Chemical fingerprint analysis for quality control of Fructus Aurantii Immaturus based on HPLC-DAD combined with chemometric methods
Xiaona
Xu
ab,
Junhui
Jiang
b,
Yizeng
Liang
*a,
Lunzhao
Yi
a and
Jinle
Cheng
c
aCollege of Chemistry and Chemical Engineering, Research Center of Modernization of Chinese Medicines, Central South University, 410083, Changsha, Hunan, People's Republic of China. E-mail: yizeng_liang@263.net; Fax: +86-731-8825637
bCollege of Chemistry and Chemical Engineering, University of South China, 421001, Hengyang, Hunan, People's Republic of China
cZhongzhi pharmaceutical limited company, 528415, Zhongshan, Guangdong, People's Republic of China
First published on 15th October 2010
Abstract
High-performance liquid chromatographic (HPLC) fingerprints of 12 authentic samples and 26 commercial samples of Fructus Aurantii Immaturus (FAI) from different sources of China were measured. Firstly, the main bioactive compounds in citrus herb, namely naringin and hesperidin, were identified and quantitated. Then, principal components analysis (PCA) was performed to differentiate and classify the studied samples based on the principal peaks and entire chromatograms, respectively. Hierarchical clustering analysis (HCA) and similarity analysis (SA) were further conducted to validate the clustering results. The optimized HPLC-DAD method aided by forementioned chemometric methods shows that the chemical fingerprints of 38 samples from extensive sources could be reasonably identified and systematically analyzed, which offers a new clue for the study of traditional Chinese medicines (TCM). The results obtained will provide a useful foundation for quality control of herbal medicine.
1 Introduction
Fructus Aurantii Immaturus (FAI) is an effective traditional Chinese medicine (TCM) used for more than 2000 years in the treatment of dissipating stagnant qi, removing qi stagnation, eliminating sputum and dispersing painful abdominal mass. It is the dried, unripe fruit of Citrus aurantium L., cultivated variety or citrus sinensis Osbeck of Rutaceae, often called Zhishi in Chinese and officially listed in the Chinese Pharmacopoeia.1 Flavonoids, alkaloids and essential oil components are the major bioactive compositions in this herb. Hesperidin and naringin, the important representative flavonoids in citrus herb, possess antioxidant, anti-inflammatory and anti-ulcer properties as most flavonoids do.2–4It is well known that traditional Chinese medicines (TCMs) contain multiple chemical components, each of which may be relevant to the medicine's putative activity. The various origins, different processing procedures, saving conditions and deposited year may lead to the difference in the contents of chemical constituents and compound denominations of herbal medicine, which may directly influence therapeutic effects. Therefore, discrimination of an herbal material's origin as well as determination of its bioactive ingredients is crucial in order to ensure its authenticity, quality, safety and efficacy. Until now, besides macroscopic and microscopic authentication, chemical identification of herbal materials with various pattern recognition methods, such as similarity analysis (SA), hierarchical clustering analysis (HCA) and principal component analysis (PCA), is increasingly employed to discriminate the habitats of raw herbal materials.5–12
Publications about FAI are mostly about one or several compounds' qualitation and quantification in HPLC-UV, HPLC-UV-MS and HPLC-MS-MS, mainly hesperidin, naringin and synephrine.13–18 Although there are some publications associated with the fingerprints of FAI, none of them involved systemic comparison among different herbal origins, identification using characteristic chemical compounds or a discrimination model. In the present study, a HPLC fingerprint method in combination with chemometric methods to analyze FAI from various origins was described. To begin with, the main bioactive compounds in citrus herb, namely naringin and hesperidin, were identified and quantitated. Then, principal components analysis (PCA) was performed to differentiate and classify the studied samples based on the principal peaks and entire chromatograms, respectively. Then, hierarchical clustering analysis (HCA) and similarity analysis (SA) were further conducted to validate the clustering results adjunctively. Finally, the HPLC fingerprints of 38 samples from extensive sources were roundly analyzed and identified. This investigation shows that the developed methodology can be generalized in the research of quality control of herb medicines. The results obtained may also provide a useful chemical basis for future research of this herb.
2 Experimental
2.1 Chemicals and reagents
The standards of hesperidin (batch number 0721-9909) and naringin (batch number 0722-9805) were purchased from National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Their chemical structures are shown in Fig. 1. HPLC grade methanol, acetonitrile and polyphosphoric acid were purchased from Jiangsu Hanbon Science and Technology Co., Ltd (Huaian, Jiangsu Province, China). Other reagents were of analytical grade. Water was deionized in a Milli-Q water purification system (Millipore Bedford, MA, USA). | ||
| Fig. 1 Chemical structures of naringin and hesperidin. | ||
2.2 Plant materials
Thirty-eight batches of samples from eighteen provinces and one municipality of China including 12 authentic samples and 26 commercial samples were authenticated morphologically and microscopically as Fructus Aurantii Immaturus by Mr. Xiangqian Liu, working at Central South University, Changsha, Hunan, P. R. China (see Table 1). Voucher specimens were preserved at our lab.| Sample no. | Sources | Habitat | Production time |
|---|---|---|---|
| 01 | Purchased from Xian, Shanxi | Jiangxi | 2008 |
| 02 | Purchased from Jinxiang Drug Store, Beijing | unknown | 2008 |
| 03 | Purchased from Zhonglian Drug Store, Wuhan, Hubei | Jiangxi | 2009 |
| 04 | Purchased from Hefei, Anhui | unknown | 2009 |
| 05 | Purchased from Shengyuan Drug Store, Nanning, Guangxi | Hunan | 2008 |
| 06 | Purchased from Changchun, Jilin | Hunan | 2009 |
| 07 | Purchased from Hengkang pharmaceutical company, Yiyang, Hunan | unknown | unknown |
| 08 | Hunan (authentic sample) | Hunan | 2009 |
| 09 | Hunan (authentic sample) | Hunan | 2009 |
| 10 | Purchased from, Yunnan | Yunnan | 2009 |
| 11 | Purchased from Dongbei Drug Store, Shenyang, Liaoning | Hunan | 2009 |
| 12 | Purchased from Taiyuan, Shanxi | Unknown | 2008 |
| 13 | Xinyu, Jiangxi (authentic sample) | Jiangxi | 2009 |
| 14 | Xinyu, Jiangxi (authentic sample) | Jiangxi | 2009 |
| 15 | Purchased from Guilin, Guangxi | Hunan | 2008 |
| 16 | Purchased from Haikou, Hainan | Unknown | 2008 |
| 17 | Purchased from Harbin, Heilongjiang | Unknown | 2008 |
| 18 | Purchased from Kangtai Drug Store, Hengyang, Hunan | Unknown | 2009 |
| 19 | Qingjiang, Jiangxi (authentic sample) | Jiangxi | 2009 |
| 20 | Qingjiang, Jiangxi (authentic sample) | Jiangxi | 2009 |
| 21 | Purchased from Huaihua, Hunan | Hunan | 2008 |
| 22 | Purchased from Zhuji, Zhejiang | Unknown | 2008 |
| 23 | Purchased from Xining, Qinghai | Unknown | 2009 |
| 24 | Purchased from Xinjiang | unknown | 2008 |
| 25 | Deyang, Sichuan (authentic sample) | Sichuan | 2009 |
| 26 | Deyang, Sichuan (authentic sample) | Sichuan | 2009 |
| 27 | Purchased from Xian, Shanxi | Jiangxi | 2008 |
| 28 | Purchased from Linxin, Shandong | Jiangxi | 2009 |
| 29 | Purchased from Chenzhou, Hunan | unknown | 2009 |
| 30 | Purchased from Chenzhou, Hunan | unknown | 2008 |
| 31 | Purchased from Dongguan, Guangdong | Hunan | 2008 |
| 32 | Purchased from Kunming City zhongyiyuan, Yunnan | Unknown | Unknown |
| 33 | Purchased from Yiyang, Hunan | Hunan | 2008 |
| 34 | Panxi, Yunnan (authentic sample) | Yunnan | 2009 |
| 35 | Panxi, Yunnan (authentic sample) | Yunnan | 2009 |
| 36 | Purchased from Lanzhou, Gansu | Unknown | 2009 |
| 37 | Yiyang, Hunan (authentic sample) | Hunan | 2008 |
| 38 | Yiyang, Hunan (authentic sample) | Hunan | 2008 |
2.3 Preparation of standard and sample solutions
The reference compounds were weighed accurately and dissolved in methanol in a 10 mL volumetric flask to make stock solutions. Working standard solutions were prepared from those stock solutions by further dilution with the appropriate volume of methanol. These solutions were stored away from light at 4 °C in a refrigerator.Pulverized sample (60 mesh, 0.5 g) was weighed accurately into a 150-mL round bottom flask, then extracted for 10 min at room temperature under ultrasound with 25 mL methanol and finally filtered. The extraction was repeated two additional times. The extracts were combined and preserved in refrigerator.
All standard solutions, samples and solvents used for HPLC measurements were filtered through a 0.22 μm nylon filter membrane before HPLC analysis.
2.4 Instrumentation and chromatographic conditions
An ultrasonic cleaner made in Kunshan ultrasonic Instrumentation Factory (Kunshan, Jiangsu, PR China) was used for extraction. All HPLC analyses were performed using Agilent/HP 1100 Series HPLC-DAD system consisting of a vacuum degasser, quaternary pump, manual-sampler, thermostated column compartment and DAD (Agilent, Palo Alto, CA, USA). A Sepax C18 (5 μm, 250 mm × 4.6 mm) column protected by a suitable guard column (C18, 5 μm, 7.5 mm × 4.6 mm) were used for all chromatographic separations. The mobile phase consisted of acetonitrile, methanol and 0.05% polyphosphoric acid water using a gradient program (see Table 2). The flow rate was 0.8 mL min−1, column temperature was maintained at 30 °C and the injection volume was 20 μL. The DAD detector was set at 284 nm for acquiring chromatograms. UV spectra were acquired from 200 to 400 nm.| Gradient/min | A (CH3CN)% | B (CH3OH)% | C (0.05% H3PO4–H2O)% |
|---|---|---|---|
| 0 | 6.0 | 3.0 | 91.0 |
| 10 | 16.0 | 13.0 | 71.0 |
| 30 | 16.0 | 13.0 | 71.0 |
| 35 | 18.0 | 18.0 | 64.0 |
| 45 | 25.0 | 35.0 | 40.0 |
| 70 | 25.0 | 70.0 | 5.0 |
| 80 | 25.0 | 70.0 | 5.0 |
2.5 Data analysis
HCA is an unsupervised statistical method, which provides a way to classify target objects into several groups based on measured characteristics. It starts with each sample in a separate cluster and then combines the clusters sequentially, reducing the number of clusters at each step until only one cluster left. When there are N samples, there involves N − 1 clustering steps or fusions. This hierarchical clustering process can be delineated as a tree or dendrogram, where each step in the clustering process is illustrated by a joint of the tree.31–33 In this study, the similarity measure is usually represented by the distance between two samples. The shorter distance between two samples represents their higher similarity.
All the data were pretreated including background deduction, smoothing and the chromatograms' alignment before SA, HCA and PCA.
3 Results and discussion
3.1 Optimization of extraction and chromatographic conditions
The efficiency of the extraction procedure was evaluated by using various extraction solvents and different extraction methods. Ethanol, methanol, and their respective different ratio aqueous solutions were used to extract the herb material respectively, and three extraction techniques, say maceration, reflux and ultrasound, were carried out. The results demonstrated that the optimum solvent was found to be anhydrous methanol, and ultrasound was validated as the optimal extraction method, both of which enabled less interfering peaks, and provided the biggest peak area of the principal peaks and peak number.A method involving three-factor-three-level orthogonal array design (OAD) including the components of volume of methanol (5, 15, and 25 mL), times of ultrasound (one, two and three times) and duration of extraction (10, 30, and 60 min) was developed for the optimization of the extraction. The results demonstrated that the established extraction method was adequate and appropriate for the analysis (see Table 3).
| No. | Factors | Total extraction rate (%) | ||
|---|---|---|---|---|
| A | B | C | ||
| a A: Extraction number; B: Extraction time (min); C: Ratio of liquid to solid. b K i = ∑total extraction rate at Ai; the sum of the total extraction rates for a certain factor at each level. c R is the extreme difference of Ki (i = 1, 2, 3) for a certain factor. | ||||
| 1 | 1 (1) | 1 (10) | 1 (10) | 13.63 |
| 2 | 1 | 2 (30) | 2 (30) | 16.52 |
| 3 | 1 | 3 (60) | 3 (50) | 17.11 |
| 4 | 2 (2) | 1 | 2 | 17.88 |
| 5 | 2 | 2 | 3 | 17.58 |
| 6 | 2 | 3 | 1 | 17.15 |
| 7 | 3 (3) | 1 | 3 | 19.87 |
| 8 | 3 | 2 | 1 | 17.04 |
| 9 | 3 | 3 | 2 | 17.01 |
| K 1 | 47.26 | 51.10 | 47.82 | |
| K 2 | 48.61 | 51.14 | 51.41 | |
| K 3 | 53.92 | 51.27 | 54.56 | |
| R | 7.16 | 0.17 | 6.74 | |
The optimization of the chromatographic conditions was performed by using the sample solution. The investigated compounds were tested and compared by using different analytical columns (Phenomenex LUNA C18, Agilent XDB-C8 or Sepax C18) with different mobile phases (acetonitrile–water, methanol–water, and acetonitrile–method–water) and different gradient elution programs. The results showed that Sepax C18 column with gradient elution of acetonitrile–method–water could efficiently separate the investigated markers. Considering the presence of flavonoids in the herbal extraction, 0.05% phosphoric acid was added to the mobile phase to reduce the ionization and lower the polarity of these compounds. According to the UV spectra recorded by DAD from 200 to 400 nm, 284 nm was finally chosen to achieve chromatographic fingerprint profiles and detection wavelength based on the peak area of principal peaks and peak number. The representative HPLC-DAD chromatograms of mixed standards and the extract of FAI are shown in Fig. 2.
 | ||
| Fig. 2 Representative HPLC-DAD chromatograms of mixed standards and the extract of Fructus Aurantii Immaturus (sample no. 32 in Table 1). Peak 2 and 3 are naringin and hesperidin, respectively. Peaks 1, 4 and 5 are also flavonoids compounds according to the similarities of their UV spectra with the standards naringin and hesperidin, they displaying the characteristic UV maximum absorbance of flavonoids. For terseness, the identification courses of the three peaks forementioned are omitted in this paper. | ||
3.2 Validation of the HPLC method
Each calibration curve was performed with six different concentrations of chemical markers. The square of all the correlation coefficients (r2) of these calibration curves were higher than 0.999. The limits of detection (LOD) and quantification (LOQ) were determined at a signal-to-noise ratio (S/N) of 3 and 10, respectively. The data were summarized in Table 4. Precision (inter-day and intra-day), repeatability, stability and recovery were all carried out to validate the HPLC method, following some reports on determination analysis.36–38| Compound | Regression equation | Correlation coefficient | Linear range/μg mL−1 | LOD/μg mL−1 | LOQ/μg mL−1 |
|---|---|---|---|---|---|
| hesperidin | Y = 5.048 × 104X − 175.3 | 0.9992 | 2–300 | 0.05 | 0.16 |
| naringin | Y = 5.207 × 104X − 128.1 | 0.9995 | 2–256 | 0.06 | 0.19 |
3.3 Sample analysis
The established analytical method was applied to determine the contents of naringin and hesperidin in the 38 samples, which are the main bioactive ingredients in citrus herb. Identification of the compounds forementioned of the tested samples was based on comparing their UV spectra and their retention time with those of the standards. The contents of naringin and hesperidin in the tested samples were listed in Table 5. The measurable concentration ranges of the two components were in 0.143–6.267 and 1.355–9.456 mg g−1, which demonstrated the striking concentration disparity of naringin and hesperidin in herbal materials. The highest contents of the two bioactive compounds are more than five times the lower ones, respectively. Even in some samples naringin had not been detected or existed with very small contents so that it could not be accurately quantitated. Such consequences were in line with the Ref. 39. Flavonoids are the major effective ingredients in citrus herb, of which naringin and hesperidin are of conclusive therapeutic effect.40–42 Therefore, the quantitation standards of the two compounds above-mentioned should be instituted by correlative institutions as soon as possible, which will be a great assistance to quality control of this herb. Further studies are required to find out the influence of the compounds' concentration diversity of TCM on the therapeutic effect.| Sample no. | Contents/mg g−1 ± S.D. | |
|---|---|---|
| Naringin | Hesperidin | |
| a Sample no. corresponds to Table 1. | ||
| 01 | 3.335 ± 0.011 | 6.176 ± 0.022 |
| 02 | 2.920 ± 0.003 | 5.317 ± 0.015 |
| 03 | 4.828 ± 0.007 | 1.355 ± 0.001 |
| 04 | 2.836 ± 0.005 | 1.526 ± 0.002 |
| 05 | 0.160 ± 0.001 | 7.060 ± 0.002 |
| 06 | trace | 7.907 ± 0.028 |
| 07 | 2.781 ± 0.004 | 4.693 ± 0.009 |
| 08 | 0.210 ± 0.001 | 8.198 ± 0.017 |
| 09 | 0.299 ± 0.001 | 7.873 ± 0.024 |
| 10 | trance | 6.632 ± 0.015 |
| 11 | 0.271 ± 0.002 | 8.686 ± 0.051 |
| 12 | 3.667 ± 0.017 | 2.062 ± 0.002 |
| 13 | 5.851 ± 0.021 | 1.526 ± 0.002 |
| 14 | 4.718 ± 0.004 | 1.840 ± 0.003 |
| 15 | trace | 6.946 ± 0.007 |
| 16 | 0.143 ± 0.000 | 5.577 ± 0.006 |
| 17 | trace | 2.867 ± 0.005 |
| 18 | 0.340 ± 0.001 | 6.062 ± 0.006 |
| 19 | 5.935 ± 0.001 | 2.468 ± 0.002 |
| 20 | 6.267 ± 0.006 | 1.549 ± 0.007 |
| 21 | 0.165 ± 0.000 | 7.973 ± 0.006 |
| 22 | 2.693 ± 0.003 | 2.627 ± 0.002 |
| 23 | 0.149 ± 0.000 | 9.456 ± 0.010 |
| 24 | trace | 7.000 ± 0.006 |
| 25 | 0.160 ± 0.000 | 7.060 ± 0.004 |
| 26 | 0.487 ± 0.001 | 6.886 ± 0.010 |
| 27 | 4.717 ± 0.005 | 5.634 ± 0.009 |
| 28 | 3.075 ± 0.002 | 4.966 ± 0.005 |
| 29 | 3.324 ± 0.001 | 3.637 ± 0.003 |
| 30 | 3.704 ± 0.002 | 2.624 ± 0.001 |
| 31 | trace | 6.436 ± 0.003 |
| 32 | 0.951 ± 0.000 | 4.667 ± 0.002 |
| 33 | 0.277 ± 0.001 | 6.658 ± 0.013 |
| 34 | trace | 7.427 ± 0.008 |
| 35 | trace | 6.547 ± 0.007 |
| 36 | 0.697 ± 0.005 | 7.685 ± 0.009 |
| 37 | 0.264 ± 0.001 | 7.300 ± 0.007 |
| 38 | 0.215 ± 0.000 | 7.060 ± 0.06 |
3.4 Principal component analysis (PCA)
 | ||
| Fig. 3 PCA projection plot for the 38 samples, using peak areas of 5 principally common components as input data (peaks 1–5 in Fig. 2). | ||
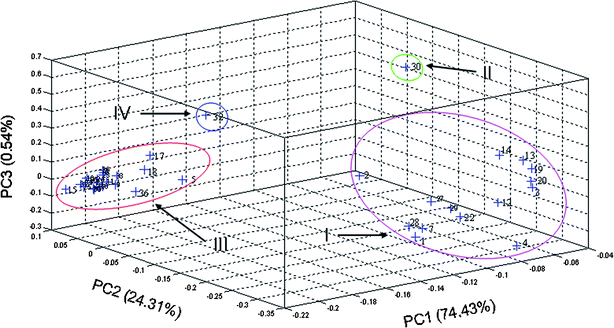 | ||
| Fig. 4 The scatter plot obtained by PCA of the tested samples based on entire chromatograms. | ||
All programs of PCA were coded in MATLAB 6.5 for windows.
3.5 Hierarchical clustering analysis (HCA)
In order to judge the reliability of the categorization results of PCA forementioned and compare them with those of other classification methods, HCA was utilized to classify those 38 fingerprints. Centroid Euclidean distance was selected as a measurement. The hierarchical clustering was done by Traditional Chinese medicine quality control system/extendable data base, which was developed by our laboratory. A dendrogram was generated (see Fig. 5), which revealed the relationships among the studied samples. Generally, the thirty-eight chromatograms were divided into two basal clusters. The authentic samples (No. 13, 14, 19 and 20) from Jiangxi together with sample nos. 1–4, 7, 12, 22 and 27–30 were in group I and the other samples including the authentic samples from Hunan were in group II, which was again divided into subgroups, respectively. Sample nos. 1–4, 7, 12–14, 19, 20, 22, and 27–29 were in subgroup A, 30 and 32 was in subgroup B and D respectively, and the rest ones centered by the authentic samples from Hunan, Sichuan and Yunnan, were in subgroup C. The results of HCA indicated that samples which had similar chemical profiles were divided into one group. Here, sample nos. 30 and 32 are again discriminated as singular points. The result of HCA is similar with that of PCA based on entire chromatograms, and further confirms the latter. Both of them were based on entire fingerprints when classification done. It is obvious that overall fingerprints have larger abilities to reveal the variability of the tested samples than partial principal peaks when conducting discrimination analysis. | ||
| Fig. 5 The result of hierarchic clustering analysis. | ||
3.6 Similarity analysis of HPLC fingerprints
Based on the definition of the fingerprint of TCM, a chromatographic fingerprint is actually a chromatographic pattern of some common kinds of pharmacologically active and chemically characteristic components in the TCM. “Integrity” and “fuzziness” are two fundamental attributes of fingerprints of TCMs. With the help of chromatographic fingerprints, the authentication and identification of herbal medicines can be conducted, even if the quantity of the chemically characteristic constituents are not exactly the same among different samples. The chromatographic fingerprints reveal both the “sameness” and “differences” between various samples successfully. In this study, the 38 samples from different origins in China were analyzed under the optimum chromatographic conditions.Similarity analysis is a conventional method describing the similarity and dissimilarity among the fingerprints quantitatively. Here, similarity analysis was carried out on the 38 fingerprints by a Traditional Chinese medicine quality control system/extendable database developed by our laboratory. The correlation coefficient of similarity between each chromatographic profile of FAI samples and the simulative median chromatogram was calculated and generated, respectively (see Table 6). According to the results of Table 6, taking 0.90 as an appropriate threshold, the samples with the correlation coefficients above it will be clustered to a group, and those below it will be sorted to another group, which has been properly proved by the previous cluster results from PCA and HCA.
| Samplea | Similaritiesb |
|---|---|
| a The sample number from 01 to 38 are corresponding to the serial numbers as listed in Table 1. b The correlation coefficient was calculated based on the information obtained from the median of all chromatograms. | |
| 01 | 0.522 |
| 02 | 0.707 |
| 03 | 0.182 |
| 04 | 0.223 |
| 05 | 0.979 |
| 06 | 0.984 |
| 07 | 0.505 |
| 08 | 0.996 |
| 09 | 0.998 |
| 10 | 0.993 |
| 11 | 0.995 |
| 12 | 0.274 |
| 13 | 0.199 |
| 14 | 0.264 |
| 15 | 0.992 |
| 16 | 0.994 |
| 17 | 0.964 |
| 18 | 0.998 |
| 19 | 0.180 |
| 20 | 0.181 |
| 21 | 0.994 |
| 22 | 0.384 |
| 23 | 0.989 |
| 24 | 0.990 |
| 25 | 0.997 |
| 26 | 0.998 |
| 27 | 0.483 |
| 28 | 0.539 |
| 29 | 0.420 |
| 30 | 0.537 |
| 31 | 0.994 |
| 32 | 0.954 |
| 33 | 0.994 |
| 34 | 0.990 |
| 35 | 0.992 |
| 36 | 0.993 |
| 37 | 0.997 |
| 38 | 0.995 |
In order to see the details in the fingerprints, the median chromatograms of the two groups of FAI together with the two outliers are shown in Fig. 6. Peaks that existed in all chromatograms are considered as “common peaks”, indicating the sameness among various samples. 18 peaks coexisting in the 38 samples are indicated for visual comparison of the tested samples. Among the common components, the contents of hesperidin and narirutin are relatively high and stable, which were in correspondence with the literatures reported.11–13 Peak M and N existing in Fig. 6 (1) are not found in Fig. 6 (2), while peak P emerging in Fig. 6 (2) does not exist in Fig. 6 (1). Furthermore, there are many tiny peaks in Samples no. 30 and 32 with the retention time range 40–70 min (see Fig. 6 (3) and Fig. 6 (4)). The non-common peaks in each chromatogram represent the fuzziness among the same kind of TCM along with the different contents of the same component existing in the samples examined. It is in accordance with the theory that the secondary metabolites of plant herbs would clearly vary in different locations. Chinese medicine theory emphasizes synergism of multiple components to enhance remedy, lower toxicity and prevent side effects. Different constituents synergistically interact together and generate different curative effects. Further work should be probed on the variance of pharmacodynamic action of FAI from different origins.
 | ||
| Fig. 6 Mean chromatograms of (1) 4 authentic samples including four from Jiangxi (sample no. 12, 13, 19 and 20) and 10 commercial samples (sample no. 1–4, 7, 14, 22, 27–29); (2) 6 authentic samples including four from Hunan (sample no. 08, 09, 37 and 38) and 2 from Sichuan (sample no. 34 and 35), and 2 from Yunnan (sample no. 25 and 26), and 16 commercial samples. (3) and (4) are the chromatograms of sample no. 30 and 32 (commercial samples), respectively. The sample numbers were coincident in Table 1. The five peaks 6–9 and 11 are corresponding to peaks 1–5 in Fig. 2, respectively. The other compounds cannot be identified. | ||
4 Conclusions
The developed chromatographic method had been employed to identify and determine the two effective components, say naringin and hesperidin. With the help of chemometric methods, say PCA, HCA and SA, the 38 herbal samples of FAI from various sources can be accurately differentiated. The method developed in this paper has demonstrated that it is able to discriminate between different herbal origins, detect the content of bioactive ingredients and systematically analyze traditional Chinese medicines, which offers a new clue for the study of traditional Chinese medicine. The results obtained will be helpful for quality control, rational herbal usage and pharmacodynamic research of herbal medicine.Acknowledgements
The authors gratefully acknowledge the financial support from the International Co-operative Project of Science and Technology from the Ministry of Science and Technology of People's Republic of China (contract/grant number: 2007DFA40680), National Nature Foundation Committee of P.R. China (Grant No. 20875104 and 21075138), China Postdoctoral Science Foundation funded project (No. 20080440181), Special Foundation of China Postdoctoral Science (No. 200902481) and Central South University Science Development Foundation (No. 10SDF22).References
- Chinese Pharmacopoeia Committee. Chinese Pharmacopoeia [M]. Publishing house of chemical industry, Beijing, 2005, pp. 118–119 Search PubMed.
- Y. T. Chen, R. L. Zheng, Z. J. Jia and Y. Ju, Free Radical Biol. Med., 1990, 9, 19–21 CrossRef CAS.
- S. V. Jovanovic, S. Steenken, M. Tosic, S. Steenken, B. Marjanovic and M. G. Simic, J. Am. Chem. Soc., 1994, 116, 4846–4851 CrossRef CAS.
- M. J. Martín, E. Marhuenda, C. Pérez-Guerrero and J. M. Franco, Pharmacology, 1994, 49, 144–150 CrossRef CAS.
- P. Zou, Y. Hong and H. L. Koh, J. Pharm. Biomed. Anal., 2005, 38, 514–520 CrossRef CAS.
- L. F. Hu, S. P. Li, H. Cao, J. J. Liu, J. L. Gao, F. Q. Yang and Y. T. Wang, J. Pharm. Biomed. Anal., 2006, 42, 200–206 CrossRef CAS.
- B. Y. Li, Y. Hu, Y. Z. Liang, P. S. Xie and Y. Ozaki, Analyst, 2006, 131, 538–546 RSC.
- K. Y. L. Yap, S. Y. Chan and C. S. Lim, Food Res. Int., 2007, 40, 643–652 CrossRef CAS.
- Y. Zhao, Z. W. Li, X. Zhou, Z. W. Cai, X. J. Gong and C. Y. Zhou, J. Pharm. Biomed. Anal., 2008, 48, 1230–1236 CrossRef CAS.
- W. J. Kong, Y. L. Zhao, X. H. Xiao, C. Jin and Z. L. Li, Phytomedicine, 2009, 16, 950–959 CrossRef CAS.
- Y. Y. Zhao, Y. M. Zhang, R. C. Lin and W. J. Sun, Fitoterapia, 2009, 80, 333–338 CrossRef CAS.
- L. Z. Yi, D. L. Yuan, Y. Z. Liang, P. S. Xie and Y. Zhao, Anal. Chim. Acta, 2009, 649, 43–51 CrossRef CAS.
- P. Mouly, E. M. Gaydou and A. Auffracy, J. Chromatogr., A, 1998, 800, 171–179 CrossRef CAS.
- L. Ding, X. B. Luo, F. Tang, J. B. Yuan, Q. Liu and S. Z. Yao, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 857, 202–209 CrossRef CAS.
- X. M. Qin, Y. T. Dai, L. Z. Zhang, X. Q. Guo and H. X. Shao, Phytochem. Anal., 2009, 20, 307–313 CrossRef CAS.
- Y. Lu, C. Zhang, P. Bucheli and D. Wei, Plant Foods Hum. Nutr., 2006, 61, 57–65 CAS.
- C. Wand, Y. J. Pan, G. R. Fan, Y. F. Chai and Y. T. Wu, Biomed. Chromatogr., 2010, 24, 235–244 CAS.
- Q. Wang and Y. Dan, Heilongjiang Med. J., 2008, 21, 1–3 Search PubMed.
- T. Layloff, Pharm. Technol., 1991, 15, 146–148 Search PubMed.
- WHO, Guildlines for the assessment of herbal medicine. WHO: Geneva, 1996 Search PubMed.
- Drug Administration Bureau of China, Requirements for studying fingerprint of traditional Chinese medicine injection, Beijing, 2002 Search PubMed.
- W. J. Welsh, W. K. Lin, S. H. Tersigni, E. Collantes, R. Duta and M. S. Carey, Anal. Chem., 1996, 68, 3473–3482 CrossRef CAS.
- E. R. Collantes, R. Duta, W. J. Welsh and W. L. Zielinski, Anal. Chem., 1997, 69, 1392–1397 CrossRef CAS.
- T. I. Aksenova, I. V. Tetko, A. G. Ivakhnenko, A. E. P. Villa, W. J. Welsh and W. L. Zielinski, Anal. Chem., 1999, 71, 2423–2430 CrossRef CAS.
- L. B. A. Hansen, J. Pharm. Sci., 2001, 90, 943–948 CrossRef CAS.
- Y. Y. Cheng, M. J. Chen and W. D. Tong, J. Chem. Inf. Comput. Sci., 2003, 43, 1068–1076 CrossRef CAS.
- Y. Y. Cheng, M. J. Chen and W. J. Welsh, J. Chem. Inf. Comput. Sci., 2003, 43, 1959–1965 CrossRef CAS.
- L. J. Ni, P. Li, R. Zheng, L. G. Zhang and L. Z. Zhu, Chin. Trad. Pat. Med., 2002, 24, 79–82 Search PubMed.
- M. J. Chen, Y. Y. Cheng and R. C. Lin, Chin. Trad. Pat. Med., 2002, 24, 905–908 Search PubMed.
- X. Wang, W. Y. Wang, K. R. Zhang and K. S. Bi, J. Shenyang Pharm. Univ., 2003, 20, 360–363 Search PubMed.
- Y. P. Li, Z. Hu and L. C. He, J. Pharm. Biomed. Anal., 2007, 43, 1667–1672 CrossRef CAS.
- P. Zou, Y. Hong and H. L. Koh, J. Pharm. Biomed. Anal., 2005, 38, 514–520 CrossRef CAS.
- R. M. Yu, B. Ye, C. Y. Yan, L. Y. Song, Z. Zhang, W. Yang and Y. Zhao, J. Pharm. Biomed. Anal., 2007, 44, 818–823 CrossRef CAS.
- W. Li, Y. L. Deng, R. J. Dai, Y. H. Yu, M. K. Saeed, L. Li, W. W. Meng and X. S. Zhang, J. Pharm. Biomed. Anal., 2007, 45, 38–46 CrossRef CAS.
- X. H. Fan, Y. Wang and Y. Y. Cheng, J. Pharm. Biomed. Anal., 2006, 40, 591–597 CrossRef CAS.
- Y. X. Sheng, L. Li, Q. Wang, H. Z. Guo and D. A. Guo, J. Pharm. Biomed. Anal., 2005, 37, 805–810 CrossRef CAS.
- J. B. Xie, W. Q. Wang, Y. Q. Zhang, Y. Bai and Q. Yang, J. Pharm. Biomed. Anal., 2007, 45, 450–455 CrossRef CAS.
- X. D. Wen, L. W. Qi, P. Li, K. D. Bao, X. W. Yan, L. Yi and C. Y. Li, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 865, 99–105 CrossRef CAS.
- Y. H. Wang, Z. Chen, D. X. Xiang and Y. R. Ou, Chin. Hosp. Pharm. J., 2006, 26, 910–913 Search PubMed.
- S. Kanno, A. Shouji, R. Hirata, K. Asou and M. Ishikawa, Life Sci., 2004, 75, 353–365 CrossRef CAS.
- U. J. Jung, M. Lee, Y. B. Park, M. A. Kang and M. Choi, Int. J. Biochem. Cell Biol., 2006, 38, 1134–1145 CrossRef CAS.
- A. Balakrishnan and V. P. Menon, Toxicology, 2007, 238, 90–98 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |