Gold nanoparticles/L-cysteine/graphene composite based immobilization strategy for an electrochemical immunosensor
Guangfeng
Wang
ab,
Hao
Huang
ab,
Ge
Zhang
ab,
Xiaojun
Zhang
ab,
Bin
Fang
ab and
Lun
Wang
*ab
aCollege of Chemistry and Materials Science, Anhui Key Laboratory of Functional Molecular Solids, Anhui Key Laboratory of Chem-Biosensing, Anhui Normal UniVersity, Wuhu, 241000, P R China. E-mail: wangyusandra@yahoo.com.cn; Fax: +86-553-3869303; Tel: +86-553-3869303
bAnhui Key Laboratory of Controllable Chemistry Reaction & Material Chemical Engineering, Hefei University of Technology, Hefei, 230009, P R China
First published on 24th September 2010
Abstract
A novel immobilization strategy combining gold nanoparticles (GNP) with graphene (Gr) via crosslinker L-cysteine (L-cys) for the enhancement of a HRP-labelled sandwiched electrochemical immunosensor was developed. In the fabrication process of the immobilization platform, graphene was first modified onto glassy carbon electrode (GCE), and then L-cys was electrodeposited onto the substrate of Gr for the following GNP assembling. The characteristics of the platform surface were studied by scanning electron microscopy (SEM), ultraviolet-visible (UV-vis) spectrometry, cyclic voltammetry (CV) and electrochemical alternating current impedance spectroscopy (EIS). From the SEM, interestingly we found the substrate of Gr induced the regular growth of L-cys, which may further improve the stable conjugation of GNP, which may result in more loading amount of the biomolecules. The special GNP/L-cys/Gr composite film constructed an effective antibody immobilization matrix and made the immunosensor hold high sensitivity, good stability and bioactivity. Under the optimized experimental conditions, a wide linear range of human IgG (HIgG) concentration from 0.2 to 320 ng mL−1 and a detection limit of 70 ng mL−1 were obtained. Accurate detection of HIgG in human serum samples was satisfyingly demonstrated by comparison to the standard ELISA assays.
1. Introduction
Immunoassay techniques based on the property of highly specific molecular recognition of antigen by antibodies, have become an important analytical method in clinical examinations, biochemical, environmental and food quality analysis.1 The detection systems for immunosensors are based on optical, electrochemical or gravimetric methods.2 Because of their simple fabrication and good sensitivity, electrochemical immunosensors have been adopted for many miniaturized and microfluidic devices.3 However, the biointerface with immobilized antibodies on various substrates plays an important role in electrochemical immunosensors.4 And an efficient biointerface holds highly dense immobilization of biomolecules, long-term stability of attached biomolecules, low nonspecific binding, proper biomolecular orientation to permit simple and rapid specific interactions.5 For these purposes, various biointerfaces have been extensively studied.6 However, due to the high standard of an efficient biointerface, it is necessary to explore novel materials and methods for the fabrication of an efficient biointerface.Graphene, is a flat monolayer of carbon atoms tightly packed into a two-dimensional (2D) honeycomb lattice and can be considered as a basic building block for other kinds of graphitic materials, such as 0D fullerenes, 1D nanotubes or 3D graphite.7 Because of their novel and fascinating properties, such as exceptional thermal, mechanical properties, and high electrical conductivity, graphene, as a “rising star” material,8 has been extensively studied in synthesizing nanocomposites9 and fabricating various microelectrical devices, such as battery,10 field-effect transistors,11 ultrasensitive sensors,12 and electromechanical resonators.13 Recent investigations showed that graphene exhibits favorable electrochemical activity towards many analytes, and it minimized passivation, improved high electron transfer and avoided interferences in a wide range of electrochemical sensing and biosensing applications.14 The unique structure and remarkable advantages of graphene made it an ideal candidate in fabricating the biointerface. However, as we know, only a few reports on the application of graphene to the field of immunosensors have been recognized.15 However, as much as we know, the immobilization matrix combining Gr with gold nanoparticles (GNP), a class of nanomaterials for biointerfaces in immunosensors,16 has not been explored up to now.
Herein, we fabricated a novel antibody immobilizing biointerface combining Gr and GNP viaL-cysteine (L-cys) crosslinking and further successfully developed a sandwiched immunosensor with an enhancement of electrochemical signal for the sensitive detection of human IgG (HIgG, Ag). By electrodeposition method, L-cys grew in a regular way on the graphene surface, GNP were then self-assembled on L-cys/Gr surface to fabricate GNP/L-cys/Gr composite films, which constructed an effective antibody immobilization matrix holding high stability and bioactivity. The resulting sandwiched immunosensor showed a linear range of IgG concentration of 0.2 ∼ 320 ng mL−1, and a detection limit of 70 pg mL−1. By comparison with immunosensors with and without the graphene, the sensor with graphene was found to have a great enhancement in the sensitivity and stability. The immunosensor provided a convenient, low-cost, sensitive, and specific method for protein detection, which could be an alternative technique in the clinical laboratory. And it is a novel method to explore the unique function of graphene to develop the promising immobilization biointerface for electrochemical immunosensors.
2. Experimental
2.1. Reagents and materials
Gold(III) chloride trihydrate (HAuCl4) (99.9%), L-cys and sodium borohydride (99%), graphite and hydrazine was purchased from Shanghai Chemical Reagent Co., Ltd. Bovine serum albumin (BSA, 96–99%), goat IgG, human IgG (Ag), rabbit anti-human IgG (Ab1) and HRP labeld goat- anti-human IgG (Ab2) were purchased from Biodee Biotechnology Co., Ltd. (Beijing, China). Hydroquinone (HQ), H2O2 and all the other reagents were of analytical reagent grade and used without further purification. Phosphate-buffered saline (PBS), with various pH values was prepared by mixing the stock solutions of NaH2PO4 and Na2HPO4 and then adjusting the pH with NaOH and H3PO4. Doubly distilled water was used throughout the experiments.2.2. Apparatus
Scanning electron microscopy (SEM) images of the electrode surface were obtained using Hitachi S-4800 SEM (operated at 10 kV). Ultraviolet-visible spectrometry (UV-vis) was performed on UV-2450 (Shimazu, Japan). Centrifugation was performed using a HERMLEZ 36 HK apparatus (Wehingen, Germany). Electrochemical measurements were performed on a model CHI660B electrochemical analyser (ChenHua Instruments Co. Ltd., Shanghai, China) controlled by a personal computer.2.3. Preparation of GNP/L-cys/Gr modified electrode
Graphite oxide was synthesized from graphite by the Hummers method.17 Graphene was prepared according to the literature.18 GNP were prepared according to the method of Frens.19 The GNP/L-cys/Gr composite was prepared as follows: first, a total of 1 mg of graphene was added to 1 mL methanol solution to form a homogeneous dispersion with mild ultrasonication. A volume of 2 μL of the resulting graphene solution was dropped onto a electrochemical cleaned bare glassy carbon electrode (GCE) and allowed to dry in ambient air for 24 h and rinsed with distilled water to get the graphene modified GCE (Gr/GCE). Second, the Gr/GCE was performed by cyclic voltammetry (CV) scanning between −1.5 and 2.5 V at the scan rate of 0.10 V s−1 about 10 cycles in 0.1 mol L−1 PBS containing 1.0 mmol L−1L-cys to get L-cys/Gr/GCE. Then L-cys/Gr/GCE was rinsed thoroughly with distilled water and at last immersed into colloidal gold solution for 10 h at 4 °C and rinsed as the above, the GNP/L-cys/Gr/GCE was obtained.2.4. Antibody immobilization and immunoreaction procedure
Ab1 was immobilized onto the GNP/L-cys/Gr/GCE. Five microlitres of 200 μg mL−1 Ab1 solution (50 mmol L−1 PBS, pH 7.4) was spread onto the GNP/L-cys/Gr/GCE surface. After incubation for at least 15 h, they were rinsed with 50 mmol L−1 PBS, 0.05% Tween (PBST) to remove physically absorbed Ab1. The nonspecific binding sites of the electrodes were then blocked with 50 μL 5% BSA PBST solution for 1 h at room temperature, and washed with PBST. After aspiration, Ab1 modified electrodes (Ab1/GNP/L-cys/Gr/GCE) were incubated with 60 μL of detecting Ag samples for 120 min. Finally, with the binding reaction between Ab1 and Ag, the electrodes named Ag/Ab1/GNP/L-cys/Gr/GCE were immersed into the 25 μL of 1 mg mL−1 HRP–Ab2 solution for an incubation of 50 min to get HRP–Ab2/Ag/Ab1/GNP/L-cys/Gr/GCE. Following the specific interaction, the subsequent quantification of HIgG was realized by electrochemical detection of enzymatic product in the HQ and H2O2 system. Each incubation procedure was performed at 25 °C in order to maintain the immobilization and immunoreaction under the same experimental condition. The immobilization of Ab1 and the immunoassay procedure are shown in Scheme 1.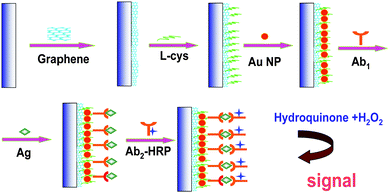 | ||
| Scheme 1 Immobilization of Ab1 and the immunoassay procedure. | ||
2.5. Electrochemical measurements
All electrochemical measurements were performed in a conventional three-electrode system with GCE or modified by different layers as working electrodes, a saturated calomel electrode (SCE) as reference electrode and a platinum foil electrode as counter electrode. All potentials reported were versus the SCE and all experiments were carried out at room temperature. Electrochemical alternating current impedance spectroscopy (EIS) behavior was obtained in the frequency range from 1 to 100000 Hz with the amplitude of the applied sine wave potential 5 mV, and the formal potential of the system 0.18 V. Alternating current impedance and cyclic voltammetry characterization was carried out in 0.1 mol L−1 KCl and 5 mmol L−1 K3[Fe(CN)6]/K4[Fe(CN)6] as a redox probe. CV and differetial pulse voltammetry (DPV) of the immunoassay were performed in 5 mL pH 7.4 50 mM PBS solution containing 1 m mol L−1 HQ with 2 m mol L−1 H2O2.3. Results and discussion
3.1. Characterization of the GNP/L-cys/Gr immunosensor
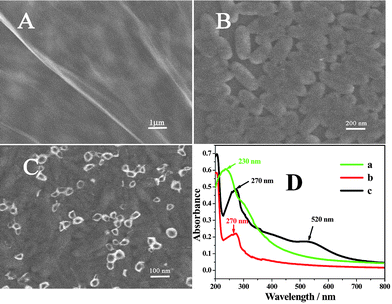 | ||
| Fig. 1 Representative SEM images of Gr/GCE (A), L-cys/Gr/GCE (B), GNP/L-cys/Gr (C), (D): UV-vis absorption spectra of (a) Gr oxides, (b) Gr and (c) GNP/L-cys/Gr in water. | ||
The successful synthesis of GNP decorated graphene was also confirmed by UV-vis spectroscopy (Fig. 1D). The UV-vis spectrum of graphene oxides (curve a) in water shows an absorption peak at 230 nm. After formation of graphene through the hydrazine reduction, the peak of graphene (curve b) is observed at 270 nm. When GNP were decorated onto the graphene and L-cys composite, the absorption peaks of the composites were observed at 270 nm and 520 nm (curve c), which was corresponding to the absorption of graphene and GNP.20 However, under the same conditions, if GNP were directly assembled onto Gr without L-cys, the peak of 520 nm is weaker (not shown here) than curve c. This proved that L-cys doping increased the amount of GNP on Gr.
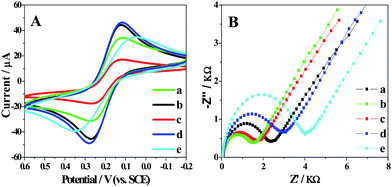 | ||
| Fig. 2 (A) CV characterization of bare GCE (a), Gr (b), L-cys/Gr (c), GNP/L-cys/Gr (d) and Ab1/GNP/L-cys/Gr (e) modified GCE at a scan rate of 100 mV s−1. (B) EIS for the L-cys/Gr/GCE (a), GNP/L-cys/Gr/GCE (b), Ab1/GNP/L-cys/Gr/GCE (c), Ag/Ab1/GNP/L-cys/Gr/GCE (d) and HRP–Ab2/Ag/Ab1/GNP/L-cys/Gr/GCE (e) in 5 mM K3Fe(CN)6 with 0.1 M KCl. | ||
Impedance spectroscopy was reported as an effective method to monitor the feature of surface allowing the understanding of chemical transformation and processes associated with the conductive electrode surface.22Fig. 2B shows the Nyquist plots of EIS for the L-cys/Gr/GCE (a), GNP/L-cys/Gr/GCE (b), Ab1/GNP/L-cys/Gr/GCE (c), Ag/Ab1/GNP/L-cys/Gr/GCE (d) and HRP–Ab2/Ag/Ab1/GNP/L-cys/Gr/GCE (e). At a Gr modified GCE, the redox process of the [Fe(CN)6]3−/4− probe showed an electron transfer resistance of 120 Ω (not shown here). With the electrodeposition of L-cys attached to the surface of Gr (curve a), the resistance increased, for L-cys is a protein blocking the electron transferring of the probe. However, GNP/L-cys/Gr/GCE (curve b) shows smaller resistance. When Ab1 molecules were assembled on the GNP/L-cys/Gr/GCE, the electron-transfer resistance increased (curve c). The results were consistent with the observation from CV as shown in Fig. 2A. And then the specific interaction of Ag and Ab1 made the resistance increase again (curve d). At last, after the HRP-labelled Ab2 interacting with Ag, the resistance increased even more because of the increasing ratio of protein (curve e).
3.2. Performance of the immunosensor
Enzyme amplification is popularly used for signal enhancement in electrochemical immunosensor; substrates such as HQ, one of the best electron transfer mediators for HRP,23 are usually added to the detection solution as a mediator to transfer the electron between HRP and H2O2 for HRP-label based electrochemical biosensors. In the presence of H2O2, the HRP molecules immobilized by immunoreaction catalyze the oxidation of HQ into benzoquinone whose subsequent reductive current can be monitored. Fig. 3A, B and C shows the CVs of GNP/L-cys/Gr/GCE, HRP–Ab–Ag complex on GNP/L-cys/GCE or GNP/L-cys/Gr/GCE in PBS (pH 7.4) solutions with and without the addition of HQ and H2O2. No redox peaks were observed at GNP/L-cys/Gr/GCE, HRP–Ab–Ag complexes covered GNP/L-cys/GCE and GNP/L-cys/Gr/GCE in pH 7.4 PBS (curves a in Fig. 3A, B and C). An oxidation/reduction peak couple, which represents the CV of HQ, was observed of curves b in Fig. 3A, B and C. However, with the addition of 2.0 mmol L−1 H2O2, on the HRP–Ab2 labelled immunosensor the CV displayed a well defined reduction peak at −0.059 V with dramatic enhancement of the cathodic peak current followed by a concomitant decrease of the anodic peak current (curves c in Fig. 3B and C), and no obvious change occurred on the GNP/L-cys/Gr/GCE (Fig. 3A curve c). That is, the HRP labelled Ab2 was captured to the electrode surface and resulted in an enzymatic amplification enhancing the reduction peak current in the presence of HQ and H2O2. Here the HRP immobilized in the immunosensor surface retained high enzymatic catalytic activity. In addition, HRP–Ab–Ag complex on GNP/L-cys/GCE without Gr (curve c in Fig. 3B) shows about 10-fold weaker response in the same solutions comparing with the immunosensor with Gr (curve c in Fig. 3C). We suggest that Gr provided an electric substrate matrix for the adsorption of GNP by L-cys, enhanced the immobilized amount of GNP, not only facilitated more Ab1 molecules to adsorb on the GNP surface, but also improved the electron transffering of the immunosensor. We also investigated the importance of L-cys. Under the same conditions, we directly assemblied the GNP on the Gr/GCE to prepare the platform without the L-cys. Comparing with the platform with L-cys, the immunosensor without L-cys showed lower response towards the same amount of the analytes (Fig. 3). This proved that L-cys plays an important role in the platform. We suggest that regular growth of L-cys may improve the assembly amount of GNP, which may further improve the loading amount of the Ab1 and result in a higher response.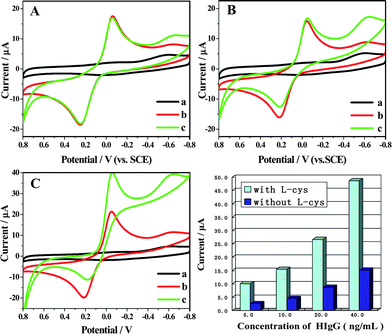 | ||
| Fig. 3 CVs of different modified electrodes in 50 mmol L−1 pH 7.4 PBS (a), 50 mmol L−1 pH 7.4 PBS containing 2.0 mmol L−1 HQ (b) and 50 mmol L−1 pH 7.4 PBS containing 1.0 mmol L−1 HQ and 2.0 mmol L−1 H2O2 (c), scan rate: 100 mV s−1: (A) GNP/L-cys/Gr/GCE (B) HRP–Ab–Ag complexes/GNP/L-cys/GCE (without Gr) and (C) HRP–Ab–Ag complexes/GNP/L-cys/Gr/GCE (with Gr). (D) The response of different platform (with and without the crosslinker of L-cys) of the immunosensor to different concentrations of the HIgG. | ||
3.3. Investigation of non-specific adsorption
Inhibition of nonspecific binding (NSB), which generally arises from electrostatic interaction between the sensor surface and the serum components, was critical to achieve the best sensitivity and detection limits. Omission of any of the washing steps below resulted in significantly increased NSB. Before exposure to the sample, the immunosensors were incubated for 1 h with 50 μL 5% BSA in PBST, then washed with PBST. For measurements, 60 μL different concentrations of Ag was incubated on the sensor surface, PBST was used to wash, then the sensor was incubated with HRP–Ab2. It can be seen that the response signal of the GNP/L-cys/Gr modified electrode is very low (Fig. 4 curve a, 0.538 μA), demonstrating a relatively low background current. The current response of the immunosensor without Ag (Fig. 4 curve b, 1.916 μA) is 4 times larger than that of GNP/L-cys/Gr modified electrode, illustrating physic adsorption existent in some degree. However DPV signals were enhanced in the presence of different concentrations of Ag (as follows).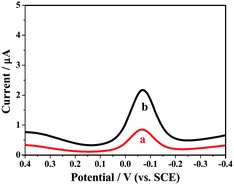 | ||
| Fig. 4 DPV of (a) GNP/L-cys/Gr/GCE, (b) as-prepared immunosensor without Ag in 50 mmol L−1 pH 7.4 PBS containing 1.0 mmol L−1 HQ and 2.0 mmol L−1 H2O2. | ||
3.4. Optimization of experimental conditions
Parameters of the assay procedure, affecting the response of the immunosensor, were investigated. To find the proper immune pH, we investigated the influence of pH on the performance of the immunosensor over the pH range of 6.0–9.0 (Fig. 5A). Highly acidic or alkaline surroundings shows poor response for they would damage the immobilized protein, especially in alkalinity. An ideal pH of 7.4 was chosen for the assay experiments, which is also a general pH value in immunology.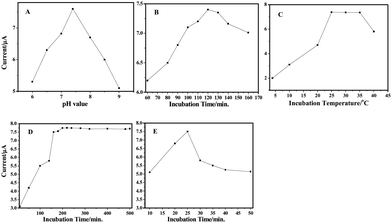 | ||
| Fig. 5 Effects of (A) pH of detection solution, (B) incubation time, (C) incubation temperature, (D) the amount of Ab1 and (E) Ab2–HRP at the immunosensor. | ||
The time-course of the IgG assembly on the hybrids was investigated by the electrochemical response signal to optimize the immobilization time (Fig. 5B). The experimental data indicate that assembly time greatly influences the response signal, which implies that the adsorption quantity of antibody relies much on the time accretion. In this work, an incubation time of 120 min could be recommended.
An additional parameter that affected the assay was the incubation temperature. Various incubation temperatures have been reported in the literature, ranging from 25 to 37 °C (Fig. 5C). As well-known, an optimal temperature of the immunoreaction would be 37 °C. The enzyme used exhibited the best activity at the temperature from 20 to 25 °C. A higher temperature would be harmful to its activity. The effect of the incubation temperature on response current was examined from 4 to 40 °C. It was found that the signal increases with an increase of temperature up to 25 °C, and then it decreases at higher temperatures. Therefore, 25 °C was chosen as the optimum temperature for the incubation of the immunosensor.
The effect of the concentration of rabbit anti-hIgG antibody (Ab1) from 10 to 500 μg mL−1 on the immobilization efficiency was investigated (Fig. 5D). Our experimental data revealed that the reductive current increased with the increasing antibody concentration till it starts to reach a plateau at 150 μg mL−1 and the highest response is observed at 200 μg mL−1; whereas, gradually increased concentration leads to a slight decrease signal. The possible reason is that only antibodies adsorbed on the hybrids with suitable orientation can participate in the immunoreaction and higher concentration might increase disorder in alignment of adsorbed antibody and steric hindrance of immunoreaction. Therefore, 200 μg mL−1 Ab1 was chosen in the present experiment.
The response signal of the immunosensor depends on the amount of HRP labelled Ab2 conjugate bound to the surface of the immunosensor, which, in turn, corresponds to the concentration of HRP in the incubating buffer. In order to obtain a maximum response with a minimum amount of HRP labelled Ab2, the immunosensor was incubated with various concentrations of HRP labelled Ab2 (Fig. 5E). The response current increased up to 25 μL of HRP–Ab2 (in 1 mL of the incubation buffer) and then tended to saturate. Consequently, 25 μL of HRP labelled Ab2 (1.0 mg mL−1) solution, added to 1 mL of final incubation solution, were routinely employed in these assays.
3.5. Analytical performance characteristics
In the present study, anti-IgG-HRP conjugate was used as a tracer and hydroquinone and H2O2 as the enzyme substrates. The amount of anti-IgG-HRP could be measured via the reduction of enzyme-catalyzed reaction product, which was proportional to the IgG concentration. Fig. 6A shows the typical DPV signals are enhanced in the presence of different concentrations of HIgG under the optimized conditions (final results were obtained following background subtraction). Fig. 6B shows the plot of background-subtracted peak current versus the logarithm of the concentration of Ag. The corresponding calibration plot was linear over the range from 0.2 to 320 ng mL−1 and was suitable for quantitative analysis. The response obtained with a protein target concentration of 0.2 ng mL−1 indicated a detection limit of around 70 pg mL−1 obtained by 3 S/N. Higher serum Ag levels could be detected with an appropriate dilution. General immunoassays such as ELISA (enzyme-linked immunosorbent assay) have detection limits in the pmol L−1 range; 1 pmol L−1 corresponds to 150 pg mL−1 for IgG.24 Our electrochemical immunosensor is highly sensitive compared to ELISA, and it is comparable to the immunosensor using funtionalized liposomes.25 The wide linear range and high sensitivity can be attributed to the large surface area of the Gr and orderly orientating L-cys inducing gold nanoparticles assembling, on which can absorb large amounts of protein molecules with spatial direction and can transfer electrons very well, thereby, improving significantly the performance of the resulting biosensing. Besides, the enzyme amplification reaction, herein, also promotes the sensitivity and extends the detection range.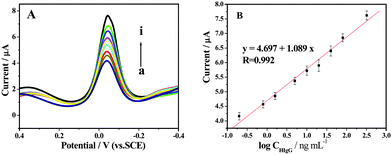 | ||
| Fig. 6 (A) Typical DPV of electrochemical immunoassay in 50 mM pH 7.4 PBS containing 1.0 mM HQ and 2.0 mM H2O2 with increasing HIgG concentration from (a) to (i) (0.2, 0.8, 1.6, 5.0, 10, 20, 40, 80 and 320 ng mL−1 HIgG, respectively). (B) The resulting calibration curve of HIgG plotted on a semi-log scale. | ||
3.6. Selectivity, regeneration, reproducibility and stability of the immunosensor
To further characterize the selectivity of the immunosensor, we detected the signal of a mixture containing 5 ng mL−1 BSA, 5 ng mL−1 CEA, and 0.5 ng mL−1 HIgG, and compared to that of the pure 0.5 ng mL−1 HIgG, then the 6.2% of RSD was obtained, indicating the immunosensor has good selectivity for HIgG. The regeneration of the proposed immunosensor was tested by washing the immunosensor with a stripping buffer of 0.1 mol L−1 pH 3.5 glycine–HCl solution for the purpose of removing HIgG and Ab2 from Ab1 after each determination. In the experiment, consecutive measurements were repeated eight times and a R.S.D. of 7.0% was acquired. The reproducibility of the immunosensor was evaluated from the response to 50 ng mL−1 HIgG Ag at eight different electrodes. A series of eight measurements from the batch resulted in a relative standard deviation of 5.1%, indicating good electrode-to-electrode reproducibility of the fabrication protocol described above. When not in use, the immunosensor was first regenerated and then Ab1/GNP/L-Cys/Gr modified electrode was stored in the refrigerator at 4 °C to be preserved. The stability was investigated over a 30-day period when the sensor was stored at 4 °C. It was measured intermittently (every 3–5 days), no obvious change of the response was found over this period, indicating the effective retention of the activity of the immobilized Ab. A long storage period of 30 days also indicated that the GNP/L-cys/Gr composite structure was very efficient for retaining the bioactivity of the immobilized Ab and preventing the Ab from leakage. And in controlling experiment with the GNP/L-cys film without graphene under the same conditions, by the detection of the response current, the stability was observed about 22-fold weaker.3.7. Application of the immunosensor in human serum
The feasibility of the immunoassay system for clinical applications was investigated by analyzing several real samples. In comparison with the ELISA method, these serum samples were diluted to different concentrations with a PBS of pH 7.4. Table 1 describes the correlation between the results obtained by the proposed immunosensor and the ELISA method. As the result shows, it is feasible for detection of antigen with this immunosensor.| Samples IgG | IgG concentration by ELISAa/ng mL−1 | IgG concentration by EIAb/ng mL−1 |
|---|---|---|
| a Enzyme-linked immunosorbent assay method. b Electrochemical immunosensing assay method. As the IgG concentration level of human serum is very high, the human serum specimens have to be diluted to the detection range of the electrochemical immunosensors by using PBS (pH 7.4) buffer according to the IgG concentration by ELISA. | ||
| 1 | 52.5 | 54.1 |
| 2 | 105.1 | 97.9 |
| 3 | 248.1 | 243.8 |
| 4 | 306.2 | 301.5 |
| 5 | 325.0 | 316.9 |
4. Conclusion
It has been demonstrated that GNP/L-cys/Gr can be used to construct an effective biointerface to immobilize the antibody for the electrochemical immunosensor. Gr as the substrate not only enlarged the surface, increased the electron transferring capability but also induced the regular growth of L-cys which further increased the conjugation of GNP, resulting in the increasing loading amount of biomolecules. And thus the immunosensor showed enhanced sensitivity, low detection limit, and high stability. It extends the excellent material, graphene, in the study of biointerfaces for immunosensors. The proposed method also provides promising potential in clinical applications.Acknowledgements
This work was financially supported by the projects (20901003, 21073001 and 21005001) from the National Natural Science Foundation of China, the Natural Science Foundation of Anhui (KJ2009B013Z) and the project of Anhui Key Laboratory of Controllable Chemistry Reaction & Material Chemical Engineering (OFCC0905).Notes and references
- M. Rieger, C. Cervino and J. Sauceda, Anal. Chem., 2009, 81, 2373 CrossRef CAS; A. Wolter, R. Niessner and M. Seidel, Anal. Chem., 2008, 80, 5854 CrossRef CAS.
- R. E. Ionescu, C. Gondran, L. A. Gheber, S. Cosnier and R. S. Marks, Anal. Chem., 2004, 76, 6808 CrossRef CAS; X. Yu, B. Munge, V. Patel, G. Jensen, A. Bhirde, J. D. Gong, S. N. Kim, J. Gillespie, J. S. Gutkind, F. Papadimitrakopoulos and F. James, J. Am. Chem. Soc., 2006, 128, 11199 CrossRef CAS; A. Bange, H. B. Halsall and W. R. Heineman, Biosens. Bioelectron., 2005, 20, 2488 CrossRef CAS; E. N. Kalmykova, E. S. Dergunova, T. N. Ermolaeva, R. P. Gorshkova and N. A. Komandrova, Appl. Biochem. Microbiol., 2008, 44, 372 Search PubMed; A. Ulman, Chem. Rev., 1996, 96, 1533 CrossRef CAS.
- M. S. Wilson, Anal. Chem., 2005, 77, 1496 CrossRef CAS; R. Ehrnstrom, Lab Chip, 2002, 2, 26 Search PubMed.
- T. H. Ha, S. O. Jung, J. M. Lee, K. Y. Lee, Y. Lee, J. S. Park and B. H. Chung, Anal. Chem., 2007, 79, 546–556 CrossRef CAS; Y. Bai, C. G. Koh, M. Boreman, Y. J. Juang, I. C. Tang, L. J. Lee and S. T. Yang, Langmuir, 2006, 22, 9458 CrossRef CAS.
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2006, 128, 3939 CrossRef CAS.
- A. Bernard, B. Michel and E. Delamarche, Anal. Chem., 2001, 73, 8 CrossRef CAS; W. Y. Yuan, H. Dong, C. M. Li, X. Q. Cui, L. Yu, Z. S. Lu and Q. Zhou, Langmuir, 2007, 23, 13046 CrossRef CAS.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS; X. L. Li, G. Y. Zhang, X. D. Bai, X. M. Sun, X. R. Wang, E. Wang and H. J. Dai, Nat. Nanotechnol., 2008, 3, 538 CrossRef CAS; Y. B. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS; G. Williams, B. Seger and P. V. Kamat, ACS Nano, 2008, 2, 1487 CrossRef CAS; Y. X. Xu, H. Bai, G. W. Lu, C. Li and G. Q. Shi, J. Am. Chem. Soc., 2008, 130, 5856 CrossRef CAS.
- T. Cassagneau and J. H. Fendler, Adv. Mater., 1998, 10, 877 CrossRef CAS.
- S. Gilje, S. Han, M. Wang, K. L. Wang and R. B. Kaner, Nano Lett., 2007, 7, 3394 CrossRef CAS.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652 CrossRef CAS.
- J. S. Bunch, A. M. van der Zande, S. S. Verbridge, I. W. Frank, D. M. Tanenbaum, J. M. Parpia, H. G. Craighead and P. L. McEuen, Science, 2007, 315, 490 CrossRef CAS.
- D. P. Tang, R. Yuan, Y. Q. Chai and Y. Z. Fu, Electrochem. Commun., 2005, 7, 177 CrossRef CAS; C. Y. Wang, D. Li, C. O. Too and G. G. Wallace, Chem. Mater., 2009, 21, 2604 CrossRef CAS; S. Alwarappan, A. Erdem, C. Liu and C. Li, J. Phys. Chem. C, 2009, 113, 8853 CrossRef CAS; E. Yoo, T. Okata, T. Akita, M. Kohyama, J. Nakamura and I. Honma, Nano Lett., 2009, 9, 2255 CrossRef CAS; X. J. Kang, J. Wang, H. Wu, I. A. Aksay, J. Liu and Y. H. Lin, Biosens. Bioelectron., 2009, 25, 901 CrossRef CAS; Y. Zhuo, R. J. Yu, R. Yuan, Y. Q. Chai and C. L. Hong, J. Electroanal. Chem., 2009, 628, 90 CrossRef CAS.
- Z. Zhong, W. Wei, D. Wang, J. L. Shan, Y. Qing and Z. M. Zhang, Biosens. Bioelectron., 2010, 25, 2379, DOI:10.1016/j.bios.2010.03.009; D. Du, Z. Zou, Y. Shin, J. Wang, H. Wu, M. H. Engelhard, J. Liu, I. A. Aksay and Y. Lin, Anal. Chem., 2010, 82, 2989 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS; N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547 CrossRef CAS.
- W. Hummers and R. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS; N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771 CrossRef CAS.
- C. S. Shan, H. F. Yang, J. F. Song, D. X. Han, A. Ivaska and L. Niu, Anal. Chem., 2009, 81, 2378 CrossRef CAS.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20 Search PubMed.
- C. S. Shan, H. F. Yang, J. F. Song, D. X. Han, Q. X. Zhang, A. Ivaska and L. Niu, Biosens. Bioelectron., 2010, 25, 1070–1074 CrossRef CAS.
- L. H. Tang, Y. Wang, Y. M. Li, H. B. Feng, J. Lu and J. H. Li, Adv. Funct. Mater., 2009, 19, 2782 CrossRef CAS; J. B. Jia, B. Q. Wang, H. Yuan and S. J. Dong, Anal. Chem., 2002, 74, 2217 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Wiley, New York, 1980 Search PubMed.
- R. E. Ionescu, S. Cosnier, S. Herrmann and R. S. Marks, Anal. Chem., 2007, 79, 8662 CrossRef CAS.
- Z. Dai, F. Yan, J. Chen and H. X. Ju, Anal. Chem., 2003, 75, 5429 CrossRef CAS; M. A. Aziz, S. Park, S. Jon and H. Yang, Chem. Commun., 2007, 2610 RSC.
- L. Alfonta, A. K. Singh and I. Willner, Anal. Chem., 2001, 73, 91 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |