Comparative analysis of protein extraction methods for the identification of seafood-borne pathogenic and spoilage bacteria by MALDI-TOF mass spectrometry
Karola
Böhme
a,
Inmaculada C.
Fernández-No
a,
Jorge
Barros-Velázquez
a,
Jose M.
Gallardo
b,
Benito
Cañas
c and
Pilar
Calo-Mata
*a
aDepartment of Analytical Chemistry, Nutrition and Food Science, School of Veterinary Sciences, University of Santiago de Compostela, E-27002, Lugo, Spain. E-mail: p.calo.mata@usc.es; Fax: +34 982252195; Tel: +34 647344274
bDepartment of Food Technology, Institute for Marine Research (IIM-CSIC), E-36208, Vigo, Spain. E-mail: gallardo@iim.csic.es
cDepartment of Analytical Chemistry, University Complutense of Madrid, E-28040, Madrid, Spain. E-mail: bcanas@quim.ucm.es
First published on 7th October 2010
Abstract
Species differentiation of food pathogenic and spoilage bacteria is important to ensure food quality and safety. Matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) has been applied to species identification of microorganisms, proving to be a rapid and cost-effective technique and allowing species differentiation due to the highly specific spectral profiles obtained. In this work, bacterial strains from our laboratory intern collection of seafood-borne pathogenic and spoilage species were studied by MALDI-TOF MS. Different sample preparation protocols were applied and compared to each other. Two methods were based on the analysis of whole bacterial cells that were suspended in an organic solvent or applied directly to the sample target. In a different sample preparation technique, cell extracts were obtained from intact bacterial cells by a dissolution/centrifugation step. The protocol applied for the study of cell extracts was shown to be very fast and simple, allowing the standardization of sample preparation. Furthermore, the analysis of cell extracts had several advantages with respect to the analysis of suspensions of whole bacterial cells. Thus, spectral profiles obtained from cell extracts showed less noise and more reproducible peaks as compared to spectra obtained from intact cells. The analysis of cell extracts by MALDI-TOF MS was also applied to create a mass spectral library of the main pathogenic and spoilage bacteria potentially present in seafood and was demonstrated to be a rapid and accurate method for microbial species differentiation, as well as for the classification of unknown strains isolated from seafood.
Introduction
Bacterial species identification is essential in the area of clinical analysis, aiming for the detection and correct treatment of human diseases. Furthermore, the differentiation of bacterial species plays an important role in the food sector. On one hand, the detection of food-borne pathogens allows one to control and minimize the microbiological hazards of food products. On the other hand, the shelf-life of food products can be predicted and enhanced by the analysis of the microflora and food-spoilage bacteria, potentially present in the food products. Traditionally, bacterial species have been identified by classic tools relying on culturing processes coupled to morphological, physiological and biochemical characterization. In the last few decades, the progress of microbiological identification has turned to more rapid and sensitive methods, including miniaturized biochemical systems, antibody-based assays and DNA-based methods, coupled with important advances in bioinformatic tools. Recently, the development of rapid and high sensitive techniques, such as real-time PCR, DNA microarrays and biosensors, has resulted in the replacement of traditional culturing methods of bacterial identification in both clinical diagnostics and in the food sector.1–3Although, phenotypic and genetic tools offer an accurate identification of bacterial species, these techniques are labor intensive, time consuming and expensive. Furthermore, some bacterial species are difficult to distinguish by DNA-based molecular tools, because of high sequence similarities.
In the last few years, proteomic tools have been applied for the identification of bacteria, finding that mass spectrometry is a competent tool for species differentiation.4,5 In particular, matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) has been reported to be a very fast and simple method for bacterial differentiation, due to its rapidness, reduced cost and minimal sample preparation, compared to traditional techniques.6–8 Spectral profiles obtained by MALDI-TOF MS are highly selective for each bacterial species, resulting in specific fingerprints that allow the differentiation of bacteria on the genus, species and even strain level.9,10 In general, characteristic high-mass ions obtained by MALDI-TOF MS are attributed to proteins.6,7 In some studies peak patterns obtained by MALDI-TOF MS were identified and confirmed that most peaks are generated by proteins11,12 and especially by ribosomal proteins.13
Two different approaches have been developed for bacterial species identification based on MALDI-TOF MS. The first technique is based on bioinformatics and identifies a bacterial species by matching the experimentally determined masses of protein biomarkers against sequence-derived masses of proteins, found together with their source organisms in proteome databases.14 However, to be applicable in the field of bacterial identification, database searches require a high mass accuracy and identification is limited to well-characterized microorganisms, where a number of sequenced biomarkers are known.7 In the second approach, bacterial identification is based on the comparison of the sample spectrum to a reference library.10,15 The so-called “fingerprint approach” has proved to be applicable for bacterial species-classification and many works have been done in constructing spectral databases of a huge number of bacterial strains.15–18 However, the critical challenge of these techniques is the limited availability of such reference databases. Furthermore, studies are mainly focused on clinical areas, whereas only a few studies have been realized on microbial food analysis.10,19,20
Many studies have been carried out to optimize the sample preparation protocol21–24 and the evolution of spectral data,25,26 facilitating spectral comparison and making MALDI-TOF MS an accurate tool for bacterial species identification. To allow the comparison of spectral profiles, special attention has to be given to the reproducibility of the applied method. Various authors observed a sensitivity of the obtained spectral profiles to little changes in the sample preparation protocol.21,23,27 Thus, a strict sample preparation protocol has to be followed, with the aim to obtain reproducible and representative fingerprints. Whereas earlier works used protein fractions isolated from bacterial cells, intact cell mass spectrometry (ICMS) was later developed to analyze whole cells directly without any sample pre-treatment.28 The aim of the present work was to establish a standardized sample preparation protocol, such that specific and reproducible spectral profiles can be obtained in a rapid and labor-saving way, allowing spectral comparison and the subsequent differentiation of bacterial species. For that, different sample preparation protocols were applied and compared to each other.
Furthermore, the study was focused on bacterial species with an interest in the seafood-sector, including the main seafood-pathogens and spoilage species, aiming towards the construction of a spectral reference library for the identification of bacterial strains isolated from seafood.
Experimental
Bacterial strains and culture media
Bacterial strains were obtained from the laboratory intern collection that included strains isolated from seafood, as well as reference strains from the Spanish Type Culture Collection (CECT) of the main seafood pathogenic and spoilage species (Table 1).| Genus | No. of | Genus | No. of | ||
|---|---|---|---|---|---|
| species | strains | species | strains | ||
| Acinetobacter | 1 | 1 | Pantoea | 1 | 2 |
| Aeromonas | 1 | 1 | Photobacterium | 2 | 2 |
| Bacillus | 6 | 20 | Proteus | 3 | 6 |
| Carnobacterium | 2 | 3 | Providencia | 2 | 2 |
| Citrobacter | 1 | 1 | Pseudomonas | 4 | 21 |
| Clostridium | 2 | 2 | Raoultella | 1 | 1 |
| Enterobacter | 4 | 4 | Serratia | 3 | 8 |
| Hafnia | 1 | 1 | Shewanella | 3 | 3 |
| Klebsiella | 2 | 2 | Staphylococcus | 4 | 5 |
| Listeria | 5 | 9 | Stenotrophomonas | 1 | 8 |
| Morganella | 1 | 2 | Vibrio | 3 | 3 |
The frozen stored strains were reactivated in Brain-Heart-Infusion (BHI) (Becton, Dickinson and Company, Le Pont de Claix, France) and incubated for 24 h at 30 °C. Afterwards, bacterial cultures were grown on Plate-Count-Agar (PCA) (Oxoid, Hampshire, England) at 30 °C and single colonies were isolated. For MALDI-TOF MS sample preparation, pure cultures were previously grown on PCA and incubated for 24 h at 30 °C.
MALDI sample preparation
Spectra acquisition
Mass spectra were obtained using a Voyager DE STR MALDI-TOF Mass Spectrometer (Applied Biosystems, Foster City, CA) operating in linear mode, extracting positive ions with an accelerating voltage of 25,000 V and a delay time of 350 ns. The grid voltage and guide wire were set to 95% and 0.05% of the extraction voltage, respectively. Spectra taken in the m/z range of 1,500–15,000 were obtained in 10 different regions of the same sample spot and each was the result of the accumulation of at least 1000 laser shots. The spectra were externally calibrated using a mixture of 2 pmol/μL oxidized B chain of insulin and 2 pmol/μL bovine insulin (Sigma-Aldrich).Data analysis
Mass spectra were processed with the DataExplorer® software (Version 4.0.0.0), baseline corrected, noise filtered and analyzed, considering the mass interval 2,000–10,000 Da, due to the good reproducibility of the spectral profile in that range. Data lists containing m/z values were extracted from mass spectral data, including signals with relative intensities higher than 2%. The final peak mass lists for each strain were obtained, extracting representative peaks present in all spectra acquired for each strain. All mass lists were further processed with the free available web-based application SPECLUST (http://bioinfo.thep.lu.se/speclust.html). The web interface calculates the mass difference between two peaks taken from different peak mass lists and determines if the two peaks are identical after taking into account a certain measurement uncertainty (σ) and peak match score (s). The peak match score represents the probability that two peaks with measured masses m and m′ have a mass difference equal or larger than |m-m′|, given that the mass difference is only due to measurement errors.29Furthermore, the mass lists of all studied strains were clustered with the clustering option available on the web interface SPECLUST. The agglomerative clustering method starts with creating one cluster for every peak list and calculates distances between the clusters. For calculating distances between two peak lists, all individual similarity scores for every pair of two peak lists were added. A correlation based metric was used and the width in peak match score was set to 5 Da.29 The two closest clusters, in this study the two clusters with the smallest average of pair wise distances (average linkage method), are then merged to a new cluster and distances are recalculated. This process is continued until only one single cluster remains.
Results and discussion
MALDI-TOF MS has been shown to be a competent tool for bacterial species differentiation, due to the resulting highly specific spectral profiles, named fingerprints.30 For bacterial identification, the spectral profile of a strain of interest is compared to a spectral library. For the comparison of spectral profiles, reproducibility plays an important role. It has been shown that spectra can display high variability depending on the sample preparation protocol.21,23,27 Therefore, the critical point of the technique of MALDI-TOF MS fingerprinting is that a strict standardized protocol for sample preparation has to be followed to obtain reproducible spectra and allow comparison of different bacterial spectra.In the present work different sample preparation protocols were carried out and compared. The first sample preparation protocol tested was based on the direct application of bacterial biomass, taken from culture plates, on the MALDI-TOF MS sample plate. Later, the bacterial cells were overlaid with the matrix solution. Although this method was the most rapid and labor-saving, the spotting of biomass directly on the sample plate had several disadvantages. In this sense, it was found to be difficult to obtain a homogenous distribution of sample and matrix in the sample spot. The major problem of this method was the difficulty in taking the correct amount of biomass to obtain good and reproducible spectra. In Fig. 1 spectral profiles that resulted from different amounts of biomass (0.7, 1.3 and 5 mg) are shown. It may be observed that the amount of biomass had a great influence on the resulting spectra and an increasing load of biomass reduced the quality of spectral profiles drastically. Furthermore, we could not obtain reproducible spectral profiles with this fast method, and spectra showed more noise and less peak resolution in comparison with the other sample preparation techniques. However, this technique was described by a number of authors and successfully applied for bacterial species identification and the construction of spectral reference databases, such as the Microbelynx™ bacterial identification system16 and the Spectral ARchive And Microbial Identification System (SARAMIS, AnagnosTec).17
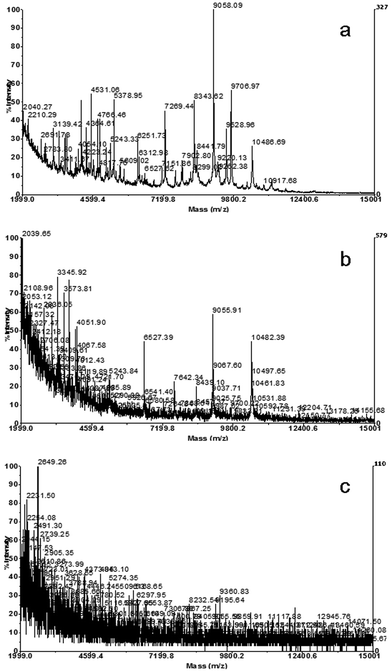 | ||
| Fig. 1 Spectra obtained by analysis of whole cells of a Morganella morganii strain, applying different amounts of biomass to the target well: a) 0.7 mg, b) 1.3 mg and c) 5 mg. | ||
In a second sample preparation approach, bacterial colonies were harvested in a solvent to obtain cell suspensions of whole bacterial cells. Most studies of MALDI-TOF MS for bacterial discrimination have been based on this method and the sample preparation usually included one or two washing steps, before the cell pellet was resuspended in the matrix solution.19,21,24,31 These washing steps are time-consuming and small soluble proteins are lost. Some authors also described a similar sample preparation method, but here no washing step was applied and the bacterial colonies were harvested in a solvent to obtain cell suspensions.32
In the present work, in order to test this method, cell suspensions were prepared without washing steps in different volumes of matrix solution (10, 20, 50 and 100 μL). The best spectral profiles were obtained when the biomass was harvested in 100 μL of matrix solution (Fig. 2).
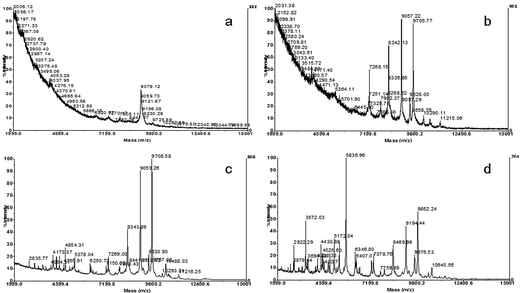 | ||
| Fig. 2 Spectra obtained by analysis of whole cell suspensions of a Morganella morganii strain, diluted in different volumes of matrix solution: a) 10 μL, b) 20 μL, c) 50 μL and d) 100 μL. | ||
In a third approach, based on the latter method, cell suspensions were centrifuged and spectra obtained by the analysis of the supernatant were compared to the spectra resulting from cell suspensions without a centrifugation step.
The analysis of cell extracts differs from commonly applied sample preparation protocols in that suspensions of whole cells are analyzed as described by Wang et al. (1998). One of the advantages of this technique is the rapid and effortless sample preparation. The extracts were obtained in just one dilution/centrifugation step and did not require time-consuming washing steps, as it was the case in sample preparation protocols applied by many other authors. Although, when working with the extracts it should be expected to find small, soluble proteins, spectral profiles showed a high number of peaks, similar or even higher than those obtained by the analysis of whole cell suspensions. In Fig. 3 spectral profiles of two Pseudomonas strains, obtained by analysis of cell extracts (a, c) and cell suspensions (b, d) are shown. As it may be observed, the spectra were very similar for both sample preparation techniques, having most peak masses in common.
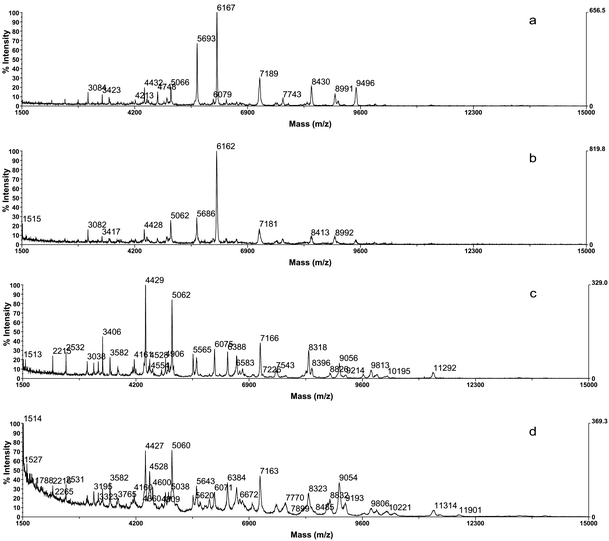 | ||
| Fig. 3 Spectra of a Pseudomonas fluorescens strain obtained from cell extract (a) and cell suspension (b); and spectra of a Pseudomonas fragi strain obtained from cell extract (c) and cell suspension (d). | ||
Thus, analyzing cell extracts instead of whole cell suspensions does not affect the number of peaks detected, neither the nature of proteins. Nevertheless, although in the obtained spectra only a few differences could be observed between the different extraction methods, the analysis of cell extracts had several advantages over the commonly applied analysis of whole cells and cell suspensions, such as a better reproducibility, a higher resolution and less noise. The decreasing of noise, lessening the background, and the increase in resolution leads to more representative and characteristic peaks for each bacterial species, improving the reproducibility. It should be mentioned that, in general terms, a more homogenous distribution of sample and matrix is expected with cell extracts, than with cell suspensions.
Table 2 gives an overview of the different sample preparation methods. In the case of cell suspensions and cell extracts the biomass can be harvested directly in the matrix solution, or in an organic solvent without matrix. Since the matrix solution is unstable, the dilution in an organic solvent without addition of the matrix has the advantage that the matrix solution can be prepared just before analysis by MALDI-TOF MS. Different samples can be stored at different times and diluted with the matrix just before analysis. In this sense, the sample solution can be mixed with the matrix solution before spotting to the target well, or can be first applied to the target well and then overlaid with the matrix solution. However, in our study we obtained a more homogenous crystallization, when mixing the sample and matrix solution before applying to the sample target.
| Whole cells | Whole cell suspension | Cell extract | |
|---|---|---|---|
| Sample preparation | |||
| (1-2 cycles of washing/centrifugation steps) | Harvest biomass in organic solvent (w/wo matrix) | ||
| Centrifugation | |||
| Direct application of biomass to target well | Harvest biomass in organic solvent (w/wo matrix) | (Supernatant mixed with matrix just before analysis) | |
| Application to target well | Application to target well | ||
| Overlaid with matrix solution | (Overlaid with matrix solution) | (Overlaid with matrix solution) | |
| Advantages and disadvantages | |||
| Very fast method | Time consuming washing steps | Very fast method | |
| Less homogenous crystallization | Homogenous crystallization | Homogenous crystallization | |
| Less reproducibility | Good reproducibility | Best reproducibility | |
| More noise | More noise | Low noise | |
| Less resolution of peaks | Good resolution of peaks | Best resolution of peaks | |
Based on these results, the protocol to obtain cell extracts was modified and the biomass was harvested in 100 μL of a solution of 50% ACN and 1% aqueous TFA without matrix. After mixing by vortex and centrifugation, the supernatant was stored frozen until analysis. As mentioned above, this approach simplifies the analysis since samples can be stored until analysis. In contrast, when the sample is mixed with the matrix, the solution cannot be stored over a long period of time and has to be prepared just before analysis. This is so, because the matrix α-CHCA is unstable and should be carefully stored under dry conditions. Remarkably, the supernatants stored without matrix showed reproducible spectral profiles after six months of frozen storage (Fig. 4). Before analysis the supernatant was mixed with the matrix solution and then spotted to the target well. Remarkably, the best results were accomplished when the sample solution was diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 with the matrix solution, resulting in highly reproducible spectral profiles with little noise and a very good resolution of peaks.
:10 with the matrix solution, resulting in highly reproducible spectral profiles with little noise and a very good resolution of peaks.
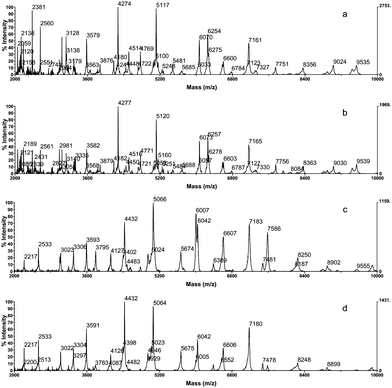 | ||
| Fig. 4 Spectra of Photobacterium damselae (a, b) and Pseudomonas fragi (c, d); directly after preparation of cell extracts (a, c) and after six months of storage at −20 °C (b, d). | ||
The described protocol was applied for the analysis of Gram-negative as well as Gram-positive strains. Various authors have applied a different protocol for Gram-positive bacterial cells, including pretreatments with lysozyme and/or sonication, to increase the number of ions detected by MALDI-TOF mass spectrometry.33,34 In our study we showed that the lysis of bacterial cells by a solution of an organic solvent (acetonitrile) and a high concentration of a strong acid (TFA) was sufficient to obtain a number of representative peaks, allowing the differentiation and classification of Gram-negative, as well as Gram-positive strains with the same protocol. As expected, due to the differences in the cell wall structure, spectra of Gram-positive strains generally showed fewer peaks and a lower mass range than Gram-negative strains, agreeing with the results of other authors.31
As mentioned before, spectral reproducibility is very important to obtain representative fingerprints for every bacterial species and to allow spectral comparison. In the present work analysis by MALDI-TOF MS was carried out in quadruplicate for every strain to demonstrate biological and technical reproducibility. For spectra analysis, the web application SPECLUST was used to examine the four spectra of each sample, extracting representative peaks that were present in all four spectra. The mass variability was less than ±3 Da in the mass range lower than 7,000 Da and less than ±5 Da in the mass range above 7,000 Da, in accordance with mass errors described by other authors.18,19,35 In this way, final peak mass lists, each including 10–35 average peak masses, were generated and represented reproducible bacterial fingerprints. Although spectra obtained by MALDI-TOF MS can only be compared when the same protocol is applied, we observed a number of peak masses for some bacterial species that had already been described by other authors through the use of different protocols. Mazzeo et al. (2006) obtained very similar spectral profiles for the genera Pseudomonas, Proteus and Listeria, even though they analyzed suspensions of intact cells. Such characteristic peak masses that are present in the spectra, independent from the different protocols applied, could serve as biomarker peaks for the identification of the corresponding bacterial species.
The established protocol was applied for the study of the main seafood pathogenic and spoilage bacterial species obtained from culture collection (CECT) as well as isolated from seafood. A reference library was created and included spectral data of 22 genera and 53 different bacterial species.10,20 Furthermore, the method was successfully applied for the classification and identification of unknown bacterial strains isolated from different seafood samples (unpublished data). The comparison of the unknown spectral profiles with the library of reference spectra was carried out with the web-application SPECLUST that determined common peak masses between the studied peak mass lists, allowing the differentiation of the studied genera and species. Furthermore, the peak mass lists of all studied strains were clustered with the clustering option also available on the web-interface SPECLUST. In a previous study, we demonstrated that the obtained dendrogram showed a clear grouping on the genus- and species-level. In addition, we found a good correlation between the proteomic cluster and the phylogenetic analysis obtained by genetic tools.20
The applied sample preparation protocol together with the web-application SPECLUST that facilitates a rapid and simple data analysis, show that MALDI-TOF MS fingerprinting proves to be a fast and accurate technique for bacterial species differentiation. This technique can discriminate bacteria in less than two hours, including preparation of the samples, loading them into the MALDI-TOF machine, and analyzing them, compared with conventional testing methods, which take at least two days. Furthermore, MALDI-TOF MS fingerprinting represents an effortless, accurate and cost-effective technique, making it a potential new platform for routine identification of bacteria in clinical bacteriology as well as in the food sector.8 In this sense, some authors consider that, taking into account the cost of materials and staff, the cost of bacterial identification by MALDI-TOF MS is around two-thirds less than conventional methods.36,37 These authors even state that this approach will soon replace conventional pathogen testing for the identification of either common pathogens, or for some bacterial species that require time to grow, such as mycobacteria. Hsieh et al. (2008) asserted that the approach can identify bacteria with low abundance even in mixed flora. This can be a great advantage for seafood pathogenic and spoilage bacterial identification to ensure the safety and quality of seafood products.
Conclusions
Different sample preparation methods for bacterial species differentiation by MALDI-TOF MS fingerprinting were compared. The best results were achieved with the analysis of cell extracts, which exhibited highly reproducible spectral profiles with low noise and a good peak resolution. The protocol involving the preparation of cell extracts proved to be very fast and labor-saving. Furthermore, cell extracts can be prepared before analysis and may be stored frozen for a long time. In addition, the established sample preparation protocol for the analysis of bacterial cell extracts can be easily standardized and can be successfully applied for the identification of bacterial strains isolated from seafood by comparison of spectral profiles with those of the reference library.Acknowledgements
This work was funded by the PGIDIT Research Program (Project PGIDIT06PXIB261164PR) of the Xunta de Galicia (Galician Council for Industry Commerce and Innovation). The work of K. Bohme and I.C. Fernandez-No is supported by a “Maria Barbeito” and “Lucas Labrada” research contract from Xunta de Galicia.Notes and references
- D. Mohania, R. Nagpal, M. Kumar, A. Bhardwaj, M. Yadav, S. Jain, F. Marotta, V. Singh, O. Parkash and H. Yadav, J. Dig. Dis., 2008, 9, 190–198 Search PubMed.
- P. Feng, Rapid Methods for the Detection of Foodborne Pathogens: Current and Next-Generation Technologies, in Food Microbiology: Fundamentals and Frontiers, ed. M. P. Doyle; L. R. Beuchat, ASM Press, Washington, D.C., 3rd edn, 2007, ch. 43, pp. 911–934 Search PubMed.
- C. P. Kolbert and D. H. Persing, Curr. Opin. Microbiol., 1999, 2, 299–305 CrossRef CAS.
- T. R. Klaenhammer; E. Pfeiler; T. Duong, Genomics and Proteomics of Foodborne Microorganisms, in Food Microbiology: Fundamentals and Frontiers, ed. M. P. Doyle; L. R. Beuchat, ASM Press, Washington, D.C., 3rd edn, 2007, ch. 44, pp. 935–951 Search PubMed.
- S. C. Russell, Mass Spectrom. Rev., 2009, 28, 376–387 CrossRef CAS.
- B. L. M. van Baar, FEMS Microbiol. Rev., 2000, 24, 193–219 CrossRef CAS.
- J. O. Lay Jr., Mass Spectrom. Rev., 2001, 20, 172–194 CrossRef CAS.
- P. Seng, M. Drancourt, F. Gouriet, B. La Scola, P.-E. Fournier, M. JeanRolain and D. Raoult, Clin. Infect. Dis., 2009, 49, 543–551 CrossRef CAS.
- J. J. Bright, M. A. Claydon, M. Soufian and D. B. Gordon, J. Microbiol. Methods, 2002, 48, 127–138 CrossRef CAS.
- I. C. Fernández-No, K. Böhme, J. M. Gallardo, J. Barros-Velázquez, B. Cañas and P. Calo-Mata, Electrophoresis, 2010, 31, 1116–1127 CAS.
- T. Krishnamurthy and P. L. Ross, Rapid Commun. Mass Spectrom., 1996, 10, 1992–1996 CrossRef CAS.
- R. D. Holland, C. R. Duffy, F. Rafii, J. B. Sutherland, T. M. Heinze, C. L. Holder, K. J. Voorhees and J. O. Lay Jr, Anal. Chem., 1999, 71, 3226–3230 CrossRef CAS.
- F. J. Pineda, M. D. Antoine, P. A. Demirev, A. B. Feldman, J. Jackman, M. Longenecker and J. S. Lin, Anal. Chem., 2003, 75, 3817–3822 CrossRef CAS.
- P. A. Demirev, A. B. Feldman and J. S. Lin, John Hopkins APL Technical Digest, 2004, 25, 27–37 Search PubMed.
- D. Dare, Rapid Bacterial Characterization and Identification by MALDI-TOF Mass Spectrometry, in Advanced Techniques in Diagnostic Microbiology, ed. Y.-W. Tang; C. W. Stratton, Springer Science + Business Media, LLC, New York, 2006, ch. 7, pp. 117–133 Search PubMed.
- C. J. Keys, D. J. Dare, H. Sutton, G. Wells, M. Lunt, T. McKenna, M. McDowall and H. N. Shah, Infect. Genet. Evol., 2004, 4, 221–242 CrossRef CAS.
- M. Erhard, U.-C. Hipler, A. Burmester, A. A. Brakhage and J. Wöstemeyer, Exp. Dermatol., 2008, 17, 356–361 CrossRef.
- N. Degand, E. Carbonnelle, B. Dauphin, J.-L. Beretti, M. Le Bourgeois, I. Sermet-Gaudelus, C. Segonds, P. Berche, X. Nassif and A. Ferroni, J. Clin. Microbiol., 2008, 46, 3361–3367 CrossRef CAS.
- M. F. Mazzeo, A. Sorrentino, M. Gaita, G. Cacace, M. Di Stasio, A. Facchiano, G. Comi, A. Malorni and R. A. Siciliano, Appl. Environ. Microbiol., 2006, 72, 1180–1189 CrossRef CAS.
- K. Böhme, I. C. Fernández-No, J. Barros-Velázquez, J. M. Gallardo, P. Calo-Mata and B. Cañas, J. Proteome Res., 2010, 9, 3169–3183 CrossRef CAS.
- N. Valentine, S. Wunschel, D. Wunschel, C. Petersen and K. Wahl, Appl. Environ. Microbiol., 2005, 71, 58–64 CrossRef CAS.
- D. S. Wunschel, E. A. Hill, J. S. McLean, K. Jarman, Y. A. Gorby, N. Valentine and K. Wahl, J. Microbiol. Methods, 2005, 62, 259–271 CrossRef CAS.
- Z. Wang, L. Russon, L. Li, D. C. Roser and S. R. Long, Rapid Commun. Mass Spectrom., 1998, 12, 456–464 CrossRef.
- R. J. Arnold, J. A. Karty, A. D. Ellington and J. P. Reilly, Anal. Chem., 1999, 71, 1990–1996 CrossRef CAS.
- R. J. Arnold and J. P. Reilly, Rapid Commun. Mass Spectrom., 1998, 12, 630–636 CrossRef CAS.
- K. H. Jarman, D. S. Daly, K. K. Anderson and K. L. Wahl, Chemom. Intell. Lab. Syst., 2003, 69, 61–76 CrossRef CAS.
- T. L. Williams, D. Andrzejewski, J. O. Lay and S. M. Musser, J. Am. Soc. Mass Spectrom., 2003, 14, 342–351 CrossRef CAS.
- R. D. Holland, J. G. Wilkes, F. Rafii, J. B. Sutherland, C. C. Persons, K. J. Voorhees and J. O. Lay Jr., Rapid Commun. Mass Spectrom., 1996, 10, 1227–1232 CrossRef CAS.
- R. Alm, P. Johansson, K. Hjerno, C. Emanuelsson, M. Ringnér and J. Häkkinen, J. Proteome Res., 2006, 5, 785–792 CrossRef CAS.
- R. Giebel, C. Worden, S. M. Rust, G. T. Kleinheinz, M. Robbins, T. R. Sandrin, I. L. Allen, S. Sima, M. G. Geoffrey, Microbial Fingerprinting using Matrix-Assisted Laser Desorption Ionization Time-Of-Flight Mass Spectrometry (MALDI-TOF MS): Applications and Challenges, in Adv. Appl. Microbiol., Academic Press, 2010, Volume 71, pp. 149–184 Search PubMed.
- M. Vargha, Z. Takáts, A. Konopka and C. H. Nakatsu, J. Microbiol. Methods, 2006, 66, 399–409 CrossRef CAS.
- E. Carbonnelle, J.-L. Beretti, S. Cottyn, G. Quesne, P. Berche, X. Nassif and A. Ferroni, J. Clin. Microbiol., 2007, 45, 2156–2161 CrossRef CAS.
- K. Bernardo, N. Pakulat, M. Macht, O. Krut, H. Seifert, S. Fleer, F. Hünger and M. Krönke, Proteomics, 2002, 2, 747–753 CrossRef CAS.
- S. C. Smole, L. A. King, P. E. Leopold and R. D. Arbeit, J. Microbiol. Methods, 2002, 48, 107–115 CrossRef CAS.
- M. J. Donohue, A. W. Smallwood, S. Pfaller, M. Rodgers and J. A. Shoemaker, J. Microbiol. Methods, 2006, 65, 380–389 CrossRef CAS.
- S.-Y. Hsieh, C.-L. Tseng, Y.-S. Lee, A.-J. Kuo, C.-F. Sun, Y.-H. Lin and J.-K. Chen, Mol. Cell. Proteomics, 2008, 7, 448–456 CAS.
- X. Nassif, Clin. Infect. Dis., 2009, 49, 552–553 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |