Validation of a passive flux sampler for on-site measurement of formaldehyde emission rates from building and furnishing materials
Alodie
Blondel
ab and
Hervé
Plaisance
*ab
aUniversité Lille Nord de France, F-59000, Lille, France
bEcole des Mines de Douai, Département Chimie et Environnement, 941 rue Charles Bourseul, BP 10838, 59508, Douai cedex, France. E-mail: herve.plaisance@mines-douai.fr
First published on 14th October 2010
Abstract
A diffusive sampling device was developed for screening indoor sources of formaldehyde and determining the emission rates of these sources on-site. It consists of a glass Petri dish containing a quartz filter coated with 2,4-dinitrophenylhydrazine (DNPH) which is placed on the tested material for sampling. Formaldehyde emitted from the material diffuses through the air column inside the sampler and is collected onto the DNPH filter. The formaldehyde emission rate is determined from the mass collected by the sampler. The dependence of the sampled formaldehyde amount on the diffusion length inside the sampler was examined and led us to select a length of 20 mm for which the diffusion in the air layer controls the mass transfer from the material to the sampling surface (filter). A linear relationship with a high correlation (r2 = 0.97) was obtained between the mass collected by the passive flux sampler and the emission rates measured by a standard chamber method, analysing a large series of indoor materials with the two methods in parallel. Neither the sampling time (ranging from 4 to 8 h) nor the temperature (from 23 to 35 °C) was observed to influence the sampler performance. Furthermore, the detection limit attained 1.2 μg m−2 h−1 for a 6h-sampling time and a satisfying precision of replicate measurements (relative standard deviation of 7.8% on average) is adequate for monitoring the emission rates of primary sources contributing to formaldehyde indoor concentrations.
1. Introduction
Formaldehyde is the most abundant aldehyde in indoor air. Formaldehyde is released by various building materials, including wood-based products assembled with urea-formaldehyde or phenol formaldehyde resins,1 and some paints2 and varnishes.3 Formaldehyde is also present in wood smoke4 and tobacco smoke.5 It was shown that formaldehyde (FA) exposure has an irritant effect on the eyes, nose and upper airways. Since 2004, it has been classified in group 1 (human carcinogen) by the International Agency for Research on Cancer based on sufficient evidence from epidemiological data.6 In France, a national survey of measurements carried out by the Observatory on Indoor Air Quality (OQAI) in 554 dwellings designed to be representative of the 24 million French dwellings. The median indoor formaldehyde concentration was found to be 19.6 μg m−3 whereas the outdoor concentration was 1.9 μg m−3 indicating the predominant contribution of indoor sources.7 In consequence, human exposure to formaldehyde is principally associated with indoor sources. An indoor air quality guideline value of 10 μg m−3 for long term exposure was recently proposed for formaldehyde by the French Agency for Environmental and Occupational Health Safety.8 A major knowledge gap identified in the set up of this guideline value was the lack of means to identify the factors associated with the presence of high levels of formaldehyde in French homes and particularly the indoor sources. However, many field studies9,10 pointed out the fact that the emissions of building and furnishing materials were mainly to blame for high levels of formaldehyde found indoors.Emission chambers, and field and laboratory emission cells (FLEC) are usually used to measure the volatile organic compounds (VOCs) emitted from building materials. The emission tests with these two standard methods involve an active sampling mode to transfer the VOCs emitted from the solid material to a sampling tube.11,12 The chamber methods cannot be used on site and FLEC13 is unsuitable for multipoint field sampling because it needs expensive equipment and it is not easy to implement.
Passive sampling represents an interesting alternative to the two standard methods mentioned above for field investigations. Sampling is made in a static mode by diffusion of chemicals inside a closed air volume from the material surface to a trap medium. Recently, various passive devices like the passive flux sampler (PFS),14 the passive emission colorimetric sensor (PECS)15 and the FLEC coupled with solid phase microextraction (FLEC-SPME)16 were developed to measure the emission rates in the field. Although the feasibility of the emission measurement was demonstrated, these passive methods were developed by tests on a limited number of materials (from 1 to 3). The objectives of this study are: to improve the existing passive device, PFS, initially proposed by Shinohara et al., (2007) for on-site measuring of the formaldehyde emission rate and to provide some missing results for the validation of this passive sampler type. A first step of this work was to define the most appropriate diffusion length of the sampler (distance between the tested material and an absorbent media) from the measurements achieved on four material types. Then, a large series of materials were tested to find a linear relationship between the collected mass by the passive flux sampler and the emission rate measured by the standard chamber method. The classical performance characteristics of the passive sampling method (blank value, detection limit, storage conditions and effects of sampling time and temperature) were estimated in specific laboratory tests. The reproducibility of the measurements was investigated in real conditions by sampling twice some furniture and building materials present in rooms.
2. Materials and methods
2.1. Passive flux sampler and analytical method
The sampler consists of a glass Petri dish (inner diameter: 35.4 mm, depth: from 5 to 60 mm) at the bottom of which a quartz fibre filter (Whatman) coated with 2,4-dinitrophenylhydrazine (DNPH) (Fig. 1) is placed. | ||
| Fig. 1 Photograph of passive flux sampler. | ||
Before sampling, 500 μL of an acidified aqueous solution of 2,4-DNPH17 is spread all over the filter area using a pipette. Then, the sampler is placed with the open face on the material to be sampled. Formaldehyde emitted from the material diffuses to the inside of the sampler and is trapped on the DNPH filter. DNPH is light sensitive so the glass dish is tinted to prevent the loss of DNPH from the filter during exposure.
After sampling, the sampler is sealed with a colour cap and stored in the dark at −20 °C. For analysis, the filter is removed from the Petri dish, and placed in a tinted glass tube. The DNPH-formaldehyde derivates are extracted from the filter in 5 mL of acetonitrile (HPLC grade, Waters) by vortex shaking for 1 min. Then, the extracted DNPH-formaldehyde derivates are analysed using a high performance liquid chromatography system (HPLC, Waters 2695, Waters) with a photodiode array detector at a wavelength of 365 nm. 20 μL of extraction solution were injected onto a Restek ULTRA C18 column (Restek, France, length 25 cm, inner diameter 4.6 mm, particle size 5 μm). The mobile phase is initially a 30% acetonitrile, 20% tetrahydrofurane and 50% water mixture which progressively evolve for 15 min to achieve a 65% acetonitrile, 5% tetrahydrofuran and 30% water mixture maintained for a further 12 minutes. The flow rate is maintained at a constant 1.5 mL min−1. The gas-phase concentration of FA was quantified from an external calibration curve made before sampler analysis. The solid standard DNPH derivative of FA was purchased from Chem-Service. Known concentrations of hydrazones were then prepared in acetonitrile and injected into the HPLC in order to set up the calibration curves (HPLC peak areas versus formaldehyde concentrations). The calibration curve was linear for the hydrazone concentration range between 0.7 and 2000 μg L−1 and the correlation coefficients were larger than 0.99. Before each set of analyses, a single point of this calibration curve with a known concentration was injected to check the accuracy of the HPLC response coefficient (deviation ≤ 5%).
2.2. Emission test chamber method
To establish a relationship between the collected mass by the passive flux sampler and the emission rate, a series of materials was tested using the passive sampling and the emission test chamber standard method.11 The experimental device set up for the tests in the chamber is presented in Fig. 2. This emission test chamber has a capacity of 36 L. The air opening in the emission test chamber is produced by a compressor and is dried and chemically filtered in an air purifier (AZ 2020, manufactured by Claind). A first air flow is produced directly by the air purifier and a second air flow comes from a humidifier system consisting of a bubbler filled up with demineralised water and flushed with purified air. These two air flows are regulated by mass flow controllers. They are mixed at the chamber inlet, generating various test atmospheres. Ambient conditions in the emission test chamber are continuously monitored and recorded by means of temperature, relative humidity and wind velocity multifunction probes (Datalogger Testo term 400 and temperature, humidity, wind velocity sensor 0635.1540). The emission test chamber is placed in a thermostatic enclosure maintained at a constant temperature. Samples are introduced or removed thanks to the opening of a removable cover. Experiments are conducted according to the international standard ISO 16000-9 (2006). The temperature was 23 °C, the relative humidity was 50% and the wind velocity was maintained at less than 0.3 m s−1. The ventilation rate was chosen to be close to that found in indoor environments, 0.5 h−1. The area of the material was 0.02 m2 corresponding to a product loading of 0.57 m2 m−3. The material sample was placed in the chamber and incubated for 6 h to achieve a steady state for formaldehyde emissions from the material. Then, the concentration of formaldehyde in the chamber was determined by active sampling using DNPH-Silica Sep-Pak cartridges (purchased from Waters, Guyancourt, France). The sampling rate was 200 mL min−1 and the sampling duration was 1 h. The used cartridges were sealed and stored at −20 °C until analysis. These cartridges were eluted with 3 mL of acetonitrile and analysed by HPLC according to the same method for analysis of passive flux samplers. | ||
| Fig. 2 Emission test chamber system. | ||
The formaldehyde emission rate was calculated by means of the following equation:
 | (Equation 1) |
2.3. Material samples
A list of 13 indoor material samples was selected for the emission tests carried out with the passive flux sampler and standard chamber method. It consisted of four medium density fibreboards (medium), two OSB (Oriented Standard Board), three chipboards, two sealing plasters, a finishing plaster and a roofing tale. To avoid bias due to the variation of emission over time, the tests in the chamber and those done with the passive flux sampler were performed successively within 24 h on the same material sample.3. Theoretical basis for determining the emission rate by the passive flux sampler
When a compound is released from a material into indoor air, two diffusion processes occur: (i) diffusion inside the material and (ii) diffusion in the gas-phase boundary layer between the material area and the indoor bulk air.In the steady state, the two mass transfers inside the material and in the gas-phase boundary layer are equal. In consequence, the emission rate from the source material is limited by the process which has the slowest transfer rate (Fig. 3).
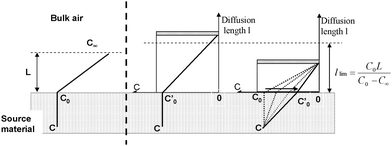 | ||
| Fig. 3 Schematic representation of formaldehyde transfer in the sampler. C∞ is the concentration in bulk indoor air, C0 is the concentration on the material surface, C is the concentration in the material, L is the thickness of the gas-phase boundary layer and l is the diffusion length of the passive flux sampler. | ||
The emission flux from the source material surface to the indoor bulk air can be obtained by the following equation (first Fick's law):
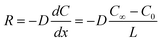 | (Equation 2) |
When a passive sampler is placed on the source material, the compound molecules diffuse inside the sampler from the material surface to the filter coated with DNPH. The emission flux inside the sampler can be expressed as follows:
 | (Equation 3) |
Then eqn (3) can be simplified:
 | (Equation 4) |
If the diffusion distance inside the sampler exceeds a limit length,  (deduced from the equality in concentration gradients), the emission flux from the source material remains unchanged (with or without sampler). In this case, the rate-determining process is the diffusion in the air layer inside the sampler and the concentration on the material surface is not modified by the presence of sampler, C′0 = C0.
(deduced from the equality in concentration gradients), the emission flux from the source material remains unchanged (with or without sampler). In this case, the rate-determining process is the diffusion in the air layer inside the sampler and the concentration on the material surface is not modified by the presence of sampler, C′0 = C0.
So,
 | (Equation 5) |
 in eqn (2), the emission rate from the source material can be written:
in eqn (2), the emission rate from the source material can be written: | (Equation 6) |
 | (Equation 7) |
In these conditions, a linear relationship should exist between the mass collected on the filter (m) and the inverse of the thickness of the diffusion zone inside the sampler (l). Similarly, there is also a linear relationship between this sampled mass (m) and the emission rate (R) as shown in eqn (7).
If the diffusion distance inside the sampler is below the limit length (llim) previously mentioned, the diffusion inside the material becomes the rate-determining process (having the slowest transfer rate). In consequence, the concentration at the material surface in the presence of sampler (C′0) tends to decrease. The emission flux decreases too and no linear relation with the sampled mass can be obtained.
4. Results and discussion
4.1. Recovery test
To validate the extraction and analysis steps, the recovery rate was determined by analysing beforehand six filters coated with DNPH which were each loaded with an aliquot of 100 μL of a solution of formaldehyde (2000 μg/aliquot) spiked using a micro pipette and allowed to diffuse for 30 min. The recovery was calculated by computing the ratio of the measured mass and the spiked mass on the filter. The mean recovery rate is 99.4 ± 1.1% and is not significantly different to 100%.4.2. Dependence of the collected mass on the inverse of diffusion length
When the diffusion in the air layer controls the mass transfer, the sampled amount should be inversely proportional to the diffusion length. To select a sampler which complies with this condition, many samplers with eight different diffusion lengths (5, 10, 15, 20, 30, 40, 50 and 60 mm) were tested to collect formaldehyde emitted from four materials (two medium density fibreboards, a chipboard and an Oriented Standard Board (OSB)). The sampling time was 6 h; the temperature was maintained at 23 °C during tests and three passive flux sampler measurements were done at each diffusion length. As shown in Fig. 4, the sampled formaldehyde amount was found to be inversely proportional to the diffusion length when it was greater than a limit length within 5 and 15 mm according to the material. | ||
| Fig. 4 Collected mass of formaldehyde on exposition time versus inverse of the diffusion length. ER: emission rate. | ||
According to the formula,  , this limit length is higher than or equal to the gas-phase boundary layer L and tends to be equal to L when the difference between C∞ and C0 is high. But in theory, these two lengths, L and llim, are not equal which is inconsistent with the theoretical considerations given by Shinohara et al. (2007) for the PFS. The range of limit lengths (from 5 to 15 mm) found in these tests on materials is close to the thickness of the gas-phase boundary layer in indoor environments that have been reported between 10 and 30 mm.18 This showed that for a length above 15 mm, the rate limiting step (the slowest process) is the diffusion in the air layer inside the sampler and in consequence the emission rate from the material should be linearly dependent on the following mass collected by the sampler.
, this limit length is higher than or equal to the gas-phase boundary layer L and tends to be equal to L when the difference between C∞ and C0 is high. But in theory, these two lengths, L and llim, are not equal which is inconsistent with the theoretical considerations given by Shinohara et al. (2007) for the PFS. The range of limit lengths (from 5 to 15 mm) found in these tests on materials is close to the thickness of the gas-phase boundary layer in indoor environments that have been reported between 10 and 30 mm.18 This showed that for a length above 15 mm, the rate limiting step (the slowest process) is the diffusion in the air layer inside the sampler and in consequence the emission rate from the material should be linearly dependent on the following mass collected by the sampler.
A diffusion length of 20 mm was retained because it belongs to the linear domains of four tested materials and provides the highest sensitivity.
Those results are inconsistent with those found by Shinohara et al. (2007) where the diffusion length chosen for PFS sampler was 10 mm. Tests detailed by Shinohara et al. (2007) were carried out on one single material, a plywood board. According to Fig. 4, a diffusion length of 10 mm does not belong to the linearity domain for all the materials. Indeed for the medium with the highest emission rate (251.4 μg m−2 h−1) the formaldehyde amount was not inversely proportional to the diffusion length when the length was 10 mm.
4.3. Calibration of the passive flux sampler
To assess the relationship between the collected mass and the emission rate, the same series of 13 indoor materials were sampled at once using the sampler with 20 mm diffusion length and the emission test chamber method. The selected materials were four different kinds of medium density fibreboard, two types of chipboard, two types of OSB, one finish plaster, one sealing plaster and one ceiling tale. For the measurements with the passive flux sampler, the sampling time was 6 h and the temperature was maintained at 23 °C (the same as for the tests in the emission chamber). A total of six measurements by passive flux samplers and two by the chamber method were made on each material.As shown in Fig. 5, a linear relationship was found between the amount collected by the passive flux sampler and the emission rates measured by the chamber method in a large range of emissions spreading from 21 to 413 μg m−2 h−1 with a high correlation (R2 > 0.97). A large variety of indoor materials were analysed (13) to set up this calibration of the passive flux sampler. In the previous works of Shinohara on the PFS, the PFS/chamber method relationship was investigated on only one type of material, a plywood board, and at four emission levels. There is a linear relation between these two methods because the sampler height (20 mm) belonged to the domain in which the emission flux of all the materials was controlled by the diffusion in the boundary layer.
 | ||
| Fig. 5 Calibration of passive flux sampler response: mass collected by the passive flux sampler versus the FA emission rates measured by the test in the emission chamber. The sampling height is 20 mm and the sampling time is 6 h. 13 materials were sampled. | ||
The equation of this linear regression will be used to convert the masses collected by the passive flux sampler into emission rates.
Note that the testing material having the highest formaldehyde emission (413 μg m−2 h−1) was a finishing plaster, which is a product widely used in construction and building restoration.
4.4. Blank value and detection limit
The blank value was determined by analysing 19 non-exposed passive flux samplers. The average mass of compound found in the blanks and its standard deviation was found to be 34 ± 12 ng. This blank value was systematically subtracted from masses measured in all exposed samplers. The detection limit was calculated as the mass corresponding to three times the standard deviation of 19 blank values. Hence, using the calibration equation shown in Fig. 5, the detection limit was expressed in the emission rate and estimated to be 1.2 μg m−2 h−1 for a 6-hour sampling time. The detection limit for a 6-hour sampling time is low enough to investigate the formaldehyde emission rates of building and furnishing materials which were usually measured between 2 and 250 μg m−2 h−1 indoors.19,204.5. Storage stability before and after sampling
The storage stability of samplers was evaluated for up to 21 days at −20 °C in a freezer before sampling and for up to 14 days after sampling (Table 1).| Time (days) | FA mass in non exposed samplers/ng n = 6 | FA mass collected by samplers on a material having an emission rate of 110 μg m−2 h−1/ng n = 6 |
|---|---|---|
| D | 28 ± 5 | 3312 ± 115 |
| D + 7 days | 21 ± 3 | 3238 ± 129 |
| D + 14 days | 26 ± 9 | 2893 ± 410 |
| D + 21 days | 33 ± 12 | |
| ANOVA test | p = 0.111 | p = 0.079 |
For the storage stability before sampling, four sets of six samplers impregnated with 2,4-DNPH solution were prepared. A first set was analysed immediately after preparation. The three other sets of six samples were stored at −20 °C and analysed 7, 14 and 21 days after.
For the storage stability after sampling, three sets of six passive flux samplers were exposed on the same material (medium) having an emission rate of 110 μg m−2 h−1 for 6 h. A first set of six exposed samplers was analysed the day after exposure, the second and the third ones, after storage at −20 °C for 8 and 15 days respectively. Analysis of variance (ANOVA) was used to determine a significant difference between the sets of samplers at a 95% confidence level (Table 1). Analysis of variance shows that there is no significant influence of storage up to 21 days for non-exposed samplers and up to 14 days for exposed samplers.
4.6. Precision of replicate measurements
In order to evaluate the precision, 9 sets of six replicate sampler measurements were carried out on the materials having different formaldehyde emission levels (from 21 and 413 μg m−2 h−1). Precision of the passive sampling method was expressed as the relative standard deviation (RSD) of the replicates and is reported in Table 2. A satisfying repeatability (RSD of 7.8% on average) was obtained for the passive flux sampler measurements of formaldehyde. As shown in Fig. 6, RSD decreases with the increase of emission rate. The highest RSD values correspond to the measurements made on the two OSB having the lowest emission rates. These two materials have a coarse structure and a polished surface suggesting heterogeneous emissions of formaldehyde at its surface. | ||
| Fig. 6 RSD versus emission rate. | ||
4.7. Influence of exposure time
Three sets of 6 replicate passive flux samplers were placed on the same material (medium) for different sampling times (4, 6 and 8 h). The results are given in Table 3. Analysis of variance shows that the sampling between 4 and 8 h has no significant effect on the emission rate measurements.| 4 h (n = 6) | 6 h (n = 6) | 8 h (n = 6) | ANOVA Test |
|---|---|---|---|
| 161.9 ± 15.5 | 166.3 ± 12.4 | 180.0 ± 6.8 | p = 0.076 |
4.8. Influence of temperature
To test if the measurement by the passive flux sampler is affected by temperature, the formaldehyde emission of the same material (medium) is analysed in parallel to two temperatures (23 and 35 °C) with a set of 6 passive flux samplers and using the test chamber method (n = 2). Results are presented in Table 4.| Passive flux sampler | Reference method | ANOVA test | |
|---|---|---|---|
| 23 °C | 243.9 ± 6.5 | 251.4 | p = 0.309 |
| 35 °C | 829.0 ± 104.5 | 739.2 | p = 0.290 |
Analysis of variance showed that there is no significant difference between the emission rates obtained by the passive flux sampler and the standard chamber method. Temperatures from 23 to 35 °C did not have an effect on the sampler performance. The passive flux sampler can be used in a large range of temperatures from 23 to 35 °C, typically encountered indoors. In the previous paper of Shinohara et al. (2007) on the development of the passive flux sampler PFS, no result was reported concerning the influence of temperature on the sampler response.
4.9. Duplication of on site passive flux sampler measurements
To report on the reproducibility of passive flux sampler measurements in real-life environmental conditions, some furniture materials (plywood, chipboard) and building surfaces (walls, floor) have been sampled twice in the studied rooms. As reported in Fig. 7, a good agreement was found between these duplicate measurements. The sensitivity of the passive flux sampler for a 6 h exposure time appears to be suitable for the measurement of emission rates in indoor environments. | ||
| Fig. 7 Comparison of duplicate passive flux sampler measurements carried out on the same materials. | ||
Conclusions
A passive flux sampler has been tested by analysing a large range of materials in various exposure conditions in order to validate it for the measurement of on-site emission rates of indoor materials. The evaluation program allowed the optimization of the diffusion length of the sampler and estimation of the performance parameters of this passive method (recovery rate, precision, detection limit, storage condition of sampler before and after exposure and calibration of the sampler response). With a detection limit of 1.2 μg m−2 h−1 for 6 h sampling time, a linearity of the sampler response in a large range of emission rates (from 1 to 413 μg m−2 h−1) and a precision of replicate measurements around 7.8% (expressed in RSD), the passive method is suitable for monitoring the emission rates of primary sources contributing to indoor concentrations of formaldehyde. A large series of 13 common indoor materials was analysed to determine the calibration curve largely completing the calibration process of this type of sampler. Moreover, the test on the influence of temperature has revealed no effect in the range of temperature typically encountered indoors. Some of the first measurements made on the furniture and building materials in rooms showed that this passive device provides reproducible results. Further studies using this passive flux sampler will be performed in various types of indoor environments and will be the subject of another publication.Acknowledgements
The authors thank G.I.P CERESTE for financial support for this study.References
- T. Kelly, D. Smith and J. Satola, Environ. Sci. Technol., 1999, 33, 81–88 CrossRef CAS.
- J. C. S. Chang, Z. Guo, R. Fortmann and H.-C. Lao, Indoor Air, 2002, 12, 10–16 CrossRef CAS.
- R. C. McCrillis, E. M. Howard, Z. S. Guo, K. A. Krebs, R. Fortman and H. C. Lao, Journal of Air and Waste Management Association, 1999, 49(1), 70–75 Search PubMed.
- N. Kagi, S. Fujii, H. Tamura and N. Namiki, Build. Environ., 2009, 44, 1199–1205 CrossRef.
- F. Grimaldi, P. Botti, M. Bouthiba, F. Gouezo and A. Viala, Pollut. Atmos., 1996, 149, 330–337 Search PubMed.
- International Agency for Research on Cancer (IARC) Overall Evaluations of Carcinogenicity to Humans. 2007 In: IARC Monographs, vol. 1–96 Search PubMed.
- OQAI, Observatoire de la qualité de l'air intérieur, Campagne nationnale logements – Etat de la qualité de l'air dans les logements français – Rapport final – 2006, p. 183 Search PubMed.
- AFSSET, Valeurs guides de qualité d'air intérieur, Le formaldéhyde, 2007, p. 78 Search PubMed.
- W. Yang, J. Sohn, J. Kim, B. Son and J. Park, J. Environ. Manage., 2009, 90, 348–354 Search PubMed.
- A. T. Hodgson and D. Beal McIlvaine, Indoor Air, 2002, 12, 235–242 CrossRef CAS.
- International Organization for Standardization (ISO), Indoor air, Part 9: Determination of the emission of volatile organic compounds from building products and furnishing – Emission test chamber method, ISO 16000–9, 2006 Search PubMed.
- International Organization for Standardization (ISO), Indoor air, Part 10: Determination of the emission of volatile organic compounds from building products and furnishing – Emission test cell method, ISO 16000–10, 2006 Search PubMed.
- E. Uhde, A. Borgschulte and T. Salthammer, Atmos. Environ., 1998, 32, 773–781 CrossRef CAS.
- N. Shinohara, M. Fujii, A. Yamasaki and Y. Yanagisawa, Atmos. Environ., 2007, 41, 4018–4028 CrossRef CAS.
- N. Shinohara, T. Kajiwara, M. Ohnishi, K. Kodama and Y. Yanagisawa, Environ. Sci. Technol., 2008, 42, 4472–4477 CrossRef CAS.
- X. Zhou and K. Mopper, Environ. Sci. Technol., 1990, 24, 1864–1869 CAS.
- J. Nicolle, V. Desauziers and P. Mocho, J. Chromatogr., A, 2008, 1208, 10–15 CrossRef CAS.
- J. S. Zhang, C. Y. Shaw, L. C. Nguyen, R. A. MacDonald and G. Kerr, ASHRAE Transactions: Research, 1995, 101, 116–124 Search PubMed.
- S. Yamashita, K. Kume, T. Horiike, N. Honma, M. Fusaya, T. Ohura and T. Amagai, J. Hazard. Mater., 2010, 178, 370 CrossRef CAS.
- T. J. Kelly, D. L. Smith and J. Satola, Environ. Sci. Technol., 1999, 33, 81–88 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |