Advances in PAHs/nitro-PAHs fractioning
Auréa
Andrade-Eiroa
*,
Valérie
Leroy
and
Philippe
Dagaut
CNRS - ICARE, 1C, Avenue de la Recherche Scientifique, 45071, Orléans cedex 2, France. E-mail: eiroa_2000@yahoo.es; andrade@cnrs-orleans.fr; Fax: (+33) 238 696004; Tel: (+33) 238 255466
First published on 21st October 2010
Abstract
The aim of this work was to develop an efficient methodology for the reliable fractioning of nitrated-polycyclic aromatic hydrocarbons (nitro-PAHs) and polycyclic aromatic hydrocarbons (PAHs). Unlike what usually occurs under pressures developed by HPLC (high performance liquid chromatography) systems (above 11 bar) we observed that when normal phase chromatographic fractioning procedures are accomplished under very low pressures (about 1 bar), dipole molecules (nitro-PAHs) elute much faster than non-polar organic molecules (PAHs). This finding allowed developing an original and very efficient methodology for fractioning nitro-PAHs and PAHs. This method is based on normal-phase liquid chromatography through a home-made phenyl column by using hexane as mobile phase at very low speed flow (0.05 ml min−1). Unlike typical HPLC methodology, the fractioning of nitro-PAHs and PAHs was accomplished as a function of their polarity (first the polar compounds as a unique peak and further, the non-polar compounds, PAHs) rather than as a function of their medium polarizability.
1. Introduction
The detection and characterization of polycyclic aromatic hydrocarbons (PAHs) and nitrated polycyclic aromatic hydrocarbons (nitro-PAH) in environmental samples is a priority since both classes of compounds are ubiquitous and well known mutagenic and carcinogenic agents.1,2 Nitro-PAHs are the class of aromatic compounds with at least one (–NO2) functional group on one aromatic ring of a PAH. They are usually produced by incomplete combustion of carbon-containing fuels.3–5As PAHs and nitro-PAHs are frequently produced together, efficient methods are needed for separation and fractioning of both classes of compounds. Most of the analytical methods proposed are based on solid-phase extraction procedures (SPE), using silica or silica-alumina sorbents and hexane or dichloromethane as elution solvents.6 However, recent papers queried the efficiency of these methods7,8 and we have found that fractioning of PAHs and nitro-PAHs is not possible by using the conventional methods aforementioned. Moreover, all the traditional methodologies are time and solvent consuming and silica might adsorb irreversibly PAHs reducing significantly their recoveries.9 Alternatively, liquid chromatographic separation procedure after SPE purification can be applied for fractioning nitro-PAHs and PAHs.10 Thus, Bamford and colleagues11 reported that the combination of SPE followed by chromatographic separation for isolation of nitro-PAHs provides much better results in terms of cleaned extracts than only SPE. These authors employed an amino/cyano column combined with a moderately polar mobile phase consisting of 20% dichloromethane (DCM) in hexane at high speed flow (5 ml min−1). This process allows to obtain a PAH fraction, and a mono-nitro-PAH fraction followed by a di-nitro-PAH fraction. The chromatographic run takes about 35 min, which means that a volume of 160 ml of mobile phase is consumed and consequently the dilution of initial sample is high.
One of the main handicaps in this kind of research, is that although liquid chromatography (LC) and solid-phase extraction (SPE) have become the most powerful and commonly used techniques for the identification, quantification and fractioning of organic compounds in all kinds of samples,12 the real retention mechanism of chromatographic and SPE columns is yet unknown.13–15 Anyway, some helpful conclusions have been stated in literature.
Thus, it has been found that the order of elution of nitro-PAHs and PAHs is supposed to be dependant on the mobile phase and the stationary phase. In fact, when phenyl or phenyl modified phases are used along with polar mobile phases (usually methanol or methanol–water), under usual chromatographic conditions (high pressure and speed flow medium or higher than 0.5 ml min−1) polar-PAHs compounds elute before non polar PAHs.16 Otherwise, when phenyl phases are used in the normal phase mode (i.e, by using unpolar mobile phases such as hexane), unpolar compounds are supposed to elute before (i.e, PAHs before nitro-PAHs). However, we found out that this apparently very well established fundamental of liquid chromatographic fractioning is somewhat inaccurate. Thus, we observed that when the pressure falls under 1 bar the elution order established switched and hence, nitro-PAHs elute before PAHs from the aforementioned columns. Besides elution order, the efficiency of separation can be improved by lowering the pressure under 1 bar.
The proposed method allows to fraction PAHs and nitro-PAHs in a reproducible, automated and easy way. This one step process needs home-made phenyl columns, hexane as the mobile phase and low speed flow (0.05 ml min−1). In these conditions, the consumption of organic solvents is reduced, and recoveries and sensitivity are increased.
2. Experimental
2.1. Material and reagents
An HPLC system from Shimadzu (Shimadzu Corporation, Kyoto, Japan) equipped with different phases, 5, 4.5, 45 and 50 μm sizes, 250 × 4.6 mm column, a column oven CTO-20A/20AC Prominence, binary pump, PDA (photodiode detector array) and a fluorimetric detector was used.The reagents used were: Acetonitrile, methanol, dichloromethane, water and n-hexane for residue analysis (Fluka, Riedel-de Haën), PAHs (16 EPA TLC Polynuclear Aromatic Hydrocabons Mix, 2000 μg mL−1 in methylene chloride![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :benzene 50:50) and solid nitro-PAHs from Supelco (Supelco Park, Bellefonte, PA, USA). PAHs included in the standard were, in alphabetic order: Acenaphthene (Ace); acenaphthylene (Acy); anthracene (Anthr); benz[a]anthracene (B[a]A); benzo[a]pyrene (B[a]P); benzo[b]fluoranthene (B[b]F); benzo[g,h,i]perylene (B[ghi]P); benzo[k]fluoranthene (B[k]F); chrysene (Chry); dibenz[a,h]anthracene (D[a,h]A); fluoranthene (Flura); fluorene (Flu); indeno[1,2,3-cd]pyrene (IP); naphthalene (naph); phenanthrene (Phen); pyrene (Pyr). These PAHs are included in EPA (Environmental Protection Agency, USA) List Priority contaminants, and were identified at high concentrations in environmental samples.17
:benzene 50:50) and solid nitro-PAHs from Supelco (Supelco Park, Bellefonte, PA, USA). PAHs included in the standard were, in alphabetic order: Acenaphthene (Ace); acenaphthylene (Acy); anthracene (Anthr); benz[a]anthracene (B[a]A); benzo[a]pyrene (B[a]P); benzo[b]fluoranthene (B[b]F); benzo[g,h,i]perylene (B[ghi]P); benzo[k]fluoranthene (B[k]F); chrysene (Chry); dibenz[a,h]anthracene (D[a,h]A); fluoranthene (Flura); fluorene (Flu); indeno[1,2,3-cd]pyrene (IP); naphthalene (naph); phenanthrene (Phen); pyrene (Pyr). These PAHs are included in EPA (Environmental Protection Agency, USA) List Priority contaminants, and were identified at high concentrations in environmental samples.17
Nitro-PAHs included in the study were: 1-nitronaphthalene, 2-nitrofluorene, 1-nitropyrene, 3-nitrofluoranthene, 9-nitroanthracene and 6-nitrochrysene from Supelco (Bellefonte, PA, USA).
Solid phases used were: Discovery-Cyano (DSC-CN), DSC-Diol, DSC-18, DSC-NH2 DSC-Ph, and alumina and silica from Sigma-Aldrich. Table 1 summarizes the physical properties of the phases according to the information provided by Sigma-Aldrich.
| Phases | Particle diameter/μm | %C Loading, end-capping | Pore size | Surface area/m2 g−1 | Matrix |
|---|---|---|---|---|---|
| Silica | 50 | Unbonded silica | 70 Å | 480 | Silica gel (irregularly shaped, acid washed) |
| Alumina A, B and N | No data | No end-capping | 60–325 mesh | No data | No data |
| DSC-C18 | 50 | 18 | 70 Å | 480 | Silica gel (irregularly shaped, acid washed) |
| DSC-NH2 | 50 | 7 C loading, end-capped | 70 Å | 480 | Silica gel (irregularly shaped, acid washed) |
| DSC-CN | 50 | 7 C loading, end-capped | 70 Å | 480 | Silica gel (irregularly shaped, acid washed) |
| DSC-diol | 50 | 7 | 70 Å | 480 | Silica gel (irregularly shaped, acid washed) |
| DSC-Ph | 50 (tube phase) and 45 (bulk phase) | 7 | 70 Å | 480 | Silica gel (irregularly shaped, acid washed) |
| General description | Retention mechanism: Reversed-phase | |
| Sample matrix compatibility: Aqueous solutions (biological fluids, water). | ||
| Similar in polarity to DSC-8. It offers improved retention of conjugated ring structures over aliphatic functional groups. | ||
| Type | DSC-Ph SPE Bulk Packing (45 μm) | DSC-Ph SPE Tube (50 μm) |
| Extent of labelling | Monomerically bonded, phenyl (7%C), end-capped | 7% C loading, end-capped |
| Matrix | Silica gel base material (irregularly shaped, acid washed) | |
| Matrix active group | 45 μm | 50 μm |
| Pore size | — | 0.9 mL g−1 pore volume, 70 Å pore diameter |
| Surface Area | — | 480 m2 g−1 |
3. Results and discussion
SPE, open chromatography and preparative chromatography are the most commonly used procedures for fractioning nitro-PAHs and PAHs. The usual SPE fractioning procedure is as follows: An extract dissolved in the minimum volume of dichloromethane is applied at the top of the SPE cartridge. The cartridge is eluted stepwise with hexane, dichloromethane and finally methanol. The hexane eluate is expected to contain aliphatic hydrocarbons and PAHs, the dichloromethane fraction is expected to contain the oxy- and nitro-PAH fraction and methanol contains the most polar compounds of the extract.17,22 However we found that the efficiency of the SPE methodology described is limited. In fact, when a mixture of 6-nitrochrysene and 16 EPA PAHs was passed through alumina-silica or silica phase, a high percentage of 6-nitrochrysene is obtained in the extract containing PAHs, even if large silica columns are used. The same results were obtained by using alumina-silica (50% w:w), different formats of columns (10 cm, 20 cm and 25 cm length and 0.5 cm and 1 cm inner diameter) and different percentages of silica activation. Other authors also found that the dichloromethane extract from alumina columns contains PAHs and nitro-PAHs and further HPLC fractioning (usually through silica) is required for the fractioning of PAHs and nitro-PAHs.24 As a summary, SPE methodology is time and solvent consuming11,24,25 and sometimes provides recoveries too low. As a consequence, SPE was ruled out as fractioning methodology for separating nitro-PAHs and PAHs.Nevertheless, preparative chromatography has important advantages over SPE. Indeed, on-line monitoring of the elution fractions makes the procedure more feasible, and on the other hand, speed flow can be controlled and the separation can be carried out against the gravity. This last point is extremely important because we think that sometimes separations are not possible due to the compounds being swept out all together when the mobile phase passes through the column in favour of gravity.
With regards to column chromatographic fractioning drawbacks, some authors have reported that normal and reverse liquid chromatographic methods are suitable for fractionating components in environmental samples because they are fast and provide higher efficiencies than open column chromatographic fractionating.26,27,28 However, as early as 1991, Claessens and colleagues9 reported that the recoveries of some PAHs from diol and silica columns in an HPLC fractioning procedure were poor. In this paper, the authors concluded that using a silica column, the recoveries of PAHs can vary between 0–100% and PAHs such as perylene, benz(a)pyrene, 3-methylcholanthrene and anthanthrene showed very low recoveries. The authors explained that although the reason for these losses is not clear, it might be assumed that these PAHs are degraded and/or absorbed on the highly active silica. Navarro and colleagues25 also found low PAHs recoveries in a SPE fractioning procedure. As a consequence of the drawbacks aforementioned, we decided to modify the preparative HPLC methodology reported in literature.
The first handicap is that the chromatographic mechanism is still unknown. If the chromatographic procedure was understood, it could be efficiently optimized avoiding the trial and error method. In the aim of gaining comprehension about the behaviour of nonpolar and dipole analytes in analytical columns, PAHs and nitro-PAHs retention times were studied under different experimental conditions. The methodology developed is based on the knowledge and conclusions drawn up in the aforementioned experiences.
As the main difference between nitro-PAHs and PAHs is their polarity, the separation methodology developed should not fractionate these compounds according to their medium polarizability as the HPLC procedures usually does but as a function of their polarity. How to get the separation of nitro-PAHs and PAHs as a function of their polarity? Under usual chromatographic conditions (pressures about 30 bar, 0.5 ml min−1 and mobile phase acetonitrile:water gradient onto C18 columns), nitro-PAHs elute mixed with PAHs. The elution order in this case is according to the medium polarizability of the analytes.
Consequently, new experimental conditions should be found for efficient fractioning of nitro-PAHs and PAHs. In this aim, the most commonly used stationary phases and mobile phases were selected and studied under different experimental conditions. DSC-C18, DSC-NH2, DSC-diol, DSC-CN and DSC-Ph, silica, alumina and silica-alumina phases (Table 1) were tested as stationary phases. All these phases were used for filling in stainless steel casings pipes according to the procedure described in the experimental part. Hexane, dichloromethane, methanol, acetonitrile and water were tested as mobile phases.
DSC-C18 was combined with hexane, dichloromethane, methanol, acetonitrile and acetonitrile–water (from 0% to 90% water) under different speed flows. In most of cases (especially when dichloromethane and hexane were used), nitro-PAHs and PAHs elute together. Surprisingly, combination of DSC-C18 with polar mobile phases (acetonitrile–water 70:30 v/v at a speed flow of 0.25 ml min−1, onto home made columns) almost provided a separation of nitro-PAHs and PAHs. In fact, very similar retention times were achieved for the less retained PAH (naphthalene) and for the one of the most retained nitro-PAHs (6-nitrochrysene), but the separation was not satisfactory. The experiences carried out by using acetonitrile:water onto commercially available columns as well as onto home made columns (non-compacted) provided the most interesting conclusions of this paper. Indeed, the results indicate that the retention times of 6-nitrochrysene decrease when the pressure inside the column falls (Table 3). Thus, as can be seen from Table 3, the retention time of 6-nitrochrysene increases when pressure increases under constant speed flow (10–89 bar, 0.25 ml min−1). The increase in retention times seems to achieve a plateau at 70 bar. Otherwise, PAHs are not significantly sensitive to pressure changes under constant speed flow (0.25 ml min−1, data not shown). All the data shown in Table 3 were obtained in commercially available columns (highly compacted).
:30 ACN/Water in conditioning and elution steps, stationary phase C18, 5 μm commercially available columns)
| Speed Flow/ml min−1 | Pressure/bar | Retention time/min (n = 3, SD 7%) |
|---|---|---|
| 0.25 | 10 | 17.6 |
| 0.25 | 19 | 39.0 |
| 0.25 | 21 | 42.6 |
| 0.25 | 65 | 46.9 |
| 0.25 | 89 | 51.0 |
In our opinion and according to the data obtained, nitro-PAHs retention is strongly sensitive to pressure. PAHs are sensitive to speed flow changes but not to pressure changes under constant speed flows. Given that the separation achieved by using DSC-C18 was not enough, as a consequence we are working with other phases and other mobile phases under different speed flows.
Silica alone and silica-alumina (Acid, Basic and neutral) were used combined with hexane and dichloromethane in different proportions at low speed flows (0.25, 0.2, 0.1, 0.05 ml min−1). When hexane was employed, compounds eluted separately into groups according to their medium polarizability even at low speed flow, that is, nitro-PAHs elute mixed with PAHs. Therefore, these sorbents are not useful. When silica was mixed with alumina, similar results were obtained but the capability of the phase for discriminating PAHs and nitro-PAHs as a function of their medium polarizability got poorer. When dichloromethane was used, all the compounds eluted together from both silica columns as well as from silica-alumina columns (50% weight).
One of the main findings of this work is that when the retention on the stationary phase is too strong the elution occurs according to the medium polarizability of the analytes. Otherwise, if the retention is too weak, all the analytes elute together. As a consequence, the main goal from now on was getting adequate retention onto the analytical column for fractioning nitro-PAHs and PAHs as a function of their polarity.
High pressures combined with high differences between the dipole moments of stationary phase and mobile phase provide strong retentions onto the analytical columns. As a consequence, DSC-C18 combined with acetonitrile or water and silica combined with hexane under high pressure should be avoided. Otherwise, DSC-NH2, DSC-Ph, DSC-Diol or DSC-CN combined with hexane at slow speed flows might work out. Decreasing the speed flow is the easiest way of decreasing the pressure and at the same time saving solvents. This is the reason why low speed flows were preferred.
DSC-NH2, DSC-CN and DSC-Diol were used combined with hexane and dichloromethane as mobile phases at the same speed flows as those used with the other stationary phases. In all cases similar retention times were achieved for nitro-PAHs and PAHs. So, they are not useful for fractioning nitro-PAHs and PAHs.
With regard to the phenyl phase, two different types of phases were studied (Tables 1 and 2). The only difference between both of them lies in the size particle. DSC-Ph bulk packing has a 45 μm size particle whereas the DSC-Ph tube packing has a 50 μm size particle. On the other hand, DSC-Ph tube packing is 0.9 mL g−1 pore volume, 70 Å pore diameter and has a surface area of 480 m2 g−1, whereas data about surface area or pore diameter for DSC-Ph bulk packing (monomerically bonded phase) were not available.
Phenyl phase is usually employed as a reversed-phase sorbent,29 however hexane exhibits a lower toxicity, lower dielectric constants and a lower viscosity than most of the polar solvents used in reversed-phase chromatography. As a consequence of its low viscosity it yields low pressure inside the column. In addition to this, it is well known that dielectric constants account for the retention time of compounds,29,30 being solvents with low dielectric constants highly recommended for shortening the retention times of non-polar compounds.
Unlike the other phases included in this work and as we expected, DSC-Ph phase combined with hexane at low speed flow provided good results and allowed the efficient fractioning of nitro-PAHs and PAHs (Fig. 1 and 2; Tables 4 and 5). As can be seen from Fig. 1 and 2, the separation is much improved when the fractioning is accomplished by using 50 μm particles. In fact, we found that it is possible to elute nitro-PAHs all together rather than grouped in different peaks as in former papers.22Fig. 1 shows the results of the separation between nitro-PAHs and PAHs through a bulk phenyl phase without previous conditioning and under a speed flow of 0.1 ml min−1. The separation of nitro-PAHs and PAHs is not satisfactory (Table 5). NitroPAHs elute after PAHs but the time gap is too short and separation is inaccurate. However, separation could be improved by lowering the speed flow until 0.05 ml min−1 and using 50 μm particles (Fig. 2).
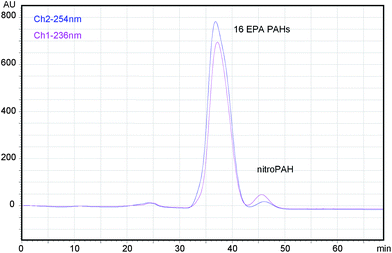 | ||
| Fig. 1 Fractioning of PAHs and nitro-PAHs with hexane on a phenyl column. Speed flow: 0.1 ml min−1, mobile phase: hexane, column oven temperature: 30 °C. 16 EPA PAHs (12 ppm), 20 ppm of 2-nitrofluorene, 2 ppm of 6-nitrochrysene. PDA data. Bulk phase (45 μm) column without previous conditioning. | ||
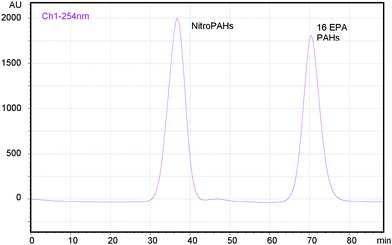 | ||
| Fig. 2 Fractioning of 16 EPA PAHs and 6 nitro-PAHs (1-nitronaphthalene, 2-nitrofluorene, 1-nitropyrene, 3-nitrofluoranthene, 6-nitrochrysene and 9-nitroanthracene) using phenyl phase (50 μm) and hexane as mobile phase. Speed flow: 0.05 ml min−1, column oven temperature: 30 °C. 16 EPA PAHs (12 ppm), ca. 2 ppm of nitro-PAHs. PDA data. Column without previous conditioning. | ||
| Speed flow/ml min−1 | Pressure/bar | R time naphthalene/min | R time 6-nitrochrysene/min | Type of phase |
|---|---|---|---|---|
| a n = 3, RSD, 7%; Rtime: retention time. b Column compacted. | ||||
| 0.05 | 0–1 | 70.5 | 31.8 | Tube phenyl phase (50 μm) |
| 0.1 | 1 | 35.7 | 29.0 | |
| 0.2 | 2 | 19.7 | 29.5 | |
| 1.0 | 10b | — | — | |
| 0.5 | 5 | 7.90 | 11.2 | |
| 0.1 | 2–3 | 34.3 | 51.3 | Bulk phenyl phase (45 μm) |
| 0.05 | 1–2 | 77.7 | 124 |
| Speed flow/ml min−1 | Pressure/bar | R time 16 EPA PAHs/min (n = 3) | R time 2-nitrofluorene/min (n = 3) | R time 6-nitrochrysene/min (n = 3) |
|---|---|---|---|---|
| 0.1 | 0–1 | 32.5–43.5 | 44.9–52.5 | 57.2–62.1 |
| 0.2 | 1–2 | 16.0–22.1 | 21.7–26.9 | 22.3–28.41 |
Discussion
The application of HPLC to nitro-PAHs and PAHs fractionation has become very popular due to its high efficiency, ease of automation, potential for column switching techniques and on-line coupling with other techniques including gas chromatography.11,20 Therefore, it is not the first time an HPLC system is proposed for the fractioning of PAHs, nitro-PAHs and other PAHs derivatives. In fact, one of the first famous HPLC nitro-PAHs/PAHs fractionation procedures was developed by Nielsen22 and it is still frequently applied in recent papers.20 This system consisted of a low pressure gradient pump, an array UV-visible detector, a thermostated oven and a column (25 cm × 8.0 mm) packed with Nucleosil Si-50-5. The mobile phase program used was cyclohexane/toluene (9:1) for 10 min followed by a linear gradient to 100% toluene and a 30 min plateau (flow rate of 3 ml min−1). The PAH fraction was collected after 6.00–12.0 min, the nitro-PAH fraction was collected after 18.5–26.0 min and the oxy-PAH fraction was typically collected after 37.5–45.0 min.
Other HPLC methodologies for fractioning nitro-PAHs and PAHs were developed by Fernández and Bayona31 or by Bamford and colleagues.11 These methodologies are normal-phase liquid chromatography fractionating procedures. However, as can be seen from Table 6, most of these methods consume high amounts of toxic organic solvents and the recoveries are frequently unsatisfactory.
| Our methodology | Three coupled normal phase HPLC columns23a | Liquid chromatography on amino/cyano column11, 32 | Gel permeation/normal phase liquid chromatography31 | |
|---|---|---|---|---|
| a Cyanopropyl, nitrophenyl, porous graphitised carbon columns. DCM: Dichloromethane, HX: Hexane. | ||||
| Order of elution | Nitro-PAHs/PAHs | PAHs/N-PAHs | PAHs/N-PAHs | PAHs/Nitro-PAHs |
| Retention times/min | N-PAHs: 32–41 | PAHs: 40–109 | PAHs: 3–6 | PAH: 4–17 |
| PAHs: 66–76 | N-PAHs: 109–121 | N-PAHs: 6–15 | N-PAHs: 17–22 | |
| Speed flow/ml min−1 | 0.05 | 10 | 5 | 4.5 |
| Mobile Phase | HX | DCM/HX | DCM/HX | DCM/HX |
| Volume of elution/ml | PAHs: 0.45 ml | PAHs: 690 | PAHs: 15 | N-PAHs: 58.5 ml |
| N-PAHs: 0.5 ml | N-PAHs: 120 | N-PAHs: 45 | PAHs: 22.5 ml | |
| Analysis time/min | PAHs: 41 | PAHs: 109 | PAHs: 17 | |
| N-PAHs: 76 | N-PAHs: 121 | N-PAHs: 22 | ||
| Recovery (%) | About 100% for both PAHs and nitro-PAHs in synthetic mixtures | PAHs: 53%–72% | PAHs: Not reported | Benzo(ghi)perylene: 98% |
| N-PAHs: 51%–71% | N-PAHs: 53%–150% (aprox.) in SRM 2975 | 1-nitrofluorene: 112% | ||
| 1-nitropyrene: 100% |
The main goal was to achieve the fractioning of nitro-PAHs and PAHs as a function of their polarity. Relating to this, we found that fractioning as a function of the polarity is easier when the retention becomes weaker than that achieved by using DSC-18/polar mobile phases or silica/alumina with hexane as mobile phase and stronger than using DSC-18 with hexane or silica with dichloromethane.
With regards to literature conclusions, important contradictions have been found. Thus, it has been recognized that the phase type, the presence or absence of end-capping32 or mobile phase29 seem to be the most important factors affecting the separation. However, in spite of the extremely high importance given in literature to the stationary phase, Smith and Cooper30 found that some nitro-PAHs exhibit a similar behaviour regardless to what type of column was used (amino, diol, cyano) and was only dependant on the mobile phase. Kayillo and colleagues29 also established that one of the phenyl columns investigated (Synergy polar-RP column) exhibited similar behaviour to C18 columns. The only difference between both phases was the particle size. Thus, Synergy Polar-RP column has a particle size of 4 μm, whereas the other phases considered by Kayillo and colleagues29 were 5 μm in size. We think that these results provide evidence that no specific interactions occur between the stationary phase and the target analytes. Although no specific interactions seem to occur between the stationary phase and the target analytes and between the mobile phase and the analyte, physical properties (especially electrical properties) of the materials used seem to determine the retention mechanism of the analytes. The retentive capacity of the DSC-Ph phase in reverse-phase chromatography is weaker than that of DSC-18, but the smaller particle size in case of DSC-Ph (and consequently the higher pressure developed inside the column) makes its retention capacity increase. These conclusions support our statements about the importance of particle size.
On the other hand, papers dealing with the isolation of PAHs and nitro-PAHs by chromatographic procedures through phenyl columns have tried unsuccessfully to explain the mechanism of retention based on the formation of π-bonding complexes between the target aromatic compounds and phenyl groups attached to the silica surface.22 However, we found that the data could be successfully explained based on the dielectric properties of both the mobile phase and the stationary phase as well as on the viscosity of the solvent (directly related with the pressure developed inside the column) confirming that non specific interactions exist between the solid phases and target analytes but that the physical properties of the mobile and stationary phases account for the retention mechanism.
Particularly, the dielectric constant of the DSC-Ph phase is expected to be higher than that of the DSC-18 and lower than that of underivatized silica. High dielectric constant phases (silica or alumina) usually require solvents with a low dielectric constant (i.e. hexane); otherwise low dielectric constant phases (DSC-18) are usually applied in combination with high dielectric constant solvents (i.e. water or acetonitrile). The higher the difference between the dielectric constants of the mobile and those of the stationary phase, the stronger the retention onto the analytical column. Thus, the retention strength of any analyte seems to depend on the difference between the dielectric constants of the mobile phase and that of the stationary phase. When this strength is too high, the analytes elute according to their medium polarizability. Otherwise, when this difference is medium, the retention times occurs mostly as a function of the polarity of the target analytes. Indeed, our experimental work showed up that for fractioning nitro-PAHs and PAHs in an efficient way, DSC-18 should be avoided because this phase combined with acetonitrile and water, retains strongly the compounds and the separation on this column is carried out according to the medium polarizability of the analyte. In this case, 6-nitrochrysene elutes after pyrene and before chrysene. Even using low pressures, the results were unsatisfactory. Similar results were achieved by using silica (and/or alumina) combined with hexane.
On the other hand, normal phases (with retention capacity similar to silica) such as DSC-Diol and DSC-NH2 employed with hexane did not provide satisfactory results. With regard to DSC-CN, it is supposed to have a polarity between silica and DSC-18 and it can be used as a normal or reversed phase, but satisfactory results were not obtained (nitro-PAHs and PAHs elute together in normal phase). And finally, DSC-Ph is supposed to have a polarity similar to that of DSC-8 and is recommended to be used in the reverse phase, but used as normal phase (as in this paper) the retentive capacity falls significantly and it make possible the efficient fractioning of nitro-PAHs and PAHs into two well differentiated groups.
As we can see from Table 4, when 45 μm particles of DSC-Ph phase are used, the 6-nitrochrysene retention times increased outstandingly compared to the 50 μm particles probably due to the fact that in this case the pressure inside the column is higher (although this increase can not be clearly noted in Table 4 due to the inaccuracy of so low pressure measures). We should take into account that measuring so low values of pressure is not easy due to the accuracy of this variable being 1. As a consequence, when we say the pressure is 1, the pressure may actually be 1.5, or 0.5. In fact, for the case of 45 μm particles although pressure most of time is 1 bar, it was actually 2 bar during some instants of the running. This could explain why when 45 μm particles are used the retention times are much higher than when 50 μm particles are used. Note that when 45 μm DSC-Ph particles are used as stationary phase, the pressure achieved at a speed flow of 0.1 ml min−1 is about 3–4 fold higher than when 50 μm silica-phenyl particles are used under the same speed flow (1–2 bar versus 0–1 bar), and consequently retention times are expected to be about 3–4 fold higher (about 120 min vs. 30 min achieved by using 50 μm particles). With regards to the values of retention times for 6-nitrochrysene at pressure 3 bar and a speed flow of 0.1 ml min−1 achieved by using 45 μm particles as the stationary phase (53.9 min), the correlation between retention time and pressure is also good. Thus, the retention time is almost twice that one achieved using 50 μm particles under the same speed flow (29.0 min, pressure 1–2 bar).
The comparative between our methodology and other methodologies is shown in Table 6. As can be seen, our method consumes much less volume of solvent and does not use dichloromethane (much more toxic than hexane). On the other hand, the separation between both classes of compounds is much clearer than in other methods and the recoveries, higher. The only drawback of our methodology is that the analysis time is a bit higher, however the dilution is less and consequently the sensitivity higher, avoiding further preconcentration steps.
Important differences between our methodology and the methodology reported in literature were found. Thus,
1. In general, literature stated that hydrophobicity is the main interaction force in RPLC. Hydrophobicity includes a group of repulsive interactions between non-polar compounds and a polar environment, such as water. These forces include electrostatic interactions such as ion–ion, ion–dipole and dipole–dipole interactions and they are strongly depending on the dielectric constant of the solvent.29,33 However, here some evidence is provided in favour of electrostatic interactions occurring inside normal-phase chromatographic procedures too. In fact, this kind of repulsion among dipole molecules such as nitro-PAHs could account for the very anomalous behaviour of those analytes inside chromatographic columns compared to their PAHs homologues.
2. According to the papers aforementioned, nitro-PAHs elute after PAHs from cyano and phenyl columns.11, 34 Literature reported that unlike PAHs, polar compounds (like nitro-PAHs) seem to be less retained using polar mobile phases (e.g. hexane:metyl-tert-butyl ether) than using unpolar solvents, although the difference of retention times is not outstanding due to the low retention capability of phenyl phases in general.30 However, we found that when hexane is used as mobile phase at low pressures, nitro-PAHs are less retained than using polar solvents. On the other hand, we observed that when the pressure falls below 1 bar (Table 4) and under low speed flow (below 0.1 ml min−1), the order of elution of these classes of compounds was switched.
3. The proposed methodology consumes a reduced volume of low toxicity solvents (hexane vs. toluene or DCM), and allows a more accurate separation of nitro-PAHs and PAHs (Table 6). The sample injected in the paper by Nielsen22 is 500–1000 fold diluted (providing an injection volume in the range of 20–50 μL as usual in HPLC) whereas in this paper the initial sample would be only 12.5 fold diluted (volume of injection: 20 μL). Consequently, sensitivity increased.
Recoveries
Pressure affects differently PAHs and nitro-PAHs retention in chromatography procedures. In fact, whereas increasing column pressure does not affect significantly the elution of PAHs, the retention times of nitro-PAHs significantly increase with increasing pressure (Table 3). Under extremely low pressures (∼1 bar), nitro-PAHs and PAHs elute as a function of their polarity into two well differentiated groups of compounds (first the polar compounds, that is, nitro-PAHs and then, PAHs).The methodology developed here (under low pressure) provided higher recoveries than the traditional methodology (under high pressures). In fact, recoveries of nitro-PAHs are about 50% when they elute after PAHs at high pressures (45 μm phenyl phase) and about 100% when they are eluted before PAHs under low pressures (50 μm phenyl phase). The results obtained for 50 μm phenyl phase were more satisfactory than those reported in the literature11 and the low dilution avoids further concentration steps and consequently shorter and more feasible analytical methodology is achieved (no loses of volatile or degradable compounds occurs).
4. Conclusions
This paper provides a very efficient alternative for the separation of nitro-PAHs and PAHs. Based on the following conclusions drawn about the behaviour of nitro-PAHs and PAHs inside analytical columns, a new separation methodology for fractioning PAHs and nitro-PAHs was developed:1. Under very low pressures, dipole molecules (nitro-PAHs) exhibit a very different behaviour from that showed by PAHs in liquid chromatographic systems.
2. Pressure influences separation processes. In fact, high pressures do not influence significantly the retention of PAHs but make difficult the elution of dipole molecules. As a consequence, decreasing the speed flow (and consequently the pressure developed inside the column) makes nitro-PAHs elute before PAHs.
3. Traditional chromatographic procedures can be adapted and modified to separate groups of compounds as a function of their polarity rather than according to their medium polarizability as is usual in HPLC.
This paper proposed a faster and more feasible way for fractioning nitro-PAHs and PAHs by reducing the pressure under 1 bar, using 50 μm DSC-Ph phase, hexane as mobile phase and a speed flow of 0.05 ml min−1.
Acknowledgements
Partial financial support from ESA through the contract 15091/01/NL/SH-CCN No. 002, MAP Project numbers AO-99-001 AO-99-085, is gratefully acknowledged. We also thank Dr Graciela de Armas (Balearic Islands University, Spain) for bibliographic assistance and Alain Quilgars (CNRS, Orléans-France) for manufacturing glass columns.5. References
- B. J. Finlayson-Pitts and J. N. J. Pitts, Chemistry of the upper and lower atmosphere: theory, experiments and applications, Academic Press, San Diego, 2000 Search PubMed.
- A. Albinet, E. Leoz-Garziandia, H. Budzinski and E. ViIlenave, Sci. Total Environ., 2007, 384, 280 CrossRef CAS.
- Z. Fan, D. Chen, P. Birla and R. M. Kamen, Atmos. Environ., 1995, 29, 1171 CrossRef CAS.
- M. Dimashki, S. Harrad and R. M. Harrison, Atmos. Environ., 2000, 34, 2459 CrossRef CAS.
- N. V. Heeb, P. Schmid, M. Kohler, E. Gujer, M. Zennegg, D. Wenger, A. Wichser, A. Ulrich, U. Gfeller, P. Honegger, K. Zeyer, L. Emmenegger, J. L. Petermann, J. Czerwinski, T. Mosimann, M. Kasper and A. Mayer, Environ. Sci. Technol., 2008, 42, 3773 CrossRef CAS.
- W. E. May and S. A. Wise, Anal. Chem., 1984, 56, 225 CrossRef CAS.
- S. Lundstedt, Y. Persson and L. Ôberg, Chemosphere, 2006, 65, 1288 CrossRef CAS.
- S. Lundstedt, P. Haglund and L. Ôberg, Anal. Chem., 2006, 78, 2993 CrossRef CAS.
- H. A. Claessens, M. M. Rhemrev, J. P. Weevers, A. A. J. Janssen and L. J. Brasser, Chromatographia, 1991, 31, 569 CAS.
- B. S. Crimmins and J. E. Baker, Atmos. Environ., 2006, 40, 6764 CrossRef CAS.
- H. A. Bamford, D. Z. Bezabeh, M. M. Schantz, S. A. Wise and J. E. Baker, Chemosphere, 2003, 50, 575 CrossRef CAS.
- A. Andrade-Eiroa, V. Leroy, P. Dagaut and Y. Bedjanian, Chemosphere, 2010, 78, 1342 CrossRef CAS.
- Y. D. Men and D. B. Marshall, Anal. Chem., 1990, 62, 2606 CrossRef CAS.
- P. T. Ying, J. G. Domy and K. A. Dill, Anal. Chem., 1989, 61, 2540 CrossRef CAS.
- C. A. Doyle, T. J. Vickers, C. K. Mann and J. G. Dorsey, J. Chromatogr., A, 2000, 877, 41 CrossRef CAS.
- T. Letzel, U. Poschl, R. Wissiak, E. Rosenberg, M. Grasserbaur and R. Niessner, Anal. Chem., 2001, 73, 1634 CrossRef CAS.
- S. V. Kakareka, T. I. Kukharchyk and V. S. Khomich, Environ. Pollut., 2005, 133, 383 CrossRef CAS.
- S. M. Rappaport, Z. L. Jin and X. B. Xu, J. Chromatogr., A, 1982, 240, 145 CrossRef CAS.
- J. Cvacka, A. G. Fogg, J. C. Moreira and J. Zima, Analyst, 1998, 123, 9R RSC.
- A. Feilberg, M. W. B. Poulsen, T. Nielsen and H. Skov, Atmos. Environ., 2001, 35, 353 CrossRef CAS.
- Applications of High Performance Liquid Chromatography, Ed. A. Pryde and M. T. Gilbert, University Press, Cambridge, 1979, pp. 162 Search PubMed.
- T. Nielsen, Anal. Chem., 1983, 55, 286 CAS.
- U. Lübcke-von Varel, G. Streck and W. Brack, J. Chromatogr., A, 2008, 1185, 31 CrossRef CAS.
- R. Brindle, K. Albert, E. D. Morgan, P. Martin and I. D. Wilson, J. Pharm. Biomed. Anal., 1995, 13, 1305 CrossRef CAS.
- P. Navarro, E. Cortazar, L. Bartolomé, M. Deusto, J. C. Raposo, O. Zuloaga, G. Arana and N. Etxebarria, J. Chromatogr., A, 2006, 1128, 10 CrossRef CAS.
- F. Marino, A. Cecinato and P. A. Siskos, Chemosphere, 2000, 40, 533 CrossRef CAS.
- W. R. West, P. A. Smith, G. M. Booth and M. L. Lee, Environ. Sci. Technol., 1988, 22, 224 CAS.
- D. W. Later, B. W. Wilson and M. L. Lee, Anal. Chem., 1985, 57, 2979 CrossRef CAS.
- S. Kayillo, G. R. Dennis and R. A. Shalliker, J. Chromatogr., A, 2006, 1126, 283 CrossRef CAS.
- P. L. Smith and W. T. Cooper, J. Chromatogr., A, 1987, 410, 249 CrossRef CAS.
- P. Fernández and J. M. Bayona, J. Chromatogr., A, 1992, 625, 141 CrossRef CAS.
- D. Z. Bezabeh, H. A. Bamford, M. M. Schantz and S. A. Wise, Anal. Bioanal. Chem., 2003, 375, 381 CAS.
- D. H. Marchand, L. R. Snyder and J. W. Dolan, J. Chromatogr., A, 2008, 1191, 2 CrossRef CAS.
- K. Croes, A. Steffens, D. H. Marchand and L. R. Snyder, J. Chromatogr., A, 2005, 1098, 123 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |