Speciation of organotin compounds in environmental samples with semi-permanent coated capillaries by capillary electrophoresis coupled with inductively coupled plasma mass spectrometry†
Jing
Sun
,
Bin
He
*,
Yongguang
Yin
,
Lu
Li
and
Guibin
Jiang
State Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, P.O. Box 2871, Beijing, 100085, China. E-mail: bhe@rcees.ac.cn; Fax: +86-10-62849179; Tel: +86-10-62849334
First published on 28th October 2010
Abstract
A novel method using capillary electrophoresis (CE) coupled with inductively coupled plasma mass spectrometry (ICP-MS) for the separation of four organotin compounds has been developed. Semi-permanent coating of dioctadecyl dimethyl ammonium chloride (DDAC) was applied and investigated to enhance separation efficiency of trimethyltin (TMT) chloride, tributyltin (TBT) chloride, dibutyltin (DBT) dichloride and monobutyltin (MBT) trichloride. The electrolyte composition and ionic strength were optimized. Running buffer containing 0.608 mol L−1 HAc and 7 mmol L−1 NaAc was selected for the separation. The detection limits of the four organotin compounds ranged from 0.037 to 0.112 mg L−1 (as Sn) and the values of the relative standard deviation (RSD) were below 3.1% for migration time, less than 7.8% for peak height and not more than 8.4% for peak area. The developed method was successfully applied for speciation analysis of four organotin compounds in one certified reference material (CRM 477, mussel tissue), and environmental samples including water samples, grape wine and biological samples (Mya arenaria Linnaeus). The determined values (as organotin compounds) of CRM 477 (1.81 ± 0.13 (TBT), 1.36 ± 0.16 (DBT), 1.17 ± 0.18 (MBT)) were in good agreement with the certified values (2.20 ± 0.19 (TBT), 1.54 ± 0.12 (DBT), 1.50 ± 0.28 (MBT)). 0.325 mg L−1 MBT and 1.02 μg g−1 (as Sn) TBT were found in grape wine and Mya arenaria Linnaeus samples.
1. Introduction
Organotin compounds are important organometallic pollutants, which are mainly used as antifouling paints, PVC stabilizers, pesticides and additives in many industrial, chemical and agricultural applications.1 The growing consumption of tin compounds in recent years result in a great amount of organotin compounds widely detected in environmental matrixes such as seafood and wine.2 In addition, high toxicity of the organotin compounds raised increasing concerns over their existence in the environment. The toxicity of organotin compounds is strongly dependent on the type and the number of alkyl groups linked to the tin atom.3,4 Although the inorganic tin is thought to be harmless, TBT (tributyltin), TPhT (triphenyltin), and some other organotin compounds have already been proved to be harmful to non-target aquatic organisms even at a low ng L−1 level.5 Thus, separation and determination of organotin compounds in various matrixes are of great importance in studies on environmental pollution and ecotoxicological concerns.The technique for the separation and quantification of organotin compounds should be selective and sensitive. There are already many methods which have been used to separate and monitor organotin compounds in environmental samples. The most common technique to separate organotin derivatives is gas chromatography (GC) coupled with flame photometric detection (FPD),6,7 atomic absorption spectrometry (AAS),8 pulsed flame photometric detection (PFPD),9 and MS.10 GC related techniques could offer excellent resolution and low detection limit. Whereas the major drawback relates to the poor volatility of the organotin compounds and a pre-column derivatization step is needed for the GC analysis, which is tedious and time consuming.11 Meanwhile, a high performance liquid chromatography (HPLC) technique has also been used to separate these organotin compounds. HPLC coupled with inductively coupled plasma mass spectrometry (ICP-MS) and mass spectrometry have been conducted for environmental organotins determination.12 Although the ICP-MS and ICP-OES detectors are sensitive, these equipments are critical for LC mobile phases so that LC separation parameters such as the concentration of the organic mobile phase and the proportion of the salt in the mobile phase should be restricted.
Over the last two decades, there are increasing concerns using capillary electrophoresis (CE) for the separation of organometallic compounds in various complex matrixes. The CE technique has the advantage of high separation efficiency, rapid analysis, and little volume of sample consumption. CE coupled to inductively coupled plasma-mass spectrometry is an attractive method for separating and quantifying organometallic compounds. ICP-MS can offer not only elemental information of the compounds, but also offers excellent sensitivity, a wide linear dynamic range, high-speed analysis and the ability to perform isotopic analysis,11 compared with other previous publications with UV and fluorescence detectors.13–16 All these advantages bring CE-ICP-MS into the central focus of element speciation, where typically the separation of species is followed by element-selective detection.
The adsorption of organotin compounds on the inner wall of capillaries is a challenge for the CE method, which might result in poor migration time reproducibility, low sample recovery and peak tailing. No obvious improvements were reported even approaches such as high ionic strength and extreme pH were applied in the separation optimization procedures.17 Capillary inner wall coating technique was recognized and widely used as an effective way to inhibit analyte adsorption on capillaries and thus offer lower method determination limits. The double-chained cationic surfactant dioctadecyl dimethyl ammonium chloride (DDAC) and didodecyldimethylammonium bromide (DDAB) have been found to form more stable coatings onto the walls of CE capillaries. These surfactants can be reversibly adsorbed onto the negatively charged wall, reverse the surface charge and affect the capillary electroosmotic flow (EOF) when their concentrations are above the critical micelle concentration. Moreover, coating of semi-permanent DDAC and DDAB are robust and simple by flushing these aqueous solutions through the capillary for only several minutes. No further adsorption materials are needed in the running buffer. The stability of the semi-permanent coatings of DDAC and DDAB has also been confirmed by the previous articles which were excellent for CE separations over a 12-day period without ever regenerating or refreshing the capillary.18 There are many applications reported using DDAB and DDAC as capillary inner wall modifiers.19,20 However, these techniques have not been applied for the CE separation of organotin compounds.
In this paper, we describe a method using DDAC coating-CE coupled with ICP-MS to separate four organotin compounds (TMT, TBT, DBT, MBT). Acetic acid and sodium acetate were introduced into the electrolyte running buffer with certain ionic strength. The effect of concentration and composition of the background electrolyte on separation and detector response were examined and the applications of the method to several environmental samples were also conducted.
2. Experimental
2.1 Materials, chemicals, and samples
Trimethyltin (TMT) chloride, tributyltin (TBT) chloride, dibutyltin (DBT) dichloride were purchased from Dr Ehrenstorfer GmbH (Augsburg, Germany), monobutyltin (MBT) trichloride was obtained from Acros (Geel, Belgium). GR grade acetic acid was from Beijing Chemical Co. (Beijing, China) and sodium acetate (NaAc) was from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Distilled deionized water (DDW) (18 MΩ.cm) was obtained from Milli-Q Advantage A10 system (Millipore, Bedford, MA). Dioctadecyl dimethyl ammonium chloride and nitric acid (69% purity) were obtained from Sigmar-Aldrich (Steinheim, Germany). The standard mussel tissue reference material (CRM 477) was obtained from the Community Bureau of Reference, Commission of the European Communities. All other compounds used in the experiment were at least of analytical-reagent grade.2.2 Preparation of buffer and standard solutions
The stock solutions of organotin compounds were prepared by dissolving TMT, TBT, DBT and MBT in HPLC-grade methanol to obtain a solution with the concentration of 1 mg mL−1 (as Sn). The working standard solutions were freshly prepared by diluting the stock solutions with DDW, and the pH of the solutions was adjusted to 2 by using concentrated HCl to ensure the stability of the solutions. Both stock solutions and working standard solutions were store at 4 °C in the dark.The electrolyte running buffer containing HAc and NaAc in series were prepared by gradually dilution with DDW. The sheath flow was composed of 1% (v/v) nitric acid. DDAC was dissolved in DDW to a concentration of 15 μmol L−1. The electrolyte running buffer, sheath flow liquid, and DDAC solutions were filtrated through 0.45 μm membrane before use.
2.3 Semi-permanent coating and capillary conditioning
Every new capillary was conditioned by flushing with 1 mol L−1 NaOH for 60 min, 0.1mol L−1 NaOH for 60 min, DDW for 10 min, and then, by electrolyte running buffer for 60 min. Then semi-permanent coating was generated by flushing a 0.1 mM DDAC water solution through the capillary for 20 min.After each run of separation, the capillary was flushed with 0.1 mol L−1 NaOH for 2 min, 15 μM DDAC solution was used for 1 min to guarantee the stability of the DDAC coating, and finally with the running buffer solution for 3 min.
2.4 CE operating conditions
All the CE separations of these four organotin compounds were operated on a HP3D CE system (Agilent, Germany) at a voltage of 20 kV. The samples were introduced into the capillary using a hydrodynamic method under a gas presser of 20 mbar for 5 s. The fused-silica capillaries used in the experiment were 60 cm long with an inner diameter of 75 μm, which were obtained from Yongnian Optical Fiber Company (Hebei, China).2.5 ICP-MS system
An Agilent 7500ce ICP-MS instrument (Agilent, USA) was coupled with the HP3D CE system and used for the quantification. A MicroMist nebulizer (AR35-1-FM005E, GE, Australia) was used to fit on the standard spray chamber. A standard torch was also used with a plasma gas flow rate of 15 L min−1, carrier gas flow rate of 0.7 L min−1 and the makeup gas flow rate of 0.4 L min−1. The plasma RF power was 1500 W. Signals from the four isotopes (117Sn, 118Sn, 119Sn, 120Sn) of tin were collected in Full-Quant mode. The integration time was 0.3 s per mass. Complete information of the ICP-MS system is shown in Table S1.†2.6 Interface of CE-ICP-MS system
Interfaces of CE coupled to ICP-MS were considered as a key part in the hyphenation technique.21,22 The interface used in the present study has been published elsewhere.23–25 In brief, a home-made interface of a cross design, introducing a Pt electrode (0.8 mm diameter), CE capillary and a sheath flow, was chosen and used throughout the sample analysis. The CE capillary was threaded through the cross fitting and was inserted into the MicroMist nebulizer. The sheath flow was used to wet the outlet of capillaries and satisfy the demands of the closing of the electrical circuit from CE. The Pt electrode was grounded, and the sheath flow was transported into the nebulizer in the self-aspiration mode with a liquid take-up of approximately 50 μL min−1. To reduce the broadening of the sample peaks, the CE capillary was inserted to the end of the MicroMist nebulizer through a 1/16-inch PEEK tube. The outlet end of the CE capillary was positioned 5 cm from the end tip of the nebulizer.2.7 Extraction procedure and sample preparation
The river water used for the experiment was obtained from Liuyang River (Hunan, China) and the tap water was collected stochastically from the laboratory. The dry red wine sample used in the experiment was obtained from a local market. All these liquid samples were filtrated through 0.45 μm membranes twice and then used for the analysis.The microwave extraction procedure for organotin compounds from mussel tissue samples was used as published elsewhere26 with little modification. Briefly, 0.5 g of lyophilized Mya arenaria Linnaeus sample and CRM 477 (mussel tissue) were weighted and supplemented in the vessels with 10 mL of methanol containing 0.5 mol L−1 acetic acid. Then the vessels were submitted to the microwave-assisted extraction system (Mars5 HP500, CEM Corporation, Matthews) at 60 W for 3 min. After cooling, the samples were transferred into glass centrifuge tubes and centrifuged at 3000 rpm/min for 5 min. The supernatant liquid were collected and then evaporated almost to dryness under a gentle stream of nitrogen. Subsequently, the residue was diluted to 4.0 mL with electrolyte buffer (HAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaAc = 608 mM:7 mM). The emulsions were broken up by subjecting the tubes to be centrifuged at 3000 rpm/min for 5 min again. The supernatant liquid was finally gathered, filtered through a 0.22 μm membrane and analyzed by CE-ICP-MS.
:NaAc = 608 mM:7 mM). The emulsions were broken up by subjecting the tubes to be centrifuged at 3000 rpm/min for 5 min again. The supernatant liquid was finally gathered, filtered through a 0.22 μm membrane and analyzed by CE-ICP-MS.
3. Results and discussion
3.1 Optimization of CE conditions
For the optimization, several operation parameters such as concentration of electrolyte and composition of electrophoretic buffer were tested in detail. Initial experiments were performed by using acidic aqueous solutions. The addition of HAc has been proved to be necessary to increase the detector response of organotin compounds. Several other electrophoretic buffers have also been examined, including tartaric acid-methanol buffer, Na2HPO4-boric acid buffer and boric acid-Tris buffer (Fig. S1).† However, it was found that only acetic buffer could offer a good electropherogram, along with the best resolution. Thus, acetic buffer was selected for the following experiments.As a cationic surfactant, DDAC coating can form a bilayer or vesicle structure at the inner walls of capillaries, changing the ionized silanol groups on the capillary inner wall into a positively charged surface, which can prevent the organotin compounds from adsorption onto the ionized silanol groups on the inner wall of the uncoated capillary. Semi-permanent coating of DDAC is simple and stable. The stability of DDAC coatings owes to the formation of the bilayer or vesicle structure at the inner wall of capillaries, which were flatter and more homogeneous, resulting in not only better stability but also greater surface coverage to prevent the adsorption of the organotin compounds. The separation efficiency of coating and uncoating capillaries could be compared in Fig. 1. It was found that with semi-permanent coating of DDAC, the migration times of organotins have been affected obviously. The four organotin compounds (TMT, TBT, DBT, and MBT) could achieve baseline separation in 12 min and the peak areas were increased when coated capillaries were used. Also shown in Table 1, the migration times of the analytes with coated capillaries were stable with the relative standard deviation ranging from 2.1% to 3.1% (intra-day) and 2.4–3.4% (inter-day).
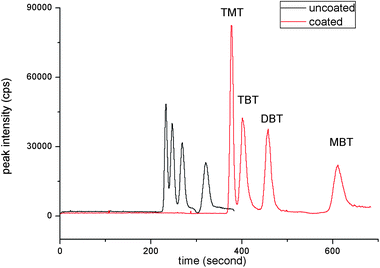 | ||
| Fig. 1 Comparison of electropherogram of semi-permanent coated and uncoated capillaries. Electropherograms: (1) separation of organotin compounds with DDAC coating; (2) separation of organotin compounds without DDAC coating. Electrolyte running buffers: 0.608 mol L−1 HAc and 7 mmol L−1 NaAc. Other conditions: 20 kV, 25 °C, 20 mbar × 5 s injection. Concentration of organotin compounds: 1 μg mL−1 as Sn. | ||
| Compound | RSD (%) | Linear range/mg L−1 | R 2 | LOD/mg L−1 | |||
|---|---|---|---|---|---|---|---|
| Height (n = 5) | Area (n = 5) | Time | |||||
| Intra-day (n = 5) | Inter-day (n = 12)a | ||||||
| a Triplicate standard solutions were injected over 4 days (n = 12). | |||||||
| TMT | 5.0 | 7.1 | 2.1 | 2.4 | 0.037–1.000 | 0.9950 | 0.037 |
| TBT | 2.8 | 6.3 | 2.3 | 2.7 | 0.062–1.000 | 0.9907 | 0.062 |
| DBT | 5.7 | 8.4 | 2.3 | 2.7 | 0.084–1.000 | 0.9886 | 0.084 |
| MBT | 7.8 | 5.7 | 3.1 | 3.4 | 0.112–1.000 | 0.9931 | 0.112 |
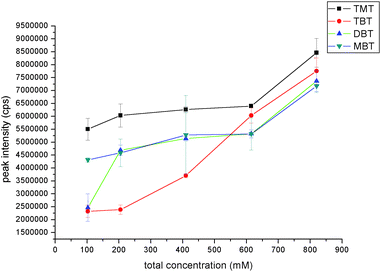 | ||
| Fig. 2 Effect of buffer concentration in mobile phase on the organotin compounds peak intensity. The data was obtained by determining 1 mg L−1 mixed solution of TMT, TBT, DBT and MBT with CE-ICP-MS under optimized condition except buffer concentration. | ||
:NaAc from 84:1 to 14:1, the migration times of the four organotin compounds were getting closer. However, when the electrolyte buffer was HAc only, migration times of TBT and DBT are closer, which could not achieve baseline separation. So the proportion of the electrolyte running buffer was chosen as 84:1 and the concentrations of NaAc and HAc were 7 mM and 608 mM, respectively.
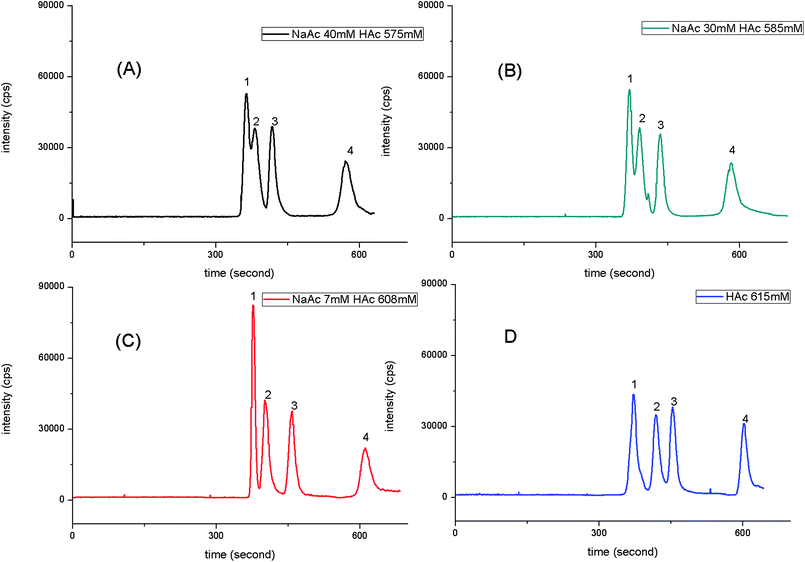 | ||
| Fig. 3 Effect of electrolyte buffer composition in mobile phase on the organotin compounds. The data was obtained by determining 1 mg L−1 mixed solution of TMT, TBT, DBT and MBT with CE-ICP-MS under optimized condition except electrolyte buffer composition. Electrolyte buffer composition: (A) NaAc: 40 mM and HAc: 575 mM; (B) NaAc: 30 mM and HAc: 585 mM; (C) NaAc: 7 mM and HAc: 608 mM; (D) HAc: 615 mM. Legend: 1, TMT; 2, TBT; 3, DBT; and 4, MBT. | ||
The effects of HNO3 concentration on the intensity of ICP-MS signals of organotins were investigated in the range of 0.1%–3% (v/v). As shown in Fig. S3,† it was obvious that the peak areas increased when the concentration of HNO3 was raised from 0.1% to 1%. Whereas the intensity decreased when the HNO3 concentration was further increased from 1% to 3%. This phenomenon could be explained by the viscosity of sheath flow solution and nebulization efficiency of the nebulizer. When HNO3 concentration rose to 1%, the increased peak intensity might be caused by the increasing viscosity of sheath flow, which made the HNO3 sheath flow slower, resulting in improvement of transfer efficiency. However, when the concentration of HNO3 increased from 1% to 3%, the viscosity still increased but nebulization efficiency was depressed significantly. Thus, the corresponding peak intensity became lower. As a result, 1% (v/v) HNO3 was chosen as the optimized sheath flow solution and also used to remove equipment memory effect.
3.2 Separation of organotin compounds under the optimized condition
With the optimized analysis parameters, TMT, TBT, DBT, and MBT were all baseline separated. A series of experiments were performed and linearity, limit of detection (LOD), precision and accuracy were validated to value the quantification performance of the method.Investigation of detection limits (LOD) of the method was based on the data used for linearity and precision. The LODs of method have been calculated based on 3 times of S/N ratio. From Table 1, it was shown that the LODs of the four organotin compounds ranged from 0.037 to 0.112 mg L−1. The developed method has lower LODs than the published CE-hyphenated techniques (Table S2).† As shown in Fig. 1, DDAC coating inhibited the adsorption of organotin compounds on the inner wall of capillaries, thus, increased the sensitivity of the method and also achieved baseline separation.
| Concentration found/mg kg−1 | Certified value/mg kg−1 | |
|---|---|---|
| TMT: Sn(CH3)3+ | — | — |
| TBT: Sn(C4H9)3+ | 1.81 ± 0.13 | 2.20 ± 0.19 |
| DBT: Sn(C4H9)22+ | 1.36 ± 0.16 | 1.54 ± 0.12 |
| MBT: Sn(C4H9)3+ | 1.17 ± 0.18 | 1.50 ± 0.28 |
3.3 Sample analysis
In order to evaluate the application of the semi-permanent coating CE-ICP-MS method, further experiments were conducted to determinate organotin compounds in real environmental samples. Water, grape wine and sea food (Mya arenaria Linnaeus) were chosen as different matrix samples.Three parallel Mya arenaria Linnaeus samples were tested, each was analyzed in triplicate. Fig. 4 is a typical chromatogram for organotins in Mya arenaria Linnaeus samples. Only TBT could be detected in the samples. The concentration level (ug g−1 as Sn) of the tin compounds contained in samples was 1.02 μg g−1. The RSD of the three parallel was 7.6%. It is the first time that organotin compounds in Mya arenaria Linnaeus samples were detected using the semi-permanent coating of the CE-ICP-MS method. Standard addition experiments also have been performed (Table S4).† Two levels of concentration (0.1, 0.5 mg L−1) of the standard mixture were spiked. The recoveries of the four organotin compounds ranged from 68.6% to 80.0% (0.5 mg L−1) and 61.7% to 81.7% (0.1 mg L−1). Higher spike recoveries could be obtained by adding agents such as tropolone and diethyldithiocarbamate, which would increase extraction efficiency in the sample preparation procedures. But it might influence the CE separation efficiency and stability. Thus, improvement of preparation section is still needed for the application of the CE-ICP-MS method to other important environmental matrixes.
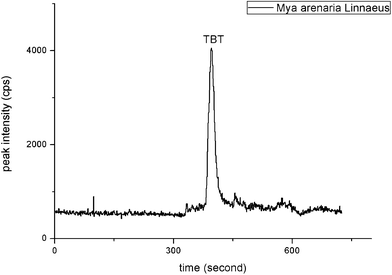 | ||
| Fig. 4 Electropherogram of Mya arenaria Linnaeus samples. The data was obtained by the developed CE-ICP-MS method under optimized condition. | ||
4 Conclusions
A DDAC coating-CE-ICP-MS method was developed for the separation of four organotin compounds, and the method was sensitive and reproducible. DDAC semi-permanent coating procedure was simple and it could effectively enhance separation efficiency and improve the LODs of organotin compounds by reducing the adsorption on the inner wall of capillaries. Key separation parameters such as ionic strength and electrolyte composition were optimized and low LODs (0.037 mg L−1 (TMT)–0.112 mg L−1 (MBT)) was obtained. The optimized method was applied for the analysis of the four organotin contaminants in the standard mussel tissue reference material (CRM 477) and several environment samples. Good agreement was achieved by determined values and certified values and 0.325 mg L−1 MBT in red wine and 1.02 μg g−1 (as Sn) TBT in Mya arenaria Linnaeus were firstly detected using the DDAC coating CE-ICP-MS system.Acknowledgements
This work was jointly supported by the National Basic Research Program of China (2009CB421605), and National Natural Science Foundation of P. R. China (20677069, 20921063 and 20937002).References
- C. J. Evans and S. Karpel, Organotin compounds in modern technology, Elsevier; Distributors for the U.S. and Canada, Elsevier Science Pub. Co., Amsterdam; New York, New York, N.Y, 1985 Search PubMed.
- G. B. Jiang, J. Y. Liu and Q. F. Zhou, Environ. Sci. Technol., 2004, 38, 4349–4352 CrossRef CAS.
- P. J. Craig, Organometallic compounds in the environment, J. Wiley, West Sussex, England; Hoboken, NJ, 2003 Search PubMed.
- F. E. Brinckman, J. M. Bellama and American Chemical Society, Division of Inorganic Chemistry, Organometals and organometalloids: occurrence and fate in the environment: based on symposium sponsored by the Division of Inorganic Chemistry at the 175th meeting of the American Chemical Society, Anaheim, California, March 13–17, 1978, American Chemical Society, Washington, 1978 Search PubMed.
- K. Fent, Crit. Rev. Toxicol., 1996, 26, 1–117 CAS.
- M. D. Muller, Anal. Chem., 1987, 59, 617–623 CrossRef.
- J. M. Liu, G. B. Jiang, J. Y. Liu, Q. F. Zhou and Z. W. Yao, Bull. Environ. Contam. Toxicol., 2003, 70, 219–225 CrossRef CAS.
- R. Ritsema, T. de Smaele, L. Moens, A. S. de Jong and O. F. X. Donard, Environ. Pollut., 1998, 99, 271–277 CrossRef CAS.
- J. Heroult, T. Zuliani, M. Bueno, L. Denaix and G. Lespes, Talanta, 2008, 75, 486–493 CrossRef CAS.
- M. G. Ikonomou, M. P. Fernandez, T. He and D. Cullon, J. Chromatogr., A, 2002, 975, 319–333 CrossRef CAS.
- G. Alvarez-Llamas, M. D. de laCampa and A. Sanz-Medel, TrAC, Trends Anal. Chem., 2005, 24, 28–36 CrossRef CAS.
- G. S. Zhai, J. F. Liu, L. Li, L. Cui, B. He, Q. F. Zhou and G. B. Jiang, Talanta, 2009, 77, 1273–1278 CrossRef CAS.
- K. S. Whang and C. W. Whang, Electrophoresis, 1997, 18, 241–246 CAS.
- Y. T. Lee and C. W. Whang, J. Chromatogr., A, 1996, 746, 269–275 CrossRef CAS.
- L. Guo, F. M. Matysik and P. Glaser, Anal. Bioanal. Chem., 2004, 380, 669–676 CrossRef CAS.
- J. L. F. Feng Han and Phyllis R. Brown, J. Chromatogr., B: Biomed. Sci. Appl., 1995, 669, 103–112 CrossRef CAS.
- E. Pobozy, B. Glod, J. Kaniewska and M. Trojanowicz, J. Chromatogr., A, 1995, 718, 329–338 CrossRef CAS.
- W. L. Wei, B. Y. Guo and J. M. Lin, J. Chromatogr., A, 2009, 1216, 1484–1489 CrossRef CAS.
- M. M. Yassine and C. A. Lucy, Anal. Chem., 2005, 77, 620–625 CrossRef CAS.
- J. E. Melanson, N. E. Baryla and C. A. Lucy, Anal. Chem., 2000, 72, 4110–4114 CrossRef CAS.
- B. Michalke, J. Anal. At. Spectrom., 1999, 14, 1297–1302 RSC.
- B. Michalke, Electrophoresis, 2005, 26, 1584–1597 CrossRef CAS.
- B. H. Li and X. P. Yan, Electrophoresis, 2007, 28, 1393–1398 CrossRef CAS.
- A. Standler, G. Koellensperger, W. Buchberger, G. Stingeder and S. Hann, Electrophoresis, 2007, 28, 3492–3499 CrossRef CAS.
- J. A. Day, S. S. Kannamkumarath, E. G. Yanes, M. Montes-Bayon and J. A. Caruso, J. Anal. At. Spectrom., 2002, 17, 27–31 RSC.
- C. Pellegrino, P. Massanisso and R. Morabito, TrAC, Trends Anal. Chem., 2000, 19, 97–106 CrossRef CAS.
- L. P. Yu, Anal. Lett., 2007, 40, 1879–1892 CrossRef CAS.
- G. B. Jiang, J. Y. Liu and K. W. Yang, Anal. Chim. Acta, 2000, 421, 67–74 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1–S4 and Fig. S1–S4. See DOI: 10.1039/c0ay00558d |
| This journal is © The Royal Society of Chemistry 2010 |