Determination of atenolol, chlorthalidone and their degradation products by TLC-densitometric and chemometric methods with application of model updating
Nada. S.
Abdelwahab
*
Analytical Chemistry Department, Faculty of Pharmacy, Benisuef University, Egypt. E-mail: nadasayed2003@yahoo.com; Fax: +082/2317950; Tel: +0163521907
First published on 26th October 2010
Abstract
In this work two well known antihypertensive drugs that are administered in combination and provide greater therapeutic effects than with either drug alone are selectively determined in the presence of their degradation products. Two chemometric methods and TLC-Densitometric one have been developed for the selective determination of Atenolol (ATE) and Chlorthalidone (CLT) along with their hydrolytic degradation products. The developed chemometric models are principal component regression (PCR) and partial least squares (PLS). These models have been updated to be used for prediction of ATE and CLT in another dosage form in which Amiloride HCl (AMH) is included. The updated models are capable of predicting the concentrations of the three components of the new dosage form with good accuracy and precision without reconstruction of the calibration set. The developed TLC-Densitometric method depends upon quantitative densitometric separation of thin layer chromatogram of ATE, CLT, Atenolol degradation product (ATE Deg) and Chlorthalidone degradation product (CLT Deg) using silica gel plates at 227 nm and chloroform: methanol: ethyl acetate: ammonia solution (75: 28: 2: 1.6, by volume) as a developing system. The suggested methods have been used for the determination of the studied drugs in their pharmaceutical formulations and the results were statistically compared to the reported RP-HPLC method.
1. Introduction
Atenolol (ATE; Fig. (1A)) has the IUPAC name (RS)2-[4-[2-hydroxy-3-(propan-2-ylamino)propoxy]phenyl]-acetamide,1 it is a β-adrenolytic, cardio selective drug, commonly used in the treatment of cardiovascular disorders especially in coronary failure, arterial hypertension and cardiac arrhythmia.2 Chlorthalidone (CLT; Fig. (1B)) has the IUPAC name 2-chloro-5-(1-hydroxy-3-oxo-2H-isoindol-1-yl)benzenesulfonamide,1 it is a diuretic with actions and uses similar to those of the thiazide diuretics even though it does not contain a thiazide ring system. It is used for hypertension and edema, including that associated with heart failure.3 The combination of ATE and CLT having the advantages of providing greater therapeutic effects than with either drug alone and permitting once a day administration only, the two drugs do not interact pharmaco-kinetically with each other or present synergic toxic effect.4 The toxicity, narrow therapeutic range of most β-blockers5 and the increasing consumption of CLT alone and in combination with other therapeutic agents,6 have resulted in a great interest in analytical methods allowing the detection and quantification of the studied drugs in pure forms and in different pharmaceutical preparations.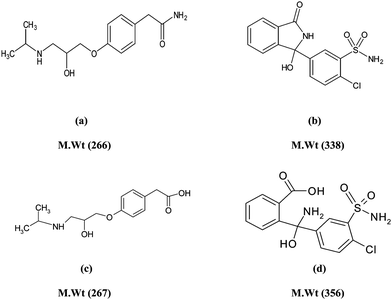 | ||
| Fig. 1 Chemical Structure of (a) ATE and (b) CLT. | ||
Several analytical methods have been used for the simultaneous determination of ATE and CLT in their binary mixture and in their mixture with other components, such as HPLC,7–16 LC-MS-MS17 and HPTLC18 methods. Different spectrophotometric techniques have been reported for the determination of the studied drugs.19–24 Also the studied mixture has been analyzed using capillary zone electrophoresis method.25
Although several methods have been developed for analysis of the binary mixture, this is the first time that the binary mixture is determined in the presence of the hydrolytic degradation products of the studied drugs. In this work sensitive, economic, rapid and selective stability indicating assay chemometric and TLC-Densitometric methods are investigated for the determination of the quaternary mixture of the drugs and the degradation products. The developed methods have the advantages of lower cost and higher selectivity than the reported RP-HPLC one.7 The suggested chemometric methods are PCR and PLS which are then updated to be used for prediction of the studied drugs along with amiloride hydrochloride (AMH) which is included in another dosage form without reconstruction of the calibration set.
2. Experimental
2. 1. Instruments
For the chemometric methods, Double beam UV-VIS spectrophotometer (Shimadzu, Kyoto-Japan), model UV-1601 PC with 1 cm quartz cells, connected IBM compatible computer. Matlab® version 6.526 was used for the proposed chemometric methods, the PLS was performed with PLS_Toolbox27 for use with Matlab® 6.5.For TLC, the samples were spotted with Sample applicator for TLC linomat V with 100μl syringe (Camage, Muttenz, Switzerland) on TLC plates (20 × 10 cm) coated with silica gel 60 F254 (Merck KgaA, Darmstad, Germany) with 200 μm thickness. Scanning was performed on TLC Scanner 3 densitometer (Camage, Muttenz, Switzerland) controlled by winCATS software (V 3.15, Camage). UV lamp with short wavelength 254 nm (VL-6.LC, MARNE LA VALLEE Cedex 1, FRANCE) was used for scanning until optimization of the proposed method.
For MS analysis, it was carried out using JEOL JMS-AX 500 system (Japan), while IR analysis was performed using VECTOR 22 system (Germany).
2. 2. Samples
Preparation of Atenolol degradation product (ATE Deg). Atenolol degradation product (ATE Deg): Was prepared following the method developed by Bayoumi et al28—An accurately weighed amount of pure ATE powder equivalent to 0.25 mg was dissolved in 50 mL 0.1 N H2SO4, refluxed for 10 h. The pH of the solution was adjusted with 0.1N NaOH to 9-10 pH units and then the solution was extracted three times, each with 50 mL diethyl ether. The etherial extraction was then evaporated to dryness.
Chlorthalidone degaradation product (CLT Deg): Accurately weighed amount of pure CLT equivalent to 0.5 gm was refluxed with 50 mL 0.1 N NaOH for five hours, cooled and then the pH was adjusted to 2 pH units for complete precipitation of the produced degradation product using 0.1 N HCl. The formed CLT Deg was filtered, washed with distilled water and then dried at 70 °C. Complete degradation was confirmed by disappearance of CLT spot using TLC plates and ethyl acetate (EtOAc)-methanol (MeOH)-ammonia solution (NH4OH) (9.5: 0.5: 0.3, by volume) as a developing system.
2. 3. Chemicals and solvents
All chemicals and solvents used were of analytical grade and were purchased from El-NASR Pharmaceutical Chemicals Co., Abu- Zabaal, Cairo, Egypt.Methanol used for chemometric methods was of HPLC grade (CHROMASOLVE®, Sigma-Aldrich Chemie GmbH, Germany).
2. 4. Solutions
Stock standard solutions of ATE and CLT and Stock solutions of ATE Deg and CLT Deg were prepared in methanol in the concentration of 1 mg ml−1.Working standard solutions of ATE and CLT and Working solutions of ATE Deg and CLT Deg were prepared in methanol in the concentration of 0.1 mg ml−1.
3. Procedure
3. 1. Chemometric methods
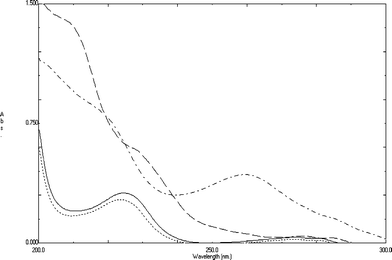 | ||
| Fig. 2 Zero order absorption spectra of 10 μg mL−1 of each of ATE (—), CLT (-----), ATE Deg (…‥) and CLT Deg (-.-.-.-) using 0.1N HCl as a solvent. | ||
| Sample No. | ATE/μg ml−1 | CLT/μg ml−1 | AT Deg/μg ml−1 | CLT Deg/μg ml−1 |
|---|---|---|---|---|
| a Samples used for model validation. | ||||
| 1 | 36.0 | 6.0 | 4.0 | 1.5 |
| 2 | 38.0 | 10.0 | 4.5 | 1.5 |
| 3 | 40.0 | 8.0 | 4.0 | 2.5 |
| 4a | 40.0 | 4.0 | 5.0 | 1.5 |
| 5 | 38.0 | 6.0 | 5.0 | 2.5 |
| 6 | 36.0 | 10.0 | 5.0 | 0.5 |
| 7a | 34.0 | 10.0 | 4.0 | 1.0 |
| 8 | 40.0 | 10.0 | 3.0 | 2.0 |
| 9 | 40.0 | 2.0 | 4.5 | 0.5 |
| 10 | 32.0 | 8.0 | 3.0 | 1.5 |
| 11 | 38.0 | 2.0 | 4.0 | 2.0 |
| 12 | 32.0 | 6.0 | 4.5 | 2.0 |
| 13a | 40.0 | 6.0 | 3.5 | 1.0 |
| 14a | 36.0 | 4.0 | 3.5 | 2.0 |
| 15 | 36.0 | 8.0 | 4.5 | 1.0 |
| 16 | 38.0 | 8.0 | 3.5 | 0.5 |
| 17 | 38.0 | 4.0 | 3.0 | 1.0 |
| 18 | 34.0 | 2.0 | 3.5 | 1.5 |
| 19 | 32.0 | 4.0 | 4.0 | 0.5 |
| 20 | 34.0 | 6.0 | 3.0 | 0.5 |
| 21 | 36.0 | 2.0 | 3.0 | 2.5 |
| 22a | 34.0 | 4.0 | 4.5 | 2.5 |
| 23 | 32.0 | 2.0 | 5.0 | 1.0 |
| 24a | 32.0 | 10.0 | 3.5 | 2.5 |
3. 2. TLC-Densitometric method
3. 3. Application to pharmaceutical preparations
Twenty tablets each of Blokium Diu®, Tenoret®, Tenedone®and Teklo® were separately weighed, powdered and mixed well. An accurately weighted portion equivalent to 100 mg ATE (and the corresponding amounts of CLT [25 mg for Blokium Diu®, Tenoret® and Teklo® tablets and 50 mg for Tenedone® tablets] and the corresponding amount of AMH [5 mg for Teklo® tablets]) were separately transferred into three 100 ml calibrated measuring flasks and then 75 ml methanol was added to each flask. The prepared solutions were sonicated for 30 min, the volume was completed with the same solvent to get 1 mg ml−1 ATE and the solution was then filtered. Appropriate dilutions of the prepared solutions were made to prepare their working solutions (0.1 mg ml−1) and then the proposed methods were followed.4. Results and discussion
Many pharmaceutical compounds undergo degradation during storage or even during the different process of their manufacture. Several chemical or physical factors can lead to the degradation of drugs,30 where hydrolysis and oxidation are the most famous chemical degradation routes.31,32 Both ATE and CLT contain amide groups that are liable to degradation upon refluxing in acidic and basic conditions, each of them gave the same hydrolytic degradation product in both acidic and basic conditions. ATE Deg was prepared by following the method developed by Bayoumi et al.28 Reviewing the literature in hand, there was no report describing the CLT degradation pathway, so after preparation, complete separation and purification of CLT Deg, the drug and the degradation products were subjected to IR and Mass analyses to confirm the structure of the produced degradation product. The assignment of IR spectrum of CLT Deg is based on comparison to that of the intact drug. It shows appearance of broad band of OH group at (3390–2559 cm−1) and at 916 cm−1 (in the finger print region) indicating the hydrolysis of amidic linkage of the drug to carboxylic acid (COOH) and amino (NH2) groups. Moreover, peak of C–H (in the five member ring) was shifted from 749 to 741 cm−1 due to conversion of cyclic NH to NH2. The obtained mass spectra (MS) for CLT (C14H11ClN2O4S) and CLT Deg (C14H13ClN2O5S) shows molecular ion peaks at m/z 338 and m/z 356, respectively, which shows an increase in the molecular weight of the drug by 18, indicating addition of a molecule of H2O to cyclic amide linkage. Chemical structure of the drugs and their degradation products are given in Fig. (1).The UV absorption spectra of ATE, CLT, ATE Deg and CLT Deg, Fig. (2), displays considerable overlap, where the application of conventional spectrophotometry, its direct derivative and derivative ratio techniques failed to resolve these overlapping spectra. This work concerns applying PCR, PLS and TLC-Densitometric methods for determination of ATE and CLT along with their degradation products either by resolving their severe spectral overlap (that can not be resolved by any other direct or indirect spectrophotometric method) or after their chromatographic separation on TLC plates. The developed work is also aimed to validate the suggested methods and to compare it with the reported method.
4. 1. Chemometric methods
For the calibration and validation sets, multilevel multifactor experimental design was used for their construction.35 A five-level, four-factor calibration design was used and 24 mixtures of the four studied components were prepared. The concentration ranges and the composition of the calibration and validation samples are given in Table (1). The spectra of the prepared mixtures in the range of 220–286 nm were recorded and the spectral data acquisition was taken with 0.4 nm interval, smoothed (using scaling factor = 1 and Δλ = 2) and transferred to Matlab® 6.5 for subsequent data manipulation, thus produced spectral data matrix has 24 rows representing different samples and 166 columns representing wavelengths (24 × 166). Eighteen samples were chosen and used for calibration and the remaining six samples were used as an external validation set. In order to determine the correct number of LVs to be used for modeling the auto scaled data, cross-validation with random subsets selection was performed to calculate the cumulative PRESS (prediction residual error sum of squares). Haaland and Thomas criterion36 was used to calculate the optimum number of LVs (latent variables) to be used in the models. The optimum number of LVs described by the constructed models was found to be eight factors for both PCR and PLS models.
The predictive ability of the developed models was evaluated by plotting the known concentrations versus predicted ones for each component for each model and the statistical parameters of the regression equations are presented in Table (2). The predictive ability of the developed models was further assessed by their application to an external validation set where good % recoveries and low RMSEP (root mean squares error of prediction) values were obtained and presented in Table (3).
| Parameters | Chemometric methods | TLC-Densitometric method | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ATE | CLT | ATE Deg | CLT Deg | ATE | CLT | ATE Deg | CLT Deg | |||||
| PCR | PLS | PCR | PLS | PCR | PLS | PCR | PLS | |||||
| a The correlation coefficient. | ||||||||||||
| Linearity | ||||||||||||
| Range | 32–40 μg ml−1 | 2–10 μg ml−1 | 3–5 μg ml−1 | 0.5–2.5 μg ml−1 | 2–10 μg band−1 | 0.2–2 μg band−1 | 2–10 μg band−1 | 0.2–1.8 μg band−1 | ||||
| Slope | 1.0174 | 0.9775 | 0.9838 | 1.0155 | 0.8738 | 0.9882 | 0.1159 | 0.2852 | 0.0338 | 0.3983 | ||
| Intercept | 0.9815 | 1.1533 | 0.0656 | 0.0604 | 0.5185 | 0.0214 | 0.2058 | 0.0519 | 0.1613 | 0.1047 | ||
| 0.9969 | 0.9966 | 0.9995 | 0.9995 | 0.9771 | 0.9995 | 0.9995 | 0.9996 | 0.9997 | 0.9997 | |||
| Accuracy | 100.11 | 100.14 | 99.91 | 99.94 | ||||||||
| Precision | ||||||||||||
| Repeatability | 0.638 | 0.804 | 1.457 | 1.029 | ||||||||
| Intermediate precision | 1.772 | 1.293 | 1.408 | 1.053 | ||||||||
| LOD | 0.5 μg band−1 | 0.08 μg band−1 | 1.0 μg band−1 | 0.1 μg band−1 | ||||||||
| LOQ | 2.0 μg band−1 | 0.2 μg band−1 | 2.0 μg band−1 | 0.2 μg band−1 | ||||||||
| Parameters | Chemometric methods | TLC-Densitometric method | Reported method7 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ATE | CLT | ATE Deg | CLT Deg | ATE | CLT | ATE | CLT | |||||
| PCR | PLS | PCR | PLS | PCR | PLS | PCR | PLS | |||||
| a : Average of 3 determinations. b : Average of 5 determinations. RMSEP: Root mean square error of prediction. c : The values in the parenthesis are the corresponding theoretical values at p = 0.05. reported method: HPLC method using mobile phase consisting of 740 mL H2O: 250 mL ACN: 8 mL 3.6 N H2SO4: 930 mg sodium octyl sulfate, λ = 275 nm. | ||||||||||||
| L.P. Mixturesa | 100.99 ± 0.821 | 100.98 ±.847 | 100.61 ± 1.308 | 100.51 ± 1.216 | 100.41 ± 4.241 | 100.41 ± 4.241 | 100.24 ± 1.081 | 100.24 ± 1.081 | ||||
| RMSEP | 0.427 | 0.428 | 0.099 | 0.099 | 0.154 | 0.154 | 0.018 | 0.018 | ||||
| Blokium Diu®b (B.N.093422) | 99.08 ± 1.087 | 99.25 ± 1.104 | 101.23 ± 1.126 | 101.75 ± 1.362 | 100.24 ± 1.910 | 99.37 ± 1.409 | 99.81 ± 1.328 | 101.84 ± 1.414 | ||||
| Standard addition a | 99.56 ± 1.712 | 99.19 ± 1.434 | 99.50 ± 1.472 | 100.13 ± 2.016 | 100.43 ± 2.013 | 100.94 ± 1.905 | ||||||
| Degree of freedom | 8 | 8 | 8 | 8 | 8 | 8 | ||||||
| F-test (6.388) c | 1.492 | 1.447 | 1.575 | 1.077 | 2.067 | 1.006 | ||||||
| Tenoret ®b (B.N.90194) | 96.58 ± 2.029 | 96.91 ± 1.797 | 100.45 ± 1.370 | 100.45 ± 1.370 | 98.07 ± 1.783 | 99.52 ± 1.396 | 97.57 ± 1.121 | 99.26 ± 1.307 | ||||
| Degree of freedomc | 8 | 8 | 8 | 8 | 8 | 8 | ||||||
| F-test (6.388) | 3.279 | 2.572 | 1.099 | 1.099 | 2.531 | 1.141 | ||||||
| Tenedone ®b (B.N.71925) | 101.16 ± 1.850 | 101.87 ± 0.966 | 97.20 ± 0.783 | 97.20 ± 0.783 | 98.37 ± 1.974 | 101.88 ± 1.130 | 96.32 ± 1.780 | 97.71 ± 0.834 | ||||
| Degree of freedom | 8 | 8 | 8 | 8 | 8 | 8 | ||||||
| F-test (6.388) c | 4.926 | 1.342 | 5.172 | 5.172 | 5.603 | 2.481 | ||||||
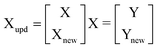 | (1) |
The model update was applied to predict the concentrations of two of the modeled compounds, ATE and CLT in another dosage form that in addition to ATE and CLT contains AMH. The minimal number of samples needed to efficiently update the developed models must be accurately determined and the influence of number of samples added to the calibration set on RMSEP was studied for each developed multivariate calibration model. The number of different samples added shows a large impact on the predictive ability of the updated models, where for ATE the RMSEP of the models updated decreases from 1.855 and 1.778 for the initial models to 0.446 and 0.419 for the updated PCR and PLS models, respectively, which represents an improvement of 75.96 and 76.43% for the two methods, respectively. While for CLT the RMSEP of the models updated decreases from 0.578 and 0.591 to 0.08 and 0.082 which represents an improvement of 86.16 and 86.13% for the two methods, respectively. The RMSEP of AMH in the updated models decreases from 0.139 and 0.122 to 0.024 and 0.022 which represents an improvement of 82.73 and 81.97% for the two methods, respectively. Four samples are found to be necessary to perform an efficient update of the developed PCR and PLS models. The results of analysis of Teklo® tablets by the updated models and its statistical comparison with those obtained by applying the reported HPLC method16 are presented in Table 4.
| Parameters | Chemometric methods | Reported method [16] | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ATE | CLT | AMH | ATE | CLT | AMH | ||||
| PCR | PLS | PCR | PLS | PCR | PLS | ||||
| a Average of 5 determinations. b The values in the parenthesis are the corresponding theoretical values at p = 0.05. reported method: HPLC method using ACN: 5 mM heptanesulfonic acid sodium salt (20: 80, v/v) pH = 4.4. λ = 274 nm. | |||||||||
| Teklo ®a (B.N.09123048) | 104.66 ± 1.154 | 104.67 ± 1.147 | 97.52 ± 0.398 | 97.56 ± 0.478 | 99.22 ± 1.218 | 99.22 ± 1.218 | 104.66 ± 0.523 | 98.42 ± 0.804 | 99.40 ± 0.804 |
| Degree of freedom | 8 | 8 | 8 | 8 | 8 | 8 | |||
| F-test (6.388) b | 4.857 | 4.801 | 4.086 | 2.841 | 3.419 | 3.419 | |||
4. 2. TLC-Densitometric method
TLC-Densitometric technique has become so well established that it is sometimes the only technique of choice for the determination of drugs in mixtures and in formulations due to its high resolution power, it has been adopted for the stability studies, pharmacokinetics and micro-quantitative analysis of a vast number of pharmaceuticals.37Different experimental conditions, such as mobile phase composition and scanning wavelength were studied and optimized in order to obtain sharp and symmetrical peaks of the studied components and to provide accurate and reproducible results. Different developing systems have been tried e.g. CHCl3-ethanol (EtOH) (6.5: 3.5, v/v), CHCl3- EtOH-acetic acid (CH3COOH) (6.5: 3.5: 0.5, by volume) CHCl3-MeOH-NH4OH (7.5: 2.5: 0.1, by volume), CHCl3-EtOAc-triethyl amine (TEA) (8.5: 1.5: 0.3, by volume), CHCl3-MeOH-EtOAc-NH4OH (7.5: 2: 0.5: 0.15, by volume) and CHCl3-MeOH: EtOAc-NH4OH (7.5: 2.8: 0.2: 0.16, by volume). Last one was found to be the best mobile phase that allowed good separation between the quaternary mixture (ATE, CTL, ATE Deg and CTL Deg) with good Rf values (where Rf of ATE Deg = 0.04, Rf of CTL Deg = 0.17, Rf of ATE = 0.49 and Rf of CTL = 0.69) and without tailing of the separated bands, Fig. (3). Also different scanning wavelengths have been studied (210, 227 and 254 nm), scanning at 227 nm gave good results for the four components at which peaks were sharp and symmetrical. Minimum noise and considerable sensitivity of the studied compounds were obtained.
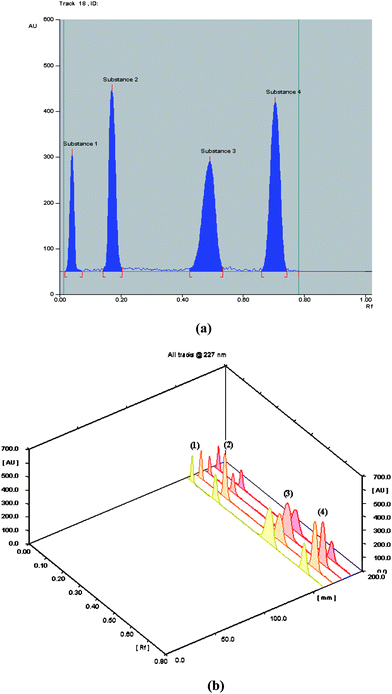 | ||
| Fig. 3 Thin layer chromatogram of ATE Deg (1), CTL Deg (2), ATE (3) and CTL (4) using chloroform: methanol: ethyl acetate: ammonia solution (75: 28: 2: 1.6, by volume). (a) Two dimension and (b) three dimension chromatograms. | ||
| Parameters | Obtained value | |||
|---|---|---|---|---|
| ATE Deg | CLT Deg | ATE | CLT | |
| Symmetry factor | 1.17 | 1 | 0.93 | 1 |
| Resolution (Rs) | 2.64 | 3.49 | 3 | |
| Capacity factor (k′) | 0.5 | 1.15 | 4.9 | 8.35 |
| Selectivity (α) | 4.3 | 2.74 | 1.58 | |
Calibration curves relating the mean integrated peak area × 10−4 to the corresponding concentration of ATE, CTL, ATE Deg and CTL Deg in the range of 2–10, 0.2–2, 2–10 and 0.2–1.8 μg band−1, respectively, were constructed. The regression equation and analytical parameters of the developed TLC-Densitometric method are given in Table (2). Method validation was performed according to ICH guidelines.38Table (2) shows results of linearity, range, accuracy, repeatability, intermediate precision, limits of detection and quantitation, selectivity and system suitability testing of the method.
The suggested chemometric and TLC-Densitometric methods have been applied for the determination of both ATE and CTL in different pharmaceutical formulations with acceptable percentage recoveries, Table (3). Furthermore, their validity was assessed by applying the standard addition technique which showed that tablet excipients did not interfere, Table (5). Statistical comparison of the developed methods and the official HPLC one7 showed no significant difference with respect to accuracy and precision, Table (3).
5. Conclusion
This work provides accurate, sensitive, economic, rapid and selective analytical methods for determination of ATE, CTL and their degradation products. The chemometric methods used in this work can be performed easily with adequate software support and they provide clean examples of the high resolving power of that technique, also they can be applied for determination of the studied drugs in different dosage form that in addition containing AMH through model updating. The disadvantage of these methods is their lower sensitivity. TLC-Densitometric method has the same advantages as chemometric methods; moreover, it can be easily applied in quality control laboratories where economy and time is essential. The disadvantage of this method is that it requires a complicated instrument. The proposed methods can be used in routine analysis of the studied drugs either in their pure bulk powders or in dosage forms without any preliminary separation step.References
- S. Budavcari. ( 2002) “The Merck Index”, An Encyclopedia of Chemicals, Drugs and Biologicals, (13th Ed), Merck and Co.Inc., Whitehouse Station, NJ Search PubMed.
- C. Vesna, M. Zvonimira, H. Hrvoje, K. Josip and K. Franjo, Analytical Profile of Drug Substances, 1984, Vol. 13 Search PubMed.
- Martindale- Extra Pharmacopoeia ( 2005) 34th Ed., “The Complete Drug References” The Pharmaceutical Press, London, UK Search PubMed.
- G. Claudio, T. Anna, C. Sergio and Z. Giovanni, J. Chromatogr., B: Biomed. Sci. Appl., 1997, 698, 187–194 CrossRef CAS.
- H. Siren, M. Saarinen, S. Hainari and M. L. Riekkola, J. Chromatogr., Biomed. Appl., 1993, 632, 215–227 CAS.
- R. Herráez-Hernández and P. Campíns-Falcó, J. Chromatogr., B: Biomed. Sci. Appl., 2000, 740, 169–177 CrossRef CAS.
- The United States Pharmacopeia ( 2007) 30 Ed., National Formulary 25, United States Pharmacopeia Convention Inc. Search PubMed.
- The British Pharmacopoeia ( 2007) Her Majesty's, The Stationary Office, London Search PubMed.
- R. Ficarra, P. Ficarra, A. Tommasini, M. L. Calabro and C. Guamiera Fench, Farmaco, Ed. Prat., 1985, 40(9), 307–312 Search PubMed.
- S. I. Sa'sa, I. M. Jalal and H. S. Khalil, J. Liq. Chromatogr. Relat. Technol., 1988, 11(8), 1673–1696 CAS.
- L. Gong and Y. Liu, Yaowu Fenxi Zazhi, 1989, 9(3), 175–176 Search PubMed.
- C. Giachetti, A. Tenconi, S. Canali and G. Zanolo, J. Chromatogr., B: Biomed. Sci. Appl., 1997, 698(1–2), 187–194 CrossRef CAS.
- H. A. Kang, H. H. Kim, S. M. Kim, H. Yoon, H. Y. Cho, S. Y. Oh, H. K. Choi and Y. B. Lee, Yakche Hakhoechi, 2006, 36(5), 331–338 Search PubMed.
- A. El-Gindy, S. Sallam and R. A. Abdel-Salam, J. Sep. Sci., 2008, 31(4), 677–682 CrossRef CAS.
- A. A. Elshanawane, M. S. Mostafa and S. M. Elgawish, J. AOAC Int, 2009, 92(2), 404–409 CAS.
- A. El-Gindy, S. Emara and A. Mostafa, Farmaco, 2005, 60(3), 269–278 CrossRef CAS.
- A. Khuroo, S. Mishra, O. Singh, S. Saxena and T. Monif, Chromatographia, 2008, 68(9–10), 721–729 CrossRef CAS.
- H. Salem, Scientica Pharmaceutica, 2004, 72(2), 157–174 Search PubMed.
- A. E.-M. I. Mohamed and H. Salem, Anal. Bioanal. Chem., 2005, 382(4), 1066–1072 CrossRef CAS.
- W. Wehner, Die Pharmazie, 2000, 55(7), 543–544 Search PubMed.
- M. C. Ferraro, P. M. Castellano and T. S. Kaufman, Anal. Bioanal. Chem., 2003, 377(7–8), 1159–1164 CAS.
- D. Bonazzi, R. Gotti, V. Andrisano and V. Cavrini, IL Farmaco, 1996, 51(11), 733–738 CAS.
- M. M. Mabrouk, H. Salem, M. F. Redwan and T. S. Kaood, Egypt. J. Anal. Sci., 2003, 12, 141–173 Search PubMed.
- C. Vetuschi and G. Ragno, Int. J. Pharm., 1990, 65(3), 177–181 CrossRef CAS.
- K. Al Azzam, K. K. Elbashir, A. M. Elbashir, B. Saad and S. Abdul Hamid, Anal. Lett., 2009, 42(10), 1458–1470 CrossRef.
- Matlab ver. 5.3. ( 1999) Mathworks inc..
- B. M. Wise, and N. B. Gallagher. ( 2001), Version 2.1.1 ed., Eigenvector Research, Inc..
- A. Bayoumi, S. Abdel-Fattah, J. Diab and M. Ellaithy, Bull. Fac. Pharm. Cairo Univ., 1999, 37(3), 33–40 Search PubMed.
- R. G. Brereton, Analyst, 1997, 122, 1521–1529 RSC.
- M. B. Henry, O. L. Charles, and R. L. Wait. ( 1963) Physical and Technical Pharmacy, Mc Graw-Hill Book Co., Inc., New York, Toronto, London, 621–644 Search PubMed.
- A. T. Florence, and D. Attwood. ( 1998) Physical Principles of Pharmacy (3rd Ed), London: Macmillan Press Search PubMed.
- Gs. Banker, and C. T. Rhodes. ( 2002) Modern Pharmaceutics (4th Ed), Marcel Dekker, Inc Search PubMed.
- H. Martens, and T. Naes. ( 1989) Multivariate Calibration, John Wiley & Sons Ltd: Chichester Search PubMed.
- M. Blanco, J. Coello, F. Gonzales, H. Iturriage and S. Maspoch, J. Pharm. Sci., 1993, 82, 834 CrossRef CAS.
- R. G. Brereton, Analyst, 1997, 122, 1521 RSC.
- D. M. Haaland and E. V. Thomas, Anal. Chem., 1988, 60, 1193 CrossRef CAS.
- C. F. Pool and S. K. Poole, J. Chromatogr., B: Biomed. Sci. Appl., 1989, 492, 539 CrossRef.
- ICH, Q2 (R1) Validation of Analytical Procedures ( 2005), Proceedings of the International Conference on Harmonization, Geneva Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |