiazolyl, 4β. The second polymorph of 4 crystallises in the tetragonal space groupI41/a and is isostructural with both 3 and 9 (Fig. 5a). The dimers are arranged in pinwheels, with close S⋯N (3.214–3.940 Å) and S⋯F contacts (3.105–3.753 Å) within the pinwheel. Both these sets of contacts are similar to those observed in 3. Exo to the pinwheel the para-H forms F⋯H (2.544 and 2.574 Å) and N⋯H (2.591 and 2.571 Å) contacts. The absence of F atoms in the meta-position (cf. radical 3) eliminates close F⋯F contacts, but also results in channels in this structure which run parallel to the c-axis (Fig. 5b). The cross-sectional area of this void is estimated at ca. 2.6 Å2 and thus is sufficiently small to preclude accommodation of guest molecules: no significant residual electron density was observed in the channel region. This should be contrasted with the comparatively diverse range of host–guest behaviour displayed by 4-(4′-trifluoromethyl-3′-fluorophenyl)-1,2,3,5-dithiadiazolyl.24
 | ||
| Fig. 5 (a) Superimposition of the isostructural DTDA derivatives 3 (red), 4β (blue) and 9 (green) viewed in the ab plane. (b) Space-filling diagram of 4β showing channels. | ||
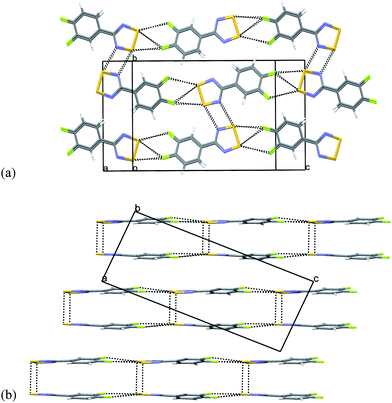 | ||
| Fig. 6 Crystal structure of 5 (a) with in-plane close S⋯F and S⋯N contacts emphasized and (b) showing stepped packing of sheets/ribbons. | ||
 | ||
| Fig. 7 Sheets of dimers in 6 viewed down the a-axis. | ||
The sheets of tetramers stack along the c-axis with alternating short intradimer (∼3.1 Å) and long interdimer (∼4.0 Å) S⋯S contacts.
The structure of 6 is closely related to the pinwheel structures of the three isomorphous radicals 3, 4β and 9 (Fig. 8). If the layer structure in 6 is dissected into slabs as shown in Fig. 8, then application of a translation as shown leads to cleavage of the two centrosymmetric S⋯N contacts (between D and E in Fig. 7) and cleavage of two sets of bifurcated S⋯F contacts. For 6, such a translation would generate two sets of (favourable) S⋯S and S⋯N pinwheel contacts but also two sets of (disfavoured) F⋯F contacts.
 | ||
| Fig. 8 Relationship between the layer-like structures of 6 (left) with isostructural 3, 4β and 9viatranslation (right, 4β shown here). | ||
Crystal structures of 4-(trifluorophenyl)-1,2,3,5-dithiadiazolyls
 | ||
| Fig. 9 (a) Layerlike structure of dimers in 7 observed in the ab plane. (b) Side-on view showing stacking of the layers (cf. 5, Fig. 6). | ||
 | ||
| Fig. 10 Packing in 11 showing (a) formation of slipped pinwheels, (b) generation of pentamers from pinwheels and their subsequent linkage to form bimolecular tapes and (c) packing of tapes in the bc plane. | ||
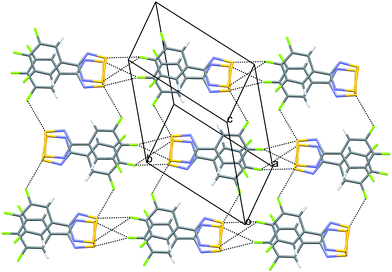 | ||
| Fig. 11 Chain-like motif in 12 generated through bifurcated S⋯F contacts. | ||
Discussion
All the radicals described in this paper are dimeric in the solid state, with singlet diamagnetic ground states due to strong radical–radical interactions, and all but one (1) form cis-cofacial dimers. Whilst some derivatives are isostructural, there is a great diversity in packing arrangements, i.e. small alterations to the fluorine substitution pattern result in significant changes to the packing motif. We now investigate these structures through an analysis of their intermolecular contacts in relation to their charge distribution determined through theoretical calculations.It is generally accepted that neutral planar aromatic molecules tend to adopt either of two packing arrangements in the solid state; π-stacked or herringbone motifs.30 Within these two broad categories there is some structural diversity e.g. slipped π-stacks, distorted π-stacks (with a commensurate doubling of the ‘short’ stacking axis), sandwich herringbone motifs etc. For neutral aromatic hydrocarbons the stronger nature of the C–H⋯π interaction over the π–π interaction favours herringbone structures. Indeed in the case of the neutral DTDA radicals (PhCNSSN˙)225a and (p-ClC6H4CNSSN˙)25c sandwich herringbone motifs are observed between dimers. Solid state structures based upon planar π-stacked arrangements become favoured when there are directional intermolecular forces which may be dipolar, covalent or electrostatic in origin. In the case of 1–12 every structure is based upon a layer-like motif consistent with the presence of strong directional in-plane forces. We have previously rationalised the intermolecular contacts in some of the simplest DTDA derivatives (HCNSSN˙ and ClCNSSN˙) through an analysis of the electrostatic contribution to bonding.7a,b We now extend this approach to this more complex family of fluoro-phenyl DTDAs.
Electrostatic contribution to bonding
It is our hypothesis that the majority of the important structure-directing intermolecular interactions involved in determining the crystal structures of these compounds are electrostatic in nature and optimise contacts between electronegative and electropositive regions in neighbouring molecules.A simple analysis of the electronegativities of S and N clearly reveals that the S–N bond should be considered to be polarized in the sense Sδ+—Nδ−. Therefore the positioning of two electropositive S atoms adjacent to each other generates a strongly electropositive region in every DTDA radical near the S atoms. As a consequence close intermolecular contacts between S and N (or indeed S and other electronegative elements, such as F) should be electrostatically favourable.
Previous theoretical and experimental analysis of intermolecular contacts in ClCNSSN˙ and HCNSSN˙ revealed four favourable in-plane modes of association between DTDA rings. These classifications are depicted in Fig. 12, and employed in this dicussion. The SN-I interaction occurs in all five polymorphs of ClCNSSN˙,7a–c and in HCNSSN˙7d–e and CF3CNSSN˙28inter alia. However in the presence of simple aryl substituents SN-I tends to become disfavoured to accommodate the sterics of the near-coplanar R group.31 As a consequence this interaction often appears less symmetric, with one contact longer than the other, generating an SN-IV motif which is more amenable to close-packing. The loss in electrostatic Sδ+⋯Nδ− attraction and increased Sδ+⋯Sδ+ repulsion may be partially balanced by a greater dispersion term between S atoms which are in close proximity.
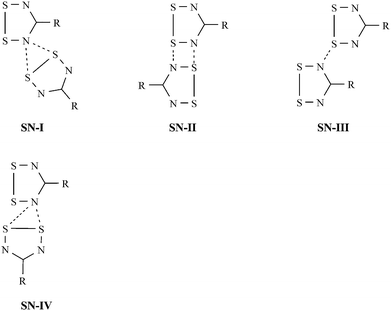 | ||
| Fig. 12 Electrostatically favourable Sδ+⋯Nδ− motifs in DTDA derivatives. | ||
Similarly with simple aryl substituents, the steric demands of R will typically disfavour SN-III in relation to SN-II. As a consequence the SN-II and SN-IV interactions are likely to play an important role in the structures of many aryl-substituted DTDA derivatives. In the current sample 3, 4α, 4β, 6, 9 and 11 all display SN-IV type interactions whereas 5, 6 and 7 display SN-II contacts. Only 1 and 12 do not appear to exhibit close in-plane contacts between DTDA rings.
The incorporation of fluorinated aromatic groups into the DTDA derivatives 1–12 leads to the additional possibility of Sδ+⋯Fδ− interactions which may compete with or additionally stabilise the favourable S⋯N interactions. The placement of F in the 2′ position of the phenyl ring is expected to enhance the stability of the SN-IV interaction described previously. Indeed, with the exception of 6, all those structures exhibiting an SN-IV contact also possess an ortho-F atom.
When two F atoms are placed ortho with respect to each other then this leads to the potential to form pairs of S⋯F contacts as in 5 and 7. Notably in 1 and 12, although these do not exhibit S⋯N contacts, they do exhibit pairs of S⋯F contacts.
Molecular electrostatic potential maps
One method to analyse the electrostatic contribution to bonding (and thereby assessing the relative strengths of Sδ+⋯Nδ−vs Sδ+⋯Fδ−) is through the use of molecular electrostatic isopotential (MEP) maps. We have previously employed this method for successfully interpreting the packing motifs in ClCNSSN˙ and HCNSSN˙7a,b as well as in dithiazolyl radicals32 and dithiatetrazocines.33 Others have utilized similar methods for other C/N/S heterocycles.34 The calculated MEP for each radical based on its geometry in the crystal is shown in Table 3. Red regions denote areas of an electropositive nature whilst blue regions have electronegative character. An analysis of these MEPs reinforces the arguments proposed above. They reveal electropositive regions near the heterocyclic S atoms and aromatic H atoms, whereas those regions near heterocyclic N and aromatic F atoms reflect a build-up of partial negative charge as anticipated from a simple consideration of electronegativity values. Favourable electrostatic interactions therefore comprise Sδ+⋯Nδ−, Sδ+⋯Fδ−, Hδ+⋯Nδ− and Hδ+⋯Fδ−. Importantly the relative positions of F atom substitution on the phenyl ring have a strong influence on both the charge distribution itself and also the relative magnitudes of the charges. Specifically, the location of electronegative F atoms on adjacent C atoms, i.e. mutually ortho with respect to each other e.g. in 1, 5, 7, 9 and 12, leads to an enhancement of the electronegative region near those F atoms. An examination of the electronegative regions in 1–12 clearly identifies that S⋯N contacts are likely to dominate the structures of 3, 4, 9 and 11 whereas S⋯F are likely to be structure directing in 1, 5, 7 and 12. Only in the case of 6 would there appear to be a fine balance between these two competing sets of interactions.| MEP | Packing summary | SN motif | MEP | Packing summary | SN motif | ||
|---|---|---|---|---|---|---|---|
| 1 |

|
Twisted dimer, stacks of molecules with closest favourable intermolecular interaction S⋯F contact. | none | 7 |

|
cis-Cofacial dimers packed in head-to-tail ribbons linked via intra-ribbon S⋯F contacts, aligned antiparallel with lateral inter-ribbon S⋯N contacts. | SN-II |
| 3 |
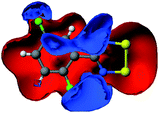
|
cis-Cofacial dimers packed in S-centred pinwheels. Isostructural with 4β and 9. | SN-IV | 9 |

|
cis-Cofacial dimers packed in S-centred pinwheels. Isostructural with 3 and 4β. | SN-IV |
| 4 |

|
4α: cis-Cofacial dimers, one-dimensional chains linked by intrachain S⋯N contacts. | 4α SN-IV | 11 |

|
cis-Cofacial dimers packed in distorted S-centred pinwheels with extra dimer in between pinwheels, S⋯F and S⋯N contacts. | SN-IV |
| 4β: cis-Cofacial dimers packed in S-centred pinwheels. Isostructural with 3 and 9. | 4β SN-IV | ||||||
| 5 |

|
cis-Cofacial dimers packed in head-to-tail ribbons linked via intra-ribbon S⋯F contacts, aligned antiparallel with lateral inter-ribbon S⋯N contacts. | SN-II | 12 |

|
cis-Cofacial dimers packed in head-to-tail chains linked viaS⋯F contacts, interchain S⋯F and long S⋯N contacts. | SN-III-type (long) |
| 6 |

|
cis-Cofacial dimers packed in off-set S-centred pinwheels, S⋯F and S⋯N contacts. | SN-II, SN-IV |
All structures presented here were examined for close contacts (less than or equal to the sum of the van der Waals radii) between S and F and between S and N. These are summarised in Table 3. It is noteworthy that 3, 4, 9 and 11 all exhibit the SN-IV motif in excellent agreement with the MEP analysis, whereas 1, 5, 7 and 12 all exhibit S⋯F contacts. Notably the chain-like motifs of 5, 6 and 7 also support some SN-II type contacts between neighbouring chains or ribbons. In addition to these general remarks, some specific observations are worthy of further comment.
Comparison of 3, 4β and 9
These three compounds are isostructural, consistent with the presence of a strongly structure-directing motif common to all molecules. The varied F atom positions on the aryl ring, coupled with positional disorder of the F atoms, suggests that here the SN-IV contact is structure-directing. This is supported by the observation that radical 4 is polymorphic, but both 4α and 4β exhibit the same SN-IV type interactions. Whilst they generate a pinwheel in 4β, they form chains in 4α. The observation of the same packing motif in two different polymorphs is also consistent with its structure-directing nature (cf. chains and hexameric rings in polymorphs of HCNSSN˙ linked viaSN-I type interactions).7On the behaviour of 11
Given the propensity for the SN-IV motif in isostructural 3, 4β and 9, it is initially surprising that 11 does not also adopt this tetragonal packing arrangement since its MEP clearly favours a structure dominated by S⋯N interactions. Indeed 11 does exhibit pinwheel motifs but these are distorted and accommodate additional molecules between the pinwheels. A comparison of the structure of 11 with isostructural 3, 4β and 9 resolves the anomalous behaviour of 11. If 11 adopted an isomorphous lattice to 4β, the additional para-F atom would be located in close proximity to the electronegative N/F ‘pocket’ of a neighbouring pinwheel resulting in structural destabilisation. Instead the pinwheel motif is retained (further evidence for the structure directing nature of SN-IV contacts), but with additional dimers packing between pinwheels. These additional dimers located between the pinwheels provide a network of C–H⋯F–C contacts to two neighbouring pinwheels and an S⋯F contact to a third pinwheel (Fig. 10).On the behaviour of 6
The MEP of radical 6 clearly reflects competition between S⋯N and S⋯F contacts. The lack of ortho-F atoms reduces the negative charge build-up associated with the N/F ‘pocket’ observed in 3, 4, 9 and 11. However in 6 the F atoms are not ortho to each other and do not offer a combined strong structure-directing influence. Earlier we emphasised the relationship between the structure of 6 and the isomorphous set of 3, 4β and 9. In 6, as in 11, the tetragonal pinwheel motif is destabilised by the presence of a repulsive F⋯F contact, this time between meta-F atoms. This results in an alternative packing strategy comprising some residual SN-IV contacts coupled with SN-II contacts.Propagation of S⋯F contacts
In the case of 1, 5, 7 and 12 the MEPs reveal that chain-like motifs with S⋯F contacts should be favoured. In the case of 5, 7 and 12 the linear or near-linear nature of the ribbon motifs favours chain formation. Interchain electrostatic contacts in 5 are few, although the SN-II motif is clearly present and favours an antiparallel arrangement of chains. Longer C–H⋯F–C and C–H⋯N contacts are also discernable. The inter-chain contacts in 7 are very similar to those observed in 5 (Fig. 13a) though the way in which the dimer distortion occurs differs in 5 and 7 (see Fig. 13b,c). | ||
| Fig. 13 Comparison of interchain interactions in 5 (red) and 7 (blue). (a) Overlay of chains. (b) Overlay of chains viewed side-on; note slippage of red chains. (c) Relationship between chain formation and dimers in 5 (top) and 7 (bottom). | ||
Conclusions
The results presented here show clearly that replacement of hydrogen by fluorine in fluorinated aromatic dithiadiazolyl derivatives has a very marked influence on solid state structure. The outcome of crystallization is extremely sensitive to the precise position and number of fluorine substituents. In the current sample of structures, the effects can be understood by considering the molecule as an entity, rather than being comprised of a series of different functionalities. Molecular electrostatic potential maps are extremely informative in identifying which of the possible combinations of electrostatic interactions are likely to dominate in the structure. In all cases the disulfide bond in the dithiadiazolyl ring bears a large partial positive charge and appears best stabilized by close contacts to large partial negative charges. The presence of fluorine substitution ortho to the dithiadiazolyl ring leads to highly electronegative regions near the dithiadiazolyl ring nitrogen. This ‘N/F pocket’ enhances S⋯N contacts which favours an SN-IV packing motif. Placement of two F atoms ortho with respect to each other generates large regions of electronegative character which can often lead to structure-directing S⋯F contacts in preference to the conventional S⋯N interaction.Importantly, whilst there is a strong tendency to examine observed crystal structures to identify structure-directing influences, much can also be gained through a study of hypothetical alternative structures or polymorphs to identify structure-destabilising motifs.
Acknowledgements
D.A.H. thanks the Cambridge Commonwealth Trust, the ORS and the Cecil Renaud Trust for funding. S.I.P. thanks the Royal Society for funding (URF). C.S.C. thanks the Natural Environment Research Council for funding.References
- J. M. Rawson, A. Alberola and A. Whalley, J. Mater. Chem., 2006, 16, 2560 RSC.
- (a) R. C. Haddon, Nature, 1975, 256, 394 CAS; (b) A. Cordes, R. Haddon and R. Oakley, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 673 CrossRef CAS; (c) R. T. Oakley, Can. J. Chem., 1993, 71, 1775 CAS see also ref. 1.
- A. J. Banister, N. Bricklebank, W. Clegg, M. R. J. Elsegood, C. I. Gregory, I. Lavender, J. M. Rawson and B. K. Tanner, J. Chem. Soc., Chem. Commun., 1995, 679 RSC.
- J. M. Rawson, A. J. Banister and I. Lavender, Adv. Heterocycl. Chem., 1995, 62, 137.
- (a) C. M. Aherne, A. J. Banister, T. G. Hibbert, A. W. Luke and J. M. Rawson, Polyhedron, 1997, 16, 4239 CrossRef CAS; (b) C. M. Aherne, A. J. Banister, I. B. Gorrell, M. I. Hansford, Z. V. Hauptman, A. W. Luke and J. M. Rawson, J. Chem. Soc., Dalton Trans., 1993, 967 RSC; (c) R. T. Boeré, K. H. Moock and M. Parvez, Z. Anorg. Allg. Chem., 1994, 620, 1589 CrossRef CAS.
- (a) A. W. Cordes, R. C. Haddon, R. G. Hicks, R. T. Oakley and T. T. M. Palstra, Inorg. Chem., 1992, 31, 1802 CrossRef CAS; (b) A. Alberola, R. J. Less, F. Palacio, C. M. Pask and J. M. Rawson, Molecules, 2004, 9, 771 Search PubMed; (c) G. Antorrena, S. Brownridge, T. S. Cameron, F. Palacio, S. Parsons, J. Passmore, L. K. Thompson and F. Zarlaida, Can. J. Chem., 2002, 80, 1568 CrossRef CAS; (d) A. W. Cordes, C. M. Chamchoumis, R. G. Hicks, R. T. Oakley, K. M. Young and R. C. Haddon, Can. J. Chem., 1992, 70, 919 CAS.
- (a) A. D. Bond, D. A. Haynes, C. M. Pask and J. M. Rawson, J. Chem. Soc., Dalton Trans., 2002, 2522 RSC; (b) C. S. Clarke, S. I. Pascu and J. M. Rawson, CrystEngComm, 2004, 6, 79 RSC; (c) C. Knapp, E. Lork, K. Gupta and R. Mews, Z. Anorg. Allg. Chem., 2005, 631, 1640 CrossRef CAS; (d) A. W. Cordes, C. D. Bryan, W. M. Davis, R. H. de Laat, S. H. Glarum, J. D. Goddard, R. C. Haddon, R. G. Hicks, D. K. Kennepohl, R. T. Oakley, S. R. Scott and N. P. C. Westwood, J. Am. Chem. Soc., 1993, 115, 7232 CrossRef CAS; (e) C. D. Bryan, A. W. Cordes, R. C. Haddon, R. G. Hicks, D. K. Kennepohl, C. D. MacKinnon, R. T. Oakley, T. T. M. Palstra, A. S. Perel, S. R. Scott, L. F. Schneemeyer and J. V. Waszczak, J. Am. Chem. Soc., 1994, 116, 1205 CrossRef CAS.
- S. A. Fairhurst, K. M. Johnson, L. H. Sutcliffe, K. F. Preston, A. J. Banister, Z. V. Hauptman and J. Passmore, J. Chem. Soc., Dalton Trans., 1986, 1465 RSC.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond, 2001, Oxford University Press Search PubMed.
- A. J. Banister, N. Bricklebank, I. Lavender, J. M. Rawson, C. I. Gregory, B. K. Tanner, W. Clegg, M. R. J. Elsegood and F. Palacio, Angew. Chem., Int. Ed. Engl., 1996, 35, 2533 CrossRef CAS.
- G. Antorrena, J. E. Davies, M. Hartley, F. Palacio, J. M. Rawson, J. N. B. Smith and A. Steiner, Chem. Commun., 1999, 1393 RSC.
- See for example J. D. Dunitz and R. Taylor, Chem.–Eur. J., 1997, 3, 89 Search PubMed; A. R. Choudhury and T. G. Row, CrystEngComm, 2006, 8, 265 CrossRef CAS.
- A. J. Banister, A. S. Batsanov, O. G. Dawe, P. L. Herbertson, J. A. K. Howard, S. Lynn, I. May, J. N. B. Smith, J. M. Rawson, T. E. Rogers, B. K. Tanner, G. Antorrena and F. Palacio, J. Chem. Soc., Dalton Trans., 1997, 2539 RSC.
- L. Beer, A. W. Cordes, D. J. T. Myles, R. T. Oakley and N. J. Taylor, CrystEngComm, 2000, 2, 109 RSC.
- In addition, K. Preuss (University of Guelph) has identified a third polymorph of 4 [personal communication].
- A. M. Bell, J. N. B. Smith, J. P. Attfield, J. M. Rawson, K. Shankland and W. I. F. David, New J. Chem., 1999, 23, 565 RSC.
- T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615 CrossRef.
- G. M. Sheldrick, University of Göttingen, Germany, 1997.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef.
- Quantum Cache, version 5.0, Fujitsu Co., 2001 Search PubMed.
- J. M. Rawson, C. S. Clarke and D. W. Bruce, Magn. Reson. Chem., 2009, 47, 3 CrossRef CAS.
- M. F. Guest, I. J. Bush, H. J. J. van Dam, P. Sherwood, J. M. H. Thomas, J. H. van Lenthe, R. W. A. Havenith and J. Kendrick, Mol. Phys., 2005, 103, 719 CrossRef CAS . GAMESS-UK is a package of ab initio programs. See: http://www.cfs.dl.ac.uk/gamess-uk/index.shtml.
- A. D. Bond, C. S. Clarke, D. A. Haynes and J. M. Rawson, Chem. Commun., 2003, 2774 RSC.
- (a) A. Vegas, A. Pérez-Salazar, A. J. Banister and R. G. Hey, J. Chem. Soc., Dalton Trans., 1980, 1812 RSC; (b) A. W. Cordes, R. C. Haddon, R. T. Oakley, L. F. Schneemeyer, J. V. Waszczak, K. M. Young and N. M. Zimmerman, J. Am. Chem. Soc., 1991, 113, 582 CrossRef CAS; (c) N. Bricklebank, S. Hargreaves and S. E. Spey, Polyhedron, 2000, 19, 1163 CrossRef CAS.
- A. Alberola, R. J. Less, C. M. Pask, J. M. Rawson, F. Palacio, P. Oliete, C. Paulsen, A. Yamaguchi, R. D. Farley and D. M. Murphy, Angew. Chem., Int. Ed., 2003, 42, 4782 CrossRef CAS.
- A. Alberola, C. S. Clarke, D. A. Haynes, S. I. Pascu and J. M. Rawson, Chem. Commun., 2005, 4726 RSC.
- H. U. Hofs, J. W. Bats, R. Gleiter, G. Hartmann, R. Mews, M. Eckert-Maksic, H. Oberhammer and G. M. Sheldrick, Chem. Ber., 1985, 118, 3781 CrossRef.
- S. C. Nyburg and C. H. Faerman, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 274 CrossRef.
- G. R. Desiraju, Crystal Engineering: The Design of Organic Solids, Materials Science Monographs54, Elsevier Press, Amsterdam, 1989 Search PubMed.
- Notably with bulky substituents such as the 2,4,6-(CF3)3C6H2CNSSN the bulky trifluoromethyl group destabilises a coplanar arrangement of phenyl and dithiadiazolylgroups and the SN-I interaction is favoured. See ref. 27.
- G. D. McManus, J. M. Rawson, N. Feeder, J. van Duijn, E. J. L. McInnes, J. Novoa, R. Burriel, F. Palacio and P. Oliete, J. Mater. Chem., 2001, 11, 1992 RSC.
- A. D. Bond, D. A. Haynes and J. M. Rawson, Can. J. Chem., 2002, 80, 1507 CrossRef CAS.
- C. Knapp, E. Lork, T. Borrmann, W. D. Stohrer and R. Mews, Eur. J. Inorg. Chem., 2003, 3211 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, analysis, tables of bond lengths and angles, details of theoretical calculations. CCDC reference numbers 736245–736251. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b911636b |
| ‡ Present address: Department of Chemistry and Polymer Science, Stellenbosch University, P. Bag X1, Matieland, 7602, Republic of South Africa. E-mail: dhaynes@sun.ac.za. |
| § Present address: Department of Chemistry, University of Bath, Bath, UK BA2 7AY. |
| This journal is © The Royal Society of Chemistry 2010 |